

Abstract
Tumour hypoxia promotes the accumulation of the otherwise oxygen-labile hypoxia-inducible factor (HIF)-α subunit whose expression is associated with cancer progression, poor prognosis and resistance to conventional radiation and chemotherapy. The oxygen-dependent degradation of HIF-α is carried out by the von Hippel–Lindau (VHL) protein-containing E3 that directly binds and ubiquitylates HIF-α for subsequent proteasomal destruction. Thus, the cellular proteins involved in the VHL–HIF pathway have been recognized as attractive molecular targets for cancer therapy. However, the various compounds designed to inhibit HIF-α or HIF-downstream targets, although promising, have shown limited success in the clinic. In the present study, we describe the bioengineering of VHL protein that removes the oxygen constraint in the recognition of HIF-α while preserving its E3 enzymatic activity. Using speckle variance–optical coherence tomography (sv–OCT), we demonstrate the dramatic inhibition of angiogenesis and growth regression of human renal cell carcinoma xenografts upon adenovirus-mediated delivery of the bioengineered VHL protein in a dorsal skin-fold window chamber model. These findings introduce the concept and feasibility of ‘bio-tailored’ enzymes in the treatment of HIF-overexpressing tumours.
Keywords: angiogenesis, ARNT, HIF, renal cell carcinoma, VHL
INTRODUCTION
Tumour growth invariably outstrips its blood supply as the diffusional capacity of oxygen from the nearest blood vessels is surpassed, leading to regions of hypoxia within the tumour mass. