

Abstract
A novel class of antibiotic acyldepsipeptides (designated ADEPs) exerts its unique antibacterial activity by targeting the peptidase caseinolytic protease P (ClpP). ClpP forms proteolytic complexes with heat shock proteins (Hsp100) that select and process substrate proteins for ClpP-mediated degradation. Here, we analyse the molecular mechanism of ADEP action and demonstrate that ADEPs abrogate ClpP interaction with cooperating Hsp100 adenosine triphosphatases (ATPases). Consequently, ADEP treated bacteria are affected in ClpP-dependent general and regulatory proteolysis. At the same time, ADEPs also activate ClpP by converting it from a tightly regulated peptidase, which can only degrade short peptides, into a proteolytic machinery that recognizes and degrades unfolded polypeptides. In vivo nascent polypeptide chains represent the putative primary target of ADEP-activated ClpP, providing a rationale for the antibacterial activity of the ADEPs. Thus, ADEPs cause a complete functional reprogramming of the Clp–protease complex.
Keywords: ADEP, antibiotic, ClpP, Hsp100, Clp proteins, proteolysis
INTRODUCTION
In view of the worldwide rise and spread of antibiotic resistance, the identification of novel targets and modes of action of antibacterial compounds is a vital objective (Levy & Marshall, 2004; Wright, 2007). In such an effort, the acyldepsipeptides (ADEPs), a new class of antibacterial compounds, were recently characterized and optimized (Hinzen et al, 2006). It could be demonstrated that ADEPs operate by targeting caseinolytic protease P (ClpP), the proteolytic core of bacterial ATP-dependent proteases, an activity that is distinct from all other antibiotic mechanisms known so far (Brötz-Oesterhelt et al, 2005). Due to this unprecedented mode of action ADEPs are not affected by resistance mechanisms compromising antibiotics in therapeutic application and their antibacterial activity against Gram-positive bacteria, including multi-resistant isolates, matches that of marketed antibiotics in vitro as well as in several rodent models of bacterial infection (Brötz-Oesterhelt et al, 2005).
ClpP is a central mediator of general and regulatory proteolysis in bacteria. General proteolysis is an important cellular process, essential for protein homeostasis and necessary for protein quality control, ensuring the proper function of proteins in their environment (Bukau et al, 2006; Hartl & Hayer-Hartl, 2002; Sauer et al, 2004; Wickner et al, 1999). Regulatory proteolysis is utilized to control many cellular adaptations or developmental processes on various levels such as transcriptional regulation or remodelling of the proteome. Examples of central cellular processes controlled by regulated proteolysis via the ClpP proteolytic machinery include cell motility, genetic competence, cell division, cell differentiation, sporulation and the expression of virulence factors (Frees et al, 2007; Gottesman, 2003; Jenal & Hengge-Aronis, 2003; Sauer et al, 2004). A prerequisite for steering such a widespread regulatory network by proteolysis is that protein degradation is highly selective and tightly controlled. This is achieved by compartmentalized ATP-dependent proteases, consisting of a proteolytic core (e.g. ClpP) and several associated adenosine triphosphatases (ATPases), often heat shock proteins (Hsp100) of the AAA+ protein family (e.g. ClpA, ClpC and ClpX). The architecture of the proteolytic ClpP core plays an important role in proteolysis regulation. The barrel-like ClpP tetradecamer forms an internal chamber decorated with proteolytic active sites. Access to this secluded chamber is restricted by narrow pores that only allow for the passage of small peptides but not of polypeptides (Wang et al, 1997). In consequence, ClpP on its own only exhibits proteolytic activity towards short peptides comprising up to six amino acids. For the degradation of polypeptides or proteins the Hsp100 component of the proteolytic complex is mandatory. The Hsp100 ATPases, which form ring-forming multimers themselves, mediate substrate selection either directly or indirectly via cooperating adaptor proteins. They unfold bound substrates and thread them into the proteolytic chamber of ClpP in an ATP-consuming reaction. The proteolytic activity of ClpP is thereby tightly regulated by the associated ATPases (Dougan et al, 2002a; Horwich et al, 1999; Lupas et al, 1997; Sauer et al, 2004; Wickner et al, 1999).
ADEPs interfere with the proteolytic activities of ClpP in both Gram-negative and -positive bacteria resulting in inhibition of cell division and finally cell death (Brötz-Oesterhelt et al, 2005). It could be demonstrated previously that ADEPs interact with Bacillus subtilis ClpP, converting ClpP in vitro from a peptidase to a protease that degrades the unfolded model polypeptide casein in the absence of a cooperating Hsp100 protein. However, the molecular mechanism of the ADEP-mediated ClpP activation and the identification of cellular targets of ADEP-activated ClpP had not been investigated. Elucidating the molecular details of the ADEP mechanism is of major interest both for the development of antibiotics acting in the same pathway and also for a better understanding of the regulation, activation, interaction and cooperation of components of the bacterial Clp–protease complex. In this work, we studied the mechanism of ADEP-mediated ClpP dysregulation at a molecular level, analysing various Hsp100–ClpP complexes of both Escherichia coli and B. subtilis in in vitro systems as well as in intact bacterial cells. We observed that ADEPs trigger a thorough functional reprogramming of the ClpP–protease complex, preventing its physiological functions and redirecting the ClpP core to uncontrolled and deleterious degradation of unfolded substrates.
RESULTS
ADEPs activate E. coli and B. subtilis ClpP to degrade casein with reduced processivity
In the present study, we used two representatives of the ADEP class which were previously characterized (Brötz-Oesterhelt et al, 2005). ADEP1 represents a natural product and ADEP2 a synthetic congener with improved activity against Gram-positive bacteria but reduced Gram-negative activity. Therefore, we employed ADEP1 for our experiments with E. coli strains and proteins and ADEP2 for the experiments with B. subtilis strains and proteins. We will subsequently use the term ADEP for both compounds.
An initial characterization showed that ADEP activates B. subtilis ClpP to degrade the unfolded model protein casein in the absence of a cooperating Hsp100 protein. To generalize this finding, we monitored the proteolytic activity of E. coli ClpP towards fluorescein isothiocyanate (FITC)-labelled casein in the absence or presence of ADEP. The casein used in this assay is heavily labelled with the fluorophore FITC, resulting in almost complete quenching of the conjugates' fluorescence. Action of the protease is detected by fluorescence increase due to a quenching reduction upon release of short peptides. ADEP also activated E. coli ClpP to degrade FITC-casein as monitored by increased substrate fluorescence (Fig 1A). Notably, the increase in FITC fluorescence was less when compared to FITC-casein degradation by ClpA–ClpP complexes (Fig 1A). The observed differences in FITC-fluorescence were not caused by different efficiencies in substrate degradation, since both ADEP-activated ClpP and ClpA/ClpP completely degraded casein with similar kinetics (Fig 1B). The reason for the differences observed became apparent when we monitored the hydrolysis of high quantities of casein by either ADEP-activated ClpP or ClpA/ClpP. While both machineries efficiently degraded full length casein, the molecular size of accumulating casein degradation products differed between the systems: degradation products of intermediate size were only observed in the case of ADEP-activated ClpP, implying reduced processivity of casein hydrolysis by ADEP-activated ClpP compared to ClpA/ClpP (Fig 1C). In the case of FITC-casein, the intermediate size degradation products still contained several fluorophores and were thus subject to a certain degree of quenching (Fig 1A). Similar findings were obtained when casein degradation by B. subtilis ClpP/ADEP and ClpC/ClpP/MecA was compared (Fig 1D). Thus, ADEP activates isolated ClpP in the absence of Clp-ATPases to degrade unfolded polypeptides albeit with reduced processivity.
Figure 1. ADEP activates ClpP to degrade casein with reduced processivity.
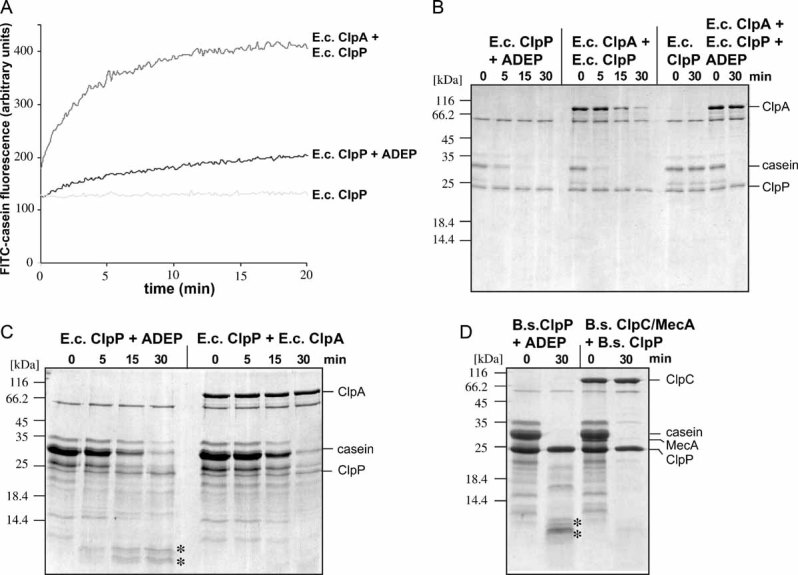
A. Degradation of FITC-labelled casein (100 nM) by E. coli ClpA/ClpP, ClpP/ADEP or ClpP only.
B. Degradation of casein by the indicated E. coli (A–C) and B. subtilis (D) components was monitored by SDS–PAGE. (B) 1 µM casein,
C, D. 20 µM casein. A protein standard is given. The positioning of casein and the respective proteins is indicated. E.c. indicates from E. coli and B.s. indicates from B. subtilis. Casein degradation products of intermediate size are labelled with ‘*’.
ADEP triggers oligomerization of B. subtilis ClpP
We have recently demonstrated that, unlike E. coli ClpP, which assembles into a double heptameric ring, ClpP of B. subtilis is a monomer and only forms the double-heptameric structure when interacting with the ClpC hexamer (Kirstein et al, 2006). A possible ADEP-mediated activation of ClpP by, e.g., assisting or influencing its assembly to the oligomeric state was therefore investigated using ClpP from B. subtilis. The results of size exclusion chromatography experiments are depicted in Fig 2 and demonstrate that in the presence of ADEP B. subtilis ClpP forms a higher oligomeric species, whose size is consistent with a double heptameric ClpP complex (Peak 1 in Fig 2A, about 300 kDa), while ClpP on its own elutes as a monomer (Peak 2 in Fig 2A, about 25 kDa). We tested the casein degradation activity of ClpP species isolated from both fractions and observed that only ADEP-treated ClpP from the higher oligomeric species (Peak 1) was degrading casein (Fig 2A). This experiment demonstrates that ADEP-binding to B. subtilis ClpP results in conformational changes allowing for ClpP oligomerization and activation.
Figure 2. ADEP binding triggers oligomerization of B. subtilis ClpP.
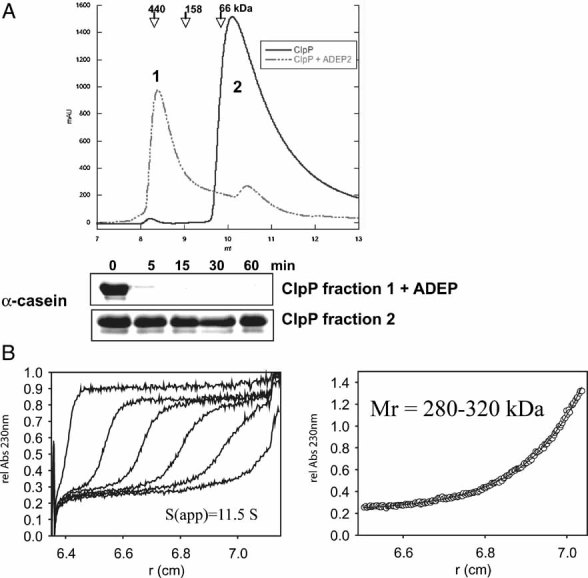
- The oligomeric state of ClpP in the absence (solid line) and presence (dashed line) of ADEP (0.5 µg/ml) was monitored by size exclusion chromatography using a Superdex 75 column. Elution positions of protein standards are given. Samples of both eluted peak fractions (as indicated in the chromatogram: 1: ClpP oligomer and 2: ClpP monomer) were incubated with 1 µM casein and tested for proteolytic activity by SDS–PAGE analysis.
- The association state of 10 µM ClpP in the presence of 12 µM ADEP was analysed by analytical ultracentrifugation. The left panel shows the sedimentation velocity at 40,000 rpm and 20 °C (scans taken after every 20 min intervals depicted). The calculated sedimentation velocity was s(app) = 11.5 S. The right panel displays the data of a sedimentation equilibrium experiment after 60 h at 6,000 rpm and 20 °C. In several independent experiments, the molecular mass of ClpP was determined over the range of 280–320 kDa, fitting well to the size of a double heptameric structure of ClpP. The offset of A230 nm = 0.23 in both sedimentation velocity and equilibrium experiments arises from non-bound ADEP, which has been used in a slight molar excess over ClpP.
We confirmed the ADEP induced oligomerization of ClpP also under equilibrium conditions by analytical ultracentrifugation (Fig 2B). Sedimentation velocity as well as sedimentation equilibrium experiments demonstrate that upon incubation with ADEP ClpP switched from a monomeric state (about 25 kDa) to a oligomer, most likely comprising 14 subunits (about 300 kDa). Titration experiments with increasing concentrations of ADEP suggested a highly cooperative oligomerization of ClpP, which was maximally induced by equimolar amounts of ADEP in relation to monomeric ClpP (data not shown). However, since E. coli ClpP forms tetradecameric complexes in the absence of ADEP, ADEP-induced oligomerization of B. subtilis ClpP cannot solely explain ClpP activation, suggesting that additional structural changes probably contribute to this process.
ADEP-activated ClpP cannot degrade folded proteins and does not cooperate with Hsp100 proteins
We next tested whether ADEP-activated ClpP can also degrade natively folded substrates that require protein unfolding prior to degradation. In contrast to the unfolded polypeptide casein, no known folded model substrates including B. subtilis MecA, McsB, ComK, green fluorescent protein (GFP)-SsrA, the N-end rule model substrate FR-linker-GFP and other natively folded proteins such as E. coli GroEL, Trigger Factor and DnaK were degraded in vitro by ADEP-activated ClpP (data not shown). Many of these substrates had been previously described as substrates for ClpP in concert with one of the Hsp100 ATPases. We therefore tested whether the addition of the substrate processing Hsp100 partner protein restores degradation of folded substrates by ADEP-activated ClpP. Surprisingly, the presence of ADEP largely prevented the degradation of the N-end rule model substrate FR-linker-GFP by the N-end rule specific ClpA/ClpP/ClpS proteolytic system, raising the possibility that ADEP not only affects ClpP but also ClpA/ClpS function (Fig 3A, left panel). To test an inactivation of substrate processing by ClpA/ClpS in the presence of ADEP, we followed the unfolding of FR-linker-GFP by ClpA/ClpS in the absence of ClpP. Such experiments were performed in the additional presence of the GroEL-D87K variant (GroEL-trap) that prevents refolding of unfolded FR-linker-GFP. Substrate unfolding by ClpA/ClpS was not influenced by ADEP addition, demonstrating that the activities of the adaptor protein ClpS and the Hsp100 protein ClpA in substrate recognition and processing are not affected (Fig 3A, right panel). Similar findings were obtained when we monitored the degradation of SsrA-tagged GFP by either ClpA/ClpP or E. coli ClpX/ClpP in the absence or presence of ADEP: ADEP exclusively prevented the ClpP-dependent degradation of GFP-SsrA but not the ClpP-independent Hsp100-mediated unfolding of the model substrate (Fig 3B, data not shown). Along the same line, the ClpC/ClpP/MecA-mediated autodegradation of MecA, the ClpC/ClpP/MecA mediated degradation of ComK and Spx as well as the B. subtilis ClpX/ClpP mediated degradation of Spx was inhibited by ADEP (Fig 3C). The addition of ADEP did not interfere with the ATPase activities of ClpC and ClpX from B. subtilis (data not shown), confirming that ADEP does not exert its effects by acting on the Hsp100 components and suggesting that ADEP abrogates the cooperation between ClpP and its Hsp100 partner proteins. Finally, we investigated the effect of ADEP on the relative amount of the ClpC/ClpP substrate MurAA from B. subtilis (Kock et al, 2004) in intact bacterial cells. ADEP addition resulted in increased MurAA levels, implying reduced ClpP-dependent proteolysis of the folded model substrate upon ADEP treatment of B. subtilis cells (Fig 3D). Together these findings demonstrate that binding of ADEP to ClpP prevents the cooperation of ClpP and Hsp100 proteins in degrading folded protein substrates.
Figure 3. ADEP abrogates the cooperation of ClpP and its Hsp100 partner proteins.

- Degradation and unfolding of the N-end rule model substrate FR-linker-GFP (100 nM) was followed by monitoring the decrease of GFP fluorescence in the presence of the indicated components (+/− ADEP).
- Degradation and unfolding of GFP-SsrA (100 nM) was followed by monitoring the decrease of GFP fluorescence in the presence of the indicated components (+/− ADEP). The rate of GFP-SsrA degradation and unfolding determined in the absence of ADEP was set as 100%.
- In vitro degradation of MecA by ClpC/ClpP and ComK by ClpC/ClpP/MecA and Spx by either ClpC/ClpP/MecA or ClpX/ClpP in the absence and presence of ADEP. Substrate degradation was analysed by SDS–PAGE (all proteins were used at 1 µM).
- Analysis of the in vivo stability of MurAA in B. subtilis cells (wild type (wt) and ΔclpP mutant). ADEP (final concentration: 1.6 µg/ml) was added upon entry of cells into stationary phase and cells were incubated for an additional period of 2 h prior to harvesting. MurAA levels were determined by immunoblot analysis using MurAA-specific antibodies.
ADEPs prevent complex formation between ClpP and cooperating Hsp100 proteins
One possible reason for the noticed loss of ClpP/Hsp100 cooperation in the presence of ADEP is that ADEP addition disturbs the association of ClpP with the Hsp100 partner proteins. We tested this hypothesis in vitro by size exclusion chromatography experiments using ClpC, MecA and ClpP from B. subtilis as described previously (Kirstein et al, 2006). To ensure stable complex formation of ClpC/MecA/ClpP, we used a ClpC variant (ClpC-DoubleWalkerB (DWB)) that harbours mutational alterations in the Walker B motif of both AAA domains and is thereby arrested in the ATP state. When ADEP was added to a preformed (MecA6ClpC-DWB6)/(ClpP7)2/(MecA6ClpC-DWB6) complex (peak 1 in Fig 4A), the high molecular weight complex was no longer observed. Instead, a peak of the size of ClpC-DWB6MecA6 (peak 2 in Fig 4A) together with a second peak, whose size is consistent with ADEP-activated ClpP ((ClpP7)2-ADEP; peak 3 in Fig 4A), could be detected (Fig 4A). These findings demonstrate that ADEP triggers the dissociation of the Hsp100/ClpP complex. As a consequence, ClpP becomes disconnected from its ATPase partner proteins and can no longer fulfil its function in the degradation of folded substrates.
Figure 4. ADEP prevents complex formation between ClpP and cooperating Hsp100 proteins both in vitro and in bacterial cells.
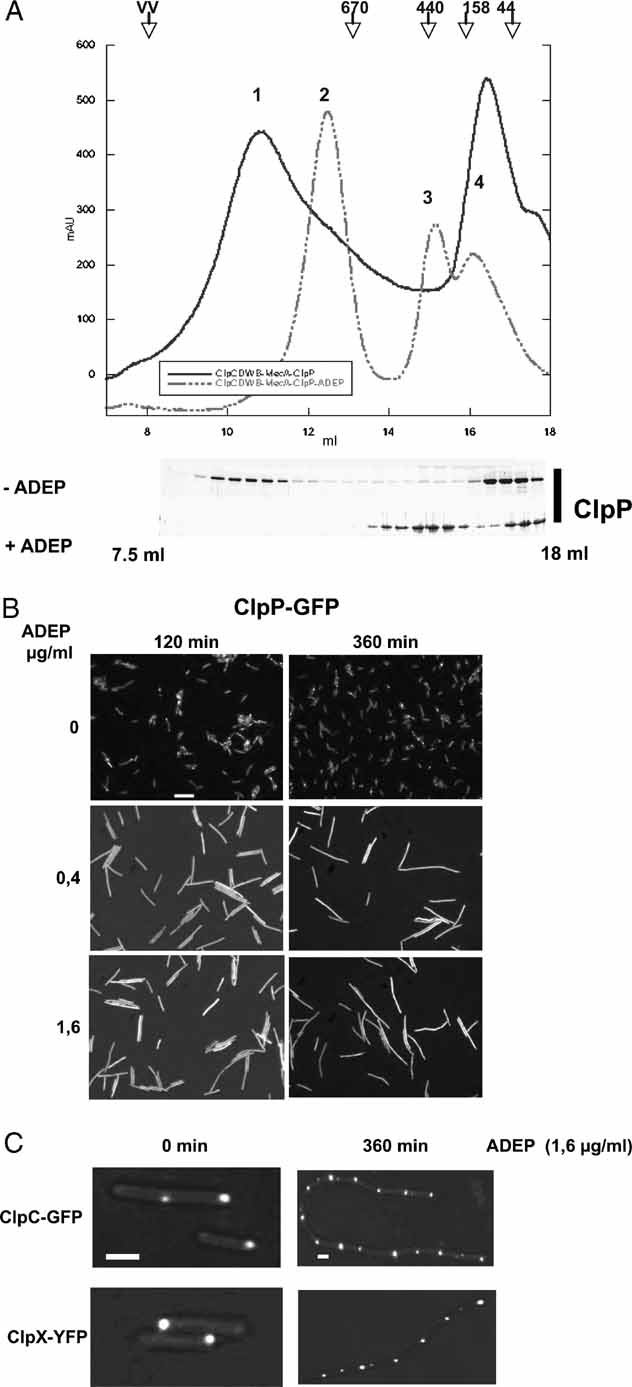
- A complex of ClpC-DWB, MecA and ClpP was preformed in the presence of 5 mM ATP at 37 °C for 10 min. ADEP (0.5 µg/ml) was subsequently added and the sample was further incubated for an additional 10 min. As a control, a sample without the addition of ADEP was analysed in parallel. The association and dissociation of the respective complexes were analysed by size exclusion chromatography using a Superose 6 column. The chromatogram of ClpC-DWB, MecA and ClpP is shown by a solid line and the chromatogram of the ADEP including sample by a dashed line. The resulting peaks (as indicated in the chromatogram) could be assigned as follows: 1: ClpC-DWB6MecA6/(ClpP7)2/ClpC-DWB6MecA6; 2: ClpC-DWB6/MecA6; 3: (ClpP7)2-ADEP and 4: ClpC_DWB/MecA heterodimer. The elution profile of ClpP is shown for both samples as a Coomassie-stained SDS gel. Elution positions of protein standards are given.
- ADEP causes a delocalization of ClpP in bacterial cells. Localization of ClpP-GFP in B. subtilis 168 in the absence (upper row) or presence of ADEP (0.4 and 1.6 µg/ml middle and bottom row, respectively) is shown. ADEP was added during the logarithmic growth phase (30 °C, OD600nm = 1) and ClpP-GFP localization was monitored 120 and 360 min after ADEP addition by fluorescence microscopy. A scale bar of 5 µm length is depicted.
- ADEP does not affect ClpC and ClpX localization in bacterial cells. Localization of ClpC-GFP and ClpX-YFP in B. subtilis 168 in the absence or presence of ADEP. Cells were treated as described in (B) and localization of fluorescent fusion proteins was analysed prior to and 360 min after the addition of ADEP. Scale bars are depicted for the left (1 µm) and right columns (2 µm).
We next investigated whether the ADEP-induced dissociation of ClpP from cooperating Hsp100 proteins can also be observed in vivo. We therefore monitored the cellular localization of ClpP, ClpC and ClpX in B. subtilis cells. To examine their distribution in vivo, we used C-terminal fusions of ClpP, ClpC and ClpX to either GFP or yellow fluorescent protein (YFP) (Kirstein et al, 2008). The gene fusions were introduced by Campbell integration into the B. subtilis chromosome, thereby replacing the original genes by the respective gfp- or yfp-fusions. All fusion strains thus created did not exhibit phenotypes that are connected to clpC, clpX or clpP B. subtilis mutant strains, demonstrating that the fusion proteins are fully functional (data not shown and Kirstein et al (2008)). By using these fluorescent fusion proteins we have shown in a previous study that the cellular localization of ClpP is determined by the cooperating Hsp100 proteins (Kirstein et al, 2008).
The results shown in Fig 4B demonstrate that ClpP, ClpC and ClpX cluster into visible foci mostly in the polar region of B. subtilis cells (Kirstein et al, 2008). ADEP treatment of cells caused the formation of large filaments, a phenotype that is reminiscent of B. subtilis ΔclpP mutant cells (Gerth et al, 1998; Msadek et al, 1998). In such ADEP-treated cells ClpP-GFP fluorescence was equally distributed throughout the elongated cell, indicating a cytosolic localization (Fig 4B). In contrast, ClpC and ClpX remained localized in foci as in the untreated cells (Fig 4C). Our results indicate that ClpC and ClpX determine the localization of the protease complex (Kirstein et al, 2006; Kirstein et al, 2008). ADEP treatment results in a loss of Hsp100-ClpP co-localization, demonstrating the observed protease complex dissociation (Fig 4) in vivo. In consequence, physiological ClpP substrates are no longer degraded, resulting in a clpP deletion phenotype.
ADEPs activate ClpP to degrade newly synthesized proteins in vitro
ADEP-bound ClpP can no longer associate with its Hsp100 partner proteins and, in consequence, cannot fulfil its function in general and regulated proteolysis. Loss of Hsp100-ClpP cooperation cannot, however, explain the bacteriocidal effect of the ADEPs since E. coli clpX, clpA or clpP null mutant cells do not exhibit a severe growth phenotype. While the corresponding B. subtilis mutants are affected in several developmental programmes, their phenotype is also far less severe than that of ADEP treated cells. Thus, ClpP activation through ADEP, rather than its inhibition, must represent the primary reason for antibacterial activity, allowing the peptidase to act independently from the associated Hsp100 proteins on novel substrates and leading to a dominant gain of function phenotype.
What are the natural targets of ADEP-activated ClpP? A first hint came from our observation that ADEP-activated ClpP can degrade casein, a soluble, misfolded model substrate, but not natively folded proteins (see Fig 1 and Fig 3). This observation suggests that newly synthesized polypeptides, prior to completion of their folding process, might act as the primary targets of ADEP-activated ClpP in vivo. To investigate whether ADEP-activated ClpP can indeed process newly synthesized proteins, we first monitored the potential of ADEP-activated E. coli ClpP to degrade nascent polypeptide chains in an E. coli in vitro transcription/translation system (t/t). First, we generated [35S]-methionine-radiolabelled nascent chains derived from different model proteins, including a truncated variant of isocitrate dehydrogenase (ICDH 1–410) and a gene fusion harbouring three copies of the folding deficient m10 variant of the α-spectrin SH3 domain (Blanco et al, 1999; Hoffmann et al, 2006). In our experimental setup, both substrates were stalled on ribosomes leading to their continuous exposure outside the ribosomal exit tunnel. ADEP and ClpP were added after translation of truncated ICDH or m10 and the stability of both exposed model substrates was determined. The simultaneous presence of both, ClpP and ADEP, caused degradation of the arrested ICDH and m10, in contrast to the presence of the single components that did not affect substrate stability (Fig 5A). These results demonstrate that nascent chains exposed at the ribosome are sensitive to proteolysis by ADEP-activated ClpP.
Figure 5. ADEP-activated ClpP degrades nascent polypeptides in vitro.
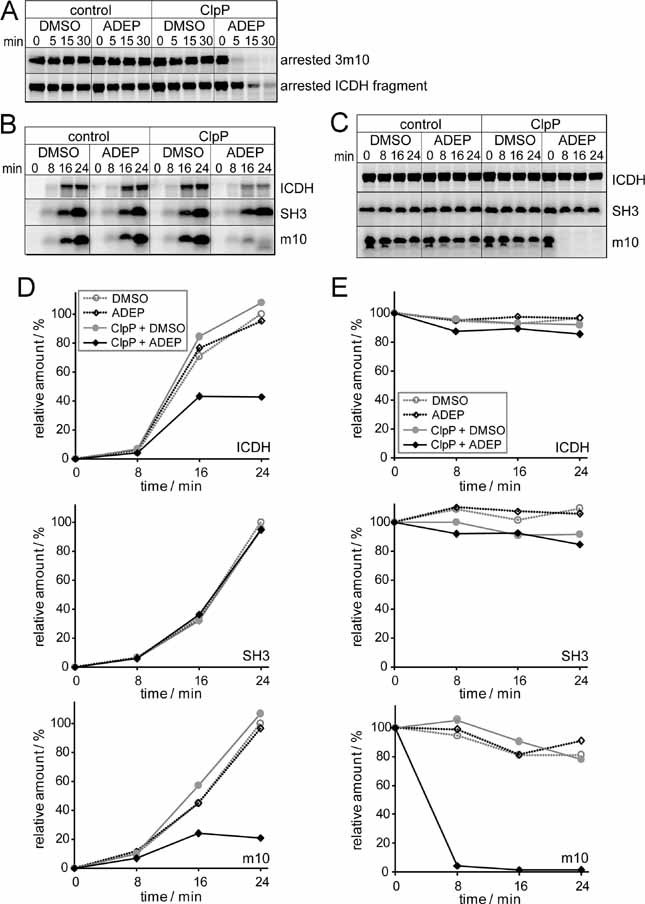
[35S] methionine-labelled model polypeptides (a ribosome-arrested ICDH fragment of 45 kDa, a ribosome-arrested 3m10-SecM chain of 26 kDa (A) and non-arrested full-length ICDH (46 kDa), SH3 and m10 (B and C) were synthesized in an E. coli-based transcription/translation system and analysed for degradation at 37 °C. Polypeptides were separated on tricine gels and visualized by autoradiography. Only the full-length products are depicted.
A. ClpP, ADEP and DMSO were added to the translation system after the generation of ribosome-arrested nascent chains.
B. ClpP, ADEP and DMSO were added to the translation system before the synthesis of full-length, non-arrested polypeptides.
C. ClpP, ADEP and DMSO added after the translation of full-length, released polypeptides.
D, E. Quantification of the degradation experiments shown in (B) and (C), respectively. The amount of full-length product at the end of translation under control conditions (DMSO without ClpP addition) was set at 100%. The data correspond to one representative experiment out of three and a second experiment, performed using another purification of the translation extract, is depicted in the Supporting Information Fig 1.
Next, we investigated whether ADEP-activated ClpP can also act on nascent polypeptides during an ongoing synthesis (Fig 5B and D). Here, full length ICDH (ICDH 1–416), SH3 wild type and its mutant form m10 were synthesized and ADEP-activated ClpP was added to the t/t system immediately before the initiation of translation (co-translational, Fig 5, Supportive Information). Synthesis of the rapidly folding SH3 wild type was only slightly affected by addition of ADEP plus ClpP, whereas its folding-deficient mutant variant m10 was massively degraded as evident from strongly reduced synthesis yields in the presence of both components. This finding demonstrates that substrate degradation by ADEP-activated ClpP is sensitive to protein conformation and confirms that ADEP-activated ClpP can only act on nascent or misfolded proteins as long as they have not reached their final folded state. In case of full length ICDH, we observed a markedly reduced synthesis yield in the co-translational presence of ADEP-activated ClpP (below 40% protein yield as compared to the control), suggesting that folding of the larger and more complex ICDH protein (416 amino acids, dimer) is retarded in relation to folding of the shorter SH3 (62 amino acids), thus exposing the nascent ICDH for a longer duration to the destructive action of ADEP-activated ClpP. In control experiments, where the nascent proteins were treated by either ClpP or ADEP separately, such a reduction in protein yield was not detected.
Furthermore, we investigated whether ADEP-activated ClpP has any effect on full-length proteins after the completion of translation. When ADEP-activated ClpP was added post-translationally to preformed ICDH (Fig 5, Supportive Information), no protein degradation was observed, indicating that ICDH is only recognized as a substrate by ADEP-activated ClpP during synthesis and de novo folding but not after finalization of the folding process. In contrast, the folding-deficient m10 mutant remained sensitive even after the completion of translation. In summary, the differences observed in the co-translational synthesis yields of full length ICDH and SH3 in the presence of ADEP-activated ClpP imply that the folding kinetics of the individual proteins affect their sensitivity towards the activated protease. Fast folding of the small-sized SH3 domain (7 kDa) will protect the substrate from degradation by ADEP-activated ClpP. Complete folding and dimerization of larger ICDH (46 kDa) requires the presence of C-terminal sequences and will be retarded until this information is available at the end of synthesis, rendering the protein more sensitive to proteolysis.
ADEP increases the proteolysis of nascent chains in E. coli
The depicted findings demonstrate that co-translational addition of ADEP activates ClpP to attack nascent polypeptides in an in vitro translation system. To analyse whether ADEP presence also causes degradation of newly synthesized proteins in intact bacterial cells, we first generated truncated ICDH polypeptides (ICDH 1–66; ICDH 1–318) harbouring the SecM peptide to induce translational arrest and additional amino acids to allow complete exposure of the nascent target substrates at the ribosomal exit site (Rutkowska et al, 2008). A triple strep tag sequence was introduced at the 5′-end of the gene constructs to allow monitoring of protein stability by Western blot analysis. The experiments were performed in an E. coli efflux pump mutant (ΔacrA) that is susceptible to ADEP (Brötz-Oesterhelt et al, 2005). Addition of ADEP to such E. coli cells led to fast degradation of both ICDH truncation variants, whereas the model nascent chains remained largely stable upon control treatment with dimethyl sulphoxide (DMSO) (Fig 6A), demonstrating that ADEP activates ClpP to attack stalled nascent polypeptides also in vivo.
Figure 6. ADEP leads to the degradation of newly synthesized proteins prior to folding in bacterial cells.
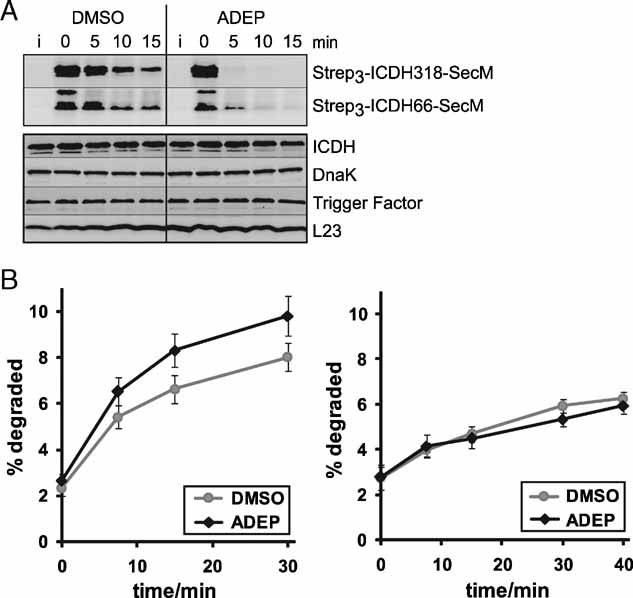
- ADEP causes the degradation of ribosome-arrested nascent polypeptides in vivo. The degradation of two ribosome-arrested nascent chains derived from ICDH (Strep3-ICDH66-SecM, Strep3-ICDH318-SecM) was monitored in ER2566 ΔacrA::kan cells at 37 °C. Two hours after induction (i) of the model constructs, translation was stopped with chloramphenicol and ADEP or DMSO was added (0 min). At the indicated time points, cells were harvested and proteins separated by SDS–PAGE followed by Western blots against the N-terminal Strep-tag (upper panel) and against ICDH, DnaK, Trigger Factor and L23 as controls (lower panel). Only full-length products are depicted.
- The degradation of newly synthesized proteins is increased in the presence of ADEP in vivo. Total protein degradation was determined in HN818 ΔacrA::kan cells in the exponential phase at 37 °C. ADEP1 and DMSO were either added before (left panel) or after (right panel) [35S] methionine pulse labelling. At the denoted time points, aliquots were TCA precipitated and the radioactivity in the TCA-soluble and insoluble fractions was determined by scintillation counting. The amount of degradation is given as percentage of the total cellular radioactivity. Mean values and standard errors of the mean of four to five independent experiments are shown.
Furthermore, we monitored the effect of ADEP on the pre-existing levels of several native proteins (full length ICDH, the chaperones Trigger Factor and DnaK and the ribosomal protein L23) in the same cells expressing the ribosome-arrested ICDH model constructs, where ADEP was added after the chloramphenicol mediated halt of protein synthesis. As the full-length proteins detected by this assay had already completed translation prior to ADEP-treatment, no ADEP/ClpP-mediated proteolysis could be observed (Fig 6A). In contrast, when in one of our previous studies with intact B. subtilis cells the co-translational effect of ADEP on de novo synthesized Trigger Factor, DnaK, GroEL and elongation factor TU had been investigated, a variety of N-terminally degraded fragments had been detected (Brötz-Oesterhelt et al, 2005). Thus, in full accordance with the results described above for the in vitro t/t system our results with intact E. coli and B. subtilis cells demonstrate that the presence of ADEP does not lead to uncontrolled proteolysis of pre-existing proteins and that degradation is rather specific for nascent, misfolded or otherwise flexible protein chains.
Next, we addressed whether ADEP addition also leads to an overall increase in proteolysis of authentic E. coli nascent chains by monitoring the global degradation rates of newly synthesized proteins in the co- and post-translational presence of ADEP (Fig 6B). Degradation of [35S]-methionine-radiolabelled polypeptides was followed at various time points after the addition of unlabelled methionine by determining the radioactivity in the soluble fraction after trichloroacetic acid (TCA) precipitation, representing short peptide products generated by proteolytic cleavage. Addition of ADEP prior to pulse-labelling (Fig 6B, left panel) increased the total degradation rates, whereas its post-translational addition after the pulse-chase procedure did no longer influence global proteolysis (Fig 6B, right panel). Here again, ADEP rendered newly made polypeptides sensitive to proteolysis when present co-translationally during synthesis, but did not affect their stability when added after finishing translation and protein folding. It should be noted that the observed increase in total degradation rates might represent an underestimation, as addition of ADEP in parts could only lead to partial substrate degradation causing the generation of larger protein fragments that would remain insoluble after TCA precipitation and therefore would not be detected by our approach. Indeed, casein is degraded with reduced processivity by ADEP-activated ClpP (Fig 1C and D) and rather large albeit deleteriously truncated variants of various newly made proteins have been previously detected in ADEP-exposed B. subtilis cells (Brötz-Oesterhelt et al, 2005).
DISCUSSION
In this study, we have investigated the molecular mechanism of action of a new class of antibacterial acyldepsipeptides designated ADEPs, which target the peptidase ClpP. ClpP forms the central proteolytic core of a major and highly conserved protease complex in bacteria and interacts with several cooperating Hsp100 proteins that select target proteins and unfold them for subsequent ClpP-mediated degradation. ClpP represents an unprecedented target that is not used by any other antibiotic known so far.
What makes the ADEP mechanism especially intriguing is that the Clp protease is not essential for cell viability in many bacterial species, including the Gram-positive and Gram-negative model species, B. subtilis and E. coli respectively, that were used in this study (Maurizi et al, 1990; Msadek et al, 1998), although it plays an important role in regulating diverse and highly complex cellular developmental programmes (Frees et al, 2007; Gottesman, 2003; Jenal & Hengge-Aronis, 2003; Sauer et al, 2004). The observation that ClpP deletion strains are viable in many bacterial species has consequences for a potential clinical application of the ADEPs. ADEPs are highly active against most of the nosocomial Gram-positive problem pathogens (e.g. staphylococci, enterococci and streptococci) with antibacterial in vitro activities surpassing those of many marketed antibiotics and impressive efficacy in animal models of bacterial infection (Brötz-Oesterhelt et al, 2005). However, previous studies have shown that ClpP deletion strains are ADEP-resistant and that ADEP resistant ClpP mutants can be selected under in vitro culture conditions (Brötz-Oesterhelt et al, 2005). Therefore, a potential clinical application of the ADEPs has to take this into careful consideration. Either the selected target organism has to have a clear requirement for the presence of functional ClpP in the course of the infection process (Frees et al, 2007) or a combination therapy with another antibiotic class has to be considered. This could allow to exploit the advantages of the ADEPs (e.g. lack of cross-resistance to marketed antibiotics, bacteriocidal activity and good penetration into eukaryotic cells) while possibly reducing the appearance of ADEP-resistance.
The fact that ADEPs act on a non-essential target opens also mechanistic questions. How is it possible that the ADEPs kill bacterial cells by targeting ClpP, if ClpP itself is dispensable? The answer is provided by our current studies. Here, we demonstrate that ADEPs do not merely inhibit the physiological functions of ClpP but reprogramme its activity in a highly sophisticated manner.
Our results show that ADEP binding to ClpP prevents complex formation between ClpP and its cooperating Hsp100 proteins and that even preformed ClpP/Hsp100 complexes dissociate upon ADEP addition. As a result, ADEP treatment prevents ClpP-dependent degradation of physiological protein substrates in vitro and in vivo, as demonstrated by, e.g., increased levels of the ClpC/ClpP substrate MurAA in ADEP-treated B. subtilis cells. Given that ClpP-mediated general and regulated proteolyses are especially important in Gram-positive bacteria for adaptation to stress conditions and also for the expression of virulence factors (Frees et al, 2007), the inhibitory effect that the ADEPs exert on ClpP probably mitigates the establishment and spread of bacterial pathogens in the infection process.
However, the growth inhibitory activity that the ADEPs exert on B. subtilis and E. coli during exponential growth in nutrient broth in vitro demands for an additional explanation, because ClpP is not essential for cell viability under these conditions. Indeed, our results demonstrate that ADEPs activate ClpP to Hsp100-independent proteolytic activity, enabling the peptidase to degrade unfolded proteins. In consequence, ADEP converts ClpP from a tightly regulated protease exhibiting high substrate specificity to an unrestrained and destructive proteolytic machinery. We provide evidence that nascent polypeptides represent the main physiological targets of ADEP-activated ClpP. Increased proteolysis of newly synthesized proteins will result in reduced amounts and, in consequence, reduced activities of target proteins causing physiological imbalance and finally cell death. The susceptibility of individual nascent chains towards ADEP will depend on several parameters including folding kinetics, association with partner proteins and guidance by molecular chaperones. Such specific details are still largely unknown and the main targets of ADEP-activated ClpP are therefore difficult to predict; however, given the antibacterial potency of the ADEPs, some substrate proteins must fulfil an essential cellular function.
It is possible that damage to the cell is caused not only by the shortage of essential cellular proteins but also by the advent of substantial amounts of diverse protein fragments. It is interesting to consider the example of certain aminoglycosides in this context. The primary mechanism of action of these antibiotics is to interfere with the proofreading activity of the bacterial ribosome leading to the synthesis of nonsense proteins that contain incorrect amino acids in their sequence. Such mistranslated non-functional proteins are presumed to be incorporated into the membrane and possibly also into other protein complexes (e.g. involved in DNA replication) and to interfere with the activity of the residual native proteins by disturbing these structures. Also, the shortened polypeptides of a normal sequence, which are prematurely released during translation by puromycin treatment, were shown to disturb the structure of the cytoplasmic membrane (Davis et al, 1986; Magnet & Blanchard, 2005). Recently, it has been demonstrated that this aminoglycoside-mediated accumulation of misfolded membrane-associated proteins leads to oxidative stress and subsequent cell death of the treated bacteria (Kohanski et al, 2007; Kohanski et al, 2008). In addition, it was demonstrated that the bactericidal effect of bleach is based on the ability of HOCl to cause misfolding and aggregation of the essential bacterial proteins (Winter et al, 2008).
It is feasible that some of the larger protein fragments that are set free by ADEP-activated ClpP have similar disturbing effects and that they contribute at least in part to the antibacterial activity of the acyldepsipeptides. In previous proteomic studies with B. subtilis, ADEP-treatment as well as incubation with aminoglycosides or puromycin triggered the heat-shock response, a further clear indication that the bacterial cells are exposed to protein damage in all three cases (Bandow et al, 2003; Brötz-Oesterhelt et al, 2005).
In the case of B. subtilis ClpP, we encountered an additional activity of the ADEPs. Here, ADEPs also trigger the oligomerization process from the free ClpP monomer to the tetradecamer, which is in this species a prerequisite for proteolytic activation. How can a small antibiotic, slightly more than 700 Da in size, mediate all of these different effects in parallel, i.e. (1) increasing the affinity of the ClpP monomers for each other to stimulate oligomerization, (2) disturbing the interaction of two large protein complexes, the ClpP tetradecamer and the Hsp100 ATPase hexamer, to prevent complex formation and (3) activation of the dormant proteolytic ClpP core? A plausible explanation is that binding of the ADEPs' to ClpP induces a conformational rearrangement in the peptidase that results in the diverse observed effects. ADEP-induced degradation of unfolded polypeptides by ClpP demands for conformational changes potentially causing an opening of the central ClpP pore that restricts the access of substrates to the inner ClpP chamber. Such ADEP-induced pore opening might create a new substrate recognition area and may allow for the passage of larger protein substrates. These potential structural changes might at the same time abrogate the interaction of ClpP with cooperating Hsp100 proteins. The association of Hsp100 proteins with ClpP is mediated by IGF loops of the ATPase components, which interact with conserved hydrophobic patches on the rim of the ClpP oligomer (Joshi et al, 2004; Kim et al, 2001; Singh et al, 1999). It is feasible that ADEP binding to ClpP causes delocalization of the IGF interaction patches thereby preventing the interaction with the IGF-loops of the Hsp100 partners.
On the ClpP side, an N-terminal located ClpP loop (Bewley et al, 2006; Gribun et al, 2005), which could interact with a pore-2-loop of the associated ATPase was identified as the second determinant for the ClpP/ATPase interaction and communication (Martin et al, 2007). ADEP binding to ClpP could also induce a conformational change involving the N-terminal loop of ClpP. This conformational change might interfere with the second mode of interaction of ClpP via these N-terminal loops with the associated ATPases (Bewley et al, 2006; Gribun et al, 2005; Martin et al, 2007). These two suggested mechanisms of ADEP-induced dissociation are of course not mutually exclusive.
In summary, we have unravelled the underlying molecular principles of the ADEP-mediated activity switch of the bacterial peptidase ClpP. While ADEP causes a halt in the degradation of physiological ClpP substrates, it concomitantly relaxes the tight activity control of ClpP, resulting in uncontrolled proteolysis of nascent polypeptide chains and further protein substrates, which are not protected by their folded conformation. The antibiotics thoroughly reprogramme ClpP function and turn the peptidase from a precisely acting regulatory instrument into an unspecific protein shredder targeting nascent polypeptides. This is the first example of an antibiotic activity, where bacterial death is mediated by deregulation and overactivation of an otherwise controlled and harmless cellular protein. The multiple effects that the ADEPs have to exert on ClpP in order to achieve such complete redirecting of function go far beyond simple inhibition of catalytic activity. The example of the ADEPs is instructive in that they are derived from natural products and that their molecular mechanism has been elucidated only after the discovery of their promising antibacterial activity. In the case of the ADEPs, nature's ingenuity has presented us with an exceptional antibacterial mechanism that would have probably surpassed our capacity for rational design due to its inherent complexity.
MATERIALS AND METHODS
Proteins, expression and purification
Monomer protein concentrations were determined by using the BioRad Bradford assay with bovine serum albumin as a standard. ClpC, B. subtilis ClpP, MecA, ComK and Spx were expressed and purified as described previously (Nakano et al, 2001; Turgay et al, 1997; Schlothauer et al, 2003). His-tagged B. subtilis ClpX was purified by Ni+-NTA-agarose chromatography according to the manufacturer's instructions (Qiagen). E. coli ClpA, ClpS, ClpX and ClpP were purified as previously described (Dougan et al, 2002a; Schlothauer et al, 2003). GFP-SsrA and FR-linker-GFP were purified as described in previous studies (Dougan et al, 2002b; Erbse et al, 2006). Pyruvate kinase and α-casein were purchased from Sigma.
Generation of B. subtilis strains and antibodies
The construction of the B. subtilis ClpP deletion strain as well as the antiserum against MurAA was performed according to previous studies (Gerth et al, 1998; Kock et al, 2004).
To construct recombinant His-tagged ClpX for the in vitro experiments, clpX was amplified by PCR and cloned into the pQE60 vector using the NcoI and BamHI restriction sites. The resulting plasmid pQE60-clpX was then transformed into M15[pREP4], which was used as the host strain for the overproduction of ClpX.
To create C-terminal fusions of genes encoding ClpC, ClpP and ClpX with gfp the 3′-(terminal 400 bp) regions were amplified by PCR and each gene fragment was cloned into the KpnI and EcoRI (clpC and clpP) or KpnI and XhoI (clpX) sites of pSG1151 (Lewis & Marston, 1999). These constructs were then transformed and integrated into the respective chromosomal locus of B. subtilis 168 via a single crossover event (Kirstein et al, 2008).
Preparation of soluble B. subtilis protein extract
For the preparation of soluble extracts of B. subtilis 168, ADEP2-treated 168 and ΔclpP mutants, cells were incubated in Luria–Bertani media under vigorous agitation at 37 °C. ADEP2 was added to one sample culture to a final concentration of 1.6 µg/ml upon entry of the stationary phase. Cells were harvested by centrifugation, resuspended in TE buffer and lysed by sonication. Cell debris was removed by centrifugation at 4 °C at the rate of 10,000g for 30 min. The supernatant containing the soluble cell extract was used for the subsequent Western blot analysis of MurAA.
In vitro degradation assays
For the in vitro degradation assays, proteins were used at a final concentration of 1 µM and incubated at 37 °C in the presence of an ATP regeneration system (3 mM ATP, 2 mM phosphoenolpyruvate and 20 ng/µl pyruvate kinase) (Kirstein et al, 2006). Buffer conditions for E. coli ClpAP: 50 mM Tris, pH 7.5; 150 mM KCl, 20 mM MgCl2, 2 mM DTT; and E. coli ClpXP: 50 mM Tris pH 7.5, 300 mM NaCl, 150 mM KCl, 20 mM MgCl2, 5% glycerol, 2 mM DTT. ADEP1 and ADEP2 were used at final concentrations of 10 and 0.5 µg/ml, respectively. Protein degradation was either monitored by SDS–PAGE analysis or by monitoring the fluorescence of GFP model substrates on a LS50B luminescence spectrometer (PerkinElmer), with excitation at 400 nm and emission at 500 nm. Degradation of FITC-casein (100 nM) was monitored using the same instrument with excitation at 490 nm and emission at 520 nm.
Size exclusion chromatography
Superose 6 and Superdex 75 columns were used on a ÄKTA fast protein liquid chromatography system (GE Healthcare). Columns were equilibrated with 50 mM Tris pH 8.0, 300 mM NaCl, 5 mM MgCl2 and 0.5 mM DTT. Proteins were used at a final concentration of 10 µM and incubated for 10 min at 37 °C. Samples (100 µl) were then applied on to the column. The samples of the gel-filtration run shown in Fig 4A were incubated in the presence of 5 mM ATP and the running buffer contained 0.5 mM ATP. Gel-filtration experiments were performed at room temperature with a flow rate of 0.5 ml/min. The eluted fractions were collected and either directly used in a subsequent in vitro degradation assay or precipitated with acetone for subsequent SDS–PAGE analysis.
Analytical ultracentrifugation
ClpP was analysed at a protein concentration of 10 µM in 50 mM Tris pH 8, 150 mM NaCl, 1 mM MgCl2, 0.1 M arginine in the presence of varying concentrations of ADEP2 using a Beckman Optima XL-A analytical ultracentrifuge and an An50Ti rotor equipped with double sector cells. Arginine was added to the buffer to prevent aggregation of ClpP which occurs even otherwise during long-term sedimentation equilibrium measurements. Data were recorded at 230 or 280 nm. Sedimentation equilibrium measurements were carried out at 20 °C and 6,000 rpm. The apparent molecular mass of the protein was calculated using a software provided by Beckman Instruments with a partial specific volume of the protein of 0.73 ml/g and a buffer density of 1.009 g/ml. Sedimentation velocity data were obtained at 40,000 rpm and 20 °C. From these data the apparent s-value was calculated according to the time derivative method.
The paper explained
PROBLEM:
Antibiotics are the first choice therapy against many infectious agents. However, as bacterial resistance against antibiotics increases worldwide, there is a pressing need for the development of novel antibiotics whose use is not limited by the existence of widespread cross-resistance to other compounds. New exploratory acyldepsipeptides, designated ADEPs, may be the first representatives of such a novel group of compounds. They are highly active against most nosocomial Gram-positive pathogens with antibacterial in vitro activities surpassing that of many marketed antibiotics and high efficacy in rodent models of bacterial infection. The ADEPs act on an unprecedented target, the bacterial protease ClpP. As yet, however their mechanism of action remained unclear.
RESULTS:
This study demonstrates that binding of the ADEPs to ClpP prevents the protease from performing its physiological tasks. Instead ClpP is directed to the ribosome, where it degrades nascent polypeptide chains in an uncontrolled manner, leading to inhibition of bacterial cell division and death.
IMPACT:
Due to this novel mechanism of action, the ADEPs are active against multi-resistant bacterial isolates, demonstrating their potential for the treatment of Gram-positive infections. As ClpP is essential for virulence factor expression in several Gram-positive species but not essential for bacterial growth per se, use of ADEPs should be considered in combination therapy to prevent rapid resistance development. As a combination partner, ADEPs may be able to exert a double role in controlling the infection process by inhibiting bacterial virulence as well as bacterial growth.
Fluorescence microscopy
B. subtilis cells were grown at 30 °C to mid-logarithmic growth phase. Next ADEP2 was added to the cells and samples were withdrawn at the indicated time points and cells were mounted on agarose covered microscope slides (SM-011, Hendley Essex) and examined with an Axiovert 135TV microscope (Zeiss). The images obtained were processed using the METAMORPH V5.0 software (Universal Imaging, Media, PA, USA) (Kirstein et al, 2008).
Nascent chain degradation in the in vitro transcription/translation system
De novo synthesis of [35S] methionine-labelled model polypeptides SH3, m10 (Blanco et al, 1999), ICDH (Deuerling et al, 2003), ribosome-arrested 3m10-SecM and ribosome-arrested ICDH fragment of 45 kDa (Hoffmann et al, 2006) was performed at 37 °C in an E. coli based transcription/translation system according to published protocols (Hoffmann et al, 2006; Müller & Blobel, 1986). In the case of the post-translational degradation assays, translation reactions were performed for 30 min and terminated with 2 mM chloramphenicol (arrested polypeptides, Fig 5A) or 2 mM puromycin (released polypeptides, Fig 5C). Subsequently, 1 µM (final concentration) ClpP monomer together with 10 µg/ml ADEP1 or the corresponding volume of DMSO was added. Aliquots were precipitated by TCA, 0–24 min after ClpP addition. Precipitated proteins were separated on tricine gels and analysed by autoradiography and phosphorimager quantification. In the case of co-translational degradation assays (Fig 5B), ClpP, ADEP1 and DMSO were present during the entire time course of translation. The translation extracts were derived from fractionated MC4100 Δtig cells (Deuerling et al, 1999) and contained insignificant amounts of ClpP (≤20 nM).
In vivo degradation assays in E. coli
The strain E. coli ER2566 ΔacrA::kan was generated from ER2566 by P1 transduction using a phage lysate derived from E. coli HN818 ΔacrA::kan (Hardy & Cozzarelli, 2005). ER2566 ΔacrA::kan cells containing the pZA4 plasmid and one of the expression plasmids pBAT-Strep3-ICDH66-SecM or pBAT-Strep3-ICDH318-SecM (Rutkowska et al, 2008) were grown at 37 °C in Luria broth medium supplemented with 100 µg/ml ampicillin to an OD600 of 0.8 and induced with 500 µM IPTG. Two hours after induction, translation was stopped with 2.5 mM chloramphenicol and, subsequently, 25 µg/ml ADEP1 or the corresponding volume of DMSO was added. Cell aliquots were harvested 0–15 min after ADEP1 addition and analysed by 15% SDS–PAGE and Western blotting using Strep-Tactin conjugated to alkaline phosphatase (IBA) and rabbit antisera specific for the E. coli proteins ICDH, DnaK, L23 and Trigger Factor.
E. coli HN818 ΔacrA::kan cells were grown at 37 °C in M9 minimal medium containing 0.4% glucose and all l-amino acids (31 µg/ml) except L-methionine (MM medium). At an OD600 of 0.6, cells were labelled with 60 µCi/ml [35S] methionine (SJ1515, Amersham) for 2 min and afterwards chased with 0.5 mg/ml unlabelled L-methionine for 3 min. Subsequently, cells were washed two times in MM medium containing unlabelled methionine and resuspended in MM medium containing unlabelled methionine and 2.5 mM chloramphenicol. Aliquots were TCA precipitated 0–40 min after resuspension and the radioactivities in the TCA-soluble and -insoluble fractions were determined by scintillation counting. The amount of protein degradation was calculated as the ratio between TCA-soluble radioactivity and the total amount of incorporated radioactivity. ADEP1 (25 µg/ml) or the corresponding volume of DMSO was added either 3–10 min before the [35S] methionine labelling or after the two wash steps.
Acknowledgments
We thank U. Gerth and M. Hecker (Universität Greifswald) for reagents, H. Paulson and S. Raddatz (Bayer HealthCare, Wuppertal) for ADEP synthesis, and L. Hamoen, J. Errington (Newcastle University) and their colleagues for hospitality and use of facilities. This work was supported by the Deutsche Forschungsgemeinschaft (SPP 1132 to KT and AM and FOR 854 to HBO), a scholarship of the Boehringer Ingelheim Fonds to AH and a short term fellowship of EMBO to JK.
Supplementary information is available at EMBO Molecular Medicine online (http://www.embomolmed.org).
The authors declare that they have no conflict of interest.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Bandow JE, Brotz H, Leichert LI, Labischinski H, Hecker M. Proteomic approach to understanding antibiotic action. Antimicrob Agents Chemother. 2003;47:948–955. doi: 10.1128/AAC.47.3.948-955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewley MC, Graziano V, Griffin K, Flanagan JM. The asymmetry in the mature amino-terminus of ClpP facilitates a local symmetry match in ClpAP and ClpXP complexes. J Struct Biol. 2006;153:113–128. doi: 10.1016/j.jsb.2005.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco FJ, Angrand I, Serrano L. Exploring the conformational properties of the sequence space between two proteins with different folds: an experimental study. J Mol Biol. 1999;285:741–753. doi: 10.1006/jmbi.1998.2333. [DOI] [PubMed] [Google Scholar]
- Brötz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, Bandow JE, Sahl HG, Labischinski H. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med. 2005;11:1082–1087. doi: 10.1038/nm1306. [DOI] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich AL. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Davis BD, Chen LL, Tai PC. Misread protein creates membrane channels: an essential step in the bactericidal action of aminoglycosides. Proc Natl Acad Sci USA. 1986;83:6164–6168. doi: 10.1073/pnas.83.16.6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuerling E, Patzelt H, Vorderwulbecke S, Rauch T, Kramer G, Schaffitzel E, Mogk A, Schulze-Specking A, Langen H, Bukau B. Trigger factor and DnaK possess overlapping substrate pools and binding specificities. Mol Microbiol. 2003;47:1317–1328. doi: 10.1046/j.1365-2958.2003.03370.x. [DOI] [PubMed] [Google Scholar]
- Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature. 1999;400:693–696. doi: 10.1038/23301. [DOI] [PubMed] [Google Scholar]
- Dougan D, Mogk A, Zeth K, Turgay K, Bukau B. AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett. 2002a;529:6–10. doi: 10.1016/s0014-5793(02)03179-4. [DOI] [PubMed] [Google Scholar]
- Dougan DA, Reid BG, Horwich AL, Bukau B. ClpS, a substrate modulator of the ClpAP machine. Mol Cell. 2002b;9:673–683. doi: 10.1016/s1097-2765(02)00485-9. [DOI] [PubMed] [Google Scholar]
- Erbse A, Schmidt R, Bornemann T, Schneider-Mergener J, Mogk A, Zahn R, Dougan DA, Bukau B. ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature. 2006;439:753–756. doi: 10.1038/nature04412. [DOI] [PubMed] [Google Scholar]
- Frees D, Savijoki K, Varmanen P, Ingmer H. Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol Microbiol. 2007;63:1285–1295. doi: 10.1111/j.1365-2958.2007.05598.x. [DOI] [PubMed] [Google Scholar]
- Gerth U, Krüger E, Derré I, Msadek T, Hecker M. Stress induction of the Bacillus subtilis clpPgene encoding a homologue of the proteolytic component of the Clp protease and the involvement of ClpP and ClpX in stress tolerance. Mol Microbiol. 1998;28:787–802. doi: 10.1046/j.1365-2958.1998.00840.x. [DOI] [PubMed] [Google Scholar]
- Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- Gribun A, Kimber MS, Ching R, Sprangers R, Fiebig KM, Houry WA. The ClpP double ring tetradecameric protease exhibits plastic ring-ring interactions, and the N termini of its subunits form flexible loops that are essential for ClpXP and ClpAP complex formation. J Biol Chem. 2005;280:16185–16196. doi: 10.1074/jbc.M414124200. [DOI] [PubMed] [Google Scholar]
- Hardy CD, Cozzarelli NR. A. genetic selection for supercoiling mutants of Escherichia coli reveals proteins implicated in chromosome structure. Mol Microbiol. 2005;57:1636–1652. doi: 10.1111/j.1365-2958.2005.04799.x. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hinzen B, Raddatz S, Paulsen H, Lampe T, Schumacher A, Habich D, Hellwig V, Benet-Buchholz J, Endermann R, Labischinski H, Brotz-Oesterhelt H. Medicinal chemistry optimization of acyldepsipeptides of the enopeptin class antibiotics. ChemMedChem. 2006;1:689–693. doi: 10.1002/cmdc.200600055. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Merz F, Rutkowska A, Zachmann-Brand B, Deuerling E, Bukau B. Trigger factor forms a protective shield for nascent polypeptides at the ribosome. J Biol Chem. 2006;281:6539–6545. doi: 10.1074/jbc.M512345200. [DOI] [PubMed] [Google Scholar]
- Horwich AL, Weber-Ban EU, Finley D. Chaperone rings in protein folding and degradation. Proc Natl Acad Sci USA. 1999;96:11033–11040. doi: 10.1073/pnas.96.20.11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal U, Hengge-Aronis R. Regulation by proteolysis in bacterial cells. Curr Opin Microbiol. 2003;6:163–172. doi: 10.1016/s1369-5274(03)00029-8. [DOI] [PubMed] [Google Scholar]
- Joshi SA, Hersch GL, Baker TA, Sauer RT. Communication between ClpX and ClpP during substrate processing and degradation. Nat Struct Mol Biol. 2004;11:404–411. doi: 10.1038/nsmb752. [DOI] [PubMed] [Google Scholar]
- Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- Kirstein J, Schlothauer T, Dougan DA, Lilie H, Tischendorf G, Mogk A, Bukau B, Turgay K. Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J. 2006;25:1481–1491. doi: 10.1038/sj.emboj.7601042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J, Strahl H, Moliere N, Hamoen LW, Turgay K. Localization of general and regulatory proteolysis in Bacillus subtiliscells. Mol Microbiol. 2008;70(3):682–694. doi: 10.1111/j.1365-2958.2008.06438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kock H, Gerth U, Hecker M. MurAA, catalysing the first committed step in peptidoglycan biosynthesis, is a target of Clp-dependent proteolysis in Bacillus subtilis. Mol Microbiol. 2004;51:1087–1102. doi: 10.1046/j.1365-2958.2003.03875.x. [DOI] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004;10:S122–129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- Lewis PJ, Marston AL. GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis. Gene. 1999;227:101–109. doi: 10.1016/s0378-1119(98)00580-0. [DOI] [PubMed] [Google Scholar]
- Lupas A, Flanagan JM, Tamura T, Baumeister W. Self-compartmentalizing proteases. Trends Biochem Sci. 1997;22:399–404. doi: 10.1016/s0968-0004(97)01117-1. [DOI] [PubMed] [Google Scholar]
- Magnet S, Blanchard JS. Molecular insights into aminoglycoside action and resistance. Chem Rev. 2005;105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol Cell. 2007;27:41–52. doi: 10.1016/j.molcel.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurizi MR, Clark WP, Katayama Y, Rudikoff S, Pumphrey J, Bowers B, Gottesman S. Sequence and structure of ClpP, the proteolytic component of the ATP-dependent Clp protease of Escherichia coli. J Biol Chem. 1990;265:12536–12545. [PubMed] [Google Scholar]
- Msadek T, Dartois V, Kunst F, Herbaud ML, Denizot F, Rapoport G. ClpP of Bacillus subtilis is required for competence development, motility, degradative enzyme synthesis, growth at high temperature and sporulation. Mol Microbiol. 1998;27:899–914. doi: 10.1046/j.1365-2958.1998.00735.x. [DOI] [PubMed] [Google Scholar]
- Müller M, Blobel G. In vitro analysis of the bacterial protein export. Curr Top Microbiol Immunol. 1986;125:33–41. doi: 10.1007/978-3-642-71251-7_4. [DOI] [PubMed] [Google Scholar]
- Nakano MM, Hajarizadeh F, Zhu Y, Zuber P. Loss-of-function mutations in yjbD result in ClpX- and ClpP-independent competence development of Bacillus subtilis. Mol Microbiol. 2001;42:383–394. doi: 10.1046/j.1365-2958.2001.02639.x. [DOI] [PubMed] [Google Scholar]
- Rutkowska A, Mayer MP, Hoffmann A, Merz F, Zachmann-Brand B, Schaffitzel C, Ban N, Deuerling E, Bukau B. Dynamics of trigger factor interaction with translating ribosomes. J Biol Chem. 2008;283:4124–4132. doi: 10.1074/jbc.M708294200. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ES, Siddiqui SM, Wah DA, Baker TA. Sculpting the proteome with AAA(+). proteases and disassembly machines. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlothauer T, Mogk A, Dougan DA, Bukau B, Turgay K. MecA, an adaptor protein necessary for ClpC chaperone activity. Proc Natl Acad Sci USA. 2003;100:2306. doi: 10.1073/pnas.0535717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Guo F, Maurizi MR. ClpA and ClpP remain associated during multiple rounds of ATP-dependent protein degradation by ClpAP protease. Biochemistry. 1999;38:14906–14915. doi: 10.1021/bi991615f. [DOI] [PubMed] [Google Scholar]
- Turgay K, Hamoen LW, Venema G, Dubnau D. Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev. 1997;11:119–128. doi: 10.1101/gad.11.1.119. [DOI] [PubMed] [Google Scholar]
- Wang J, Hartling JA, Flanagan JM. The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- Winter J, Ilbert M, Graf PC, Ozcelik D, Jakob U. Bleach activates a redox- regulated chaperone by oxidative protein unfolding. Cell. 2008;135:691–701. doi: 10.1016/j.cell.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5:175–186. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.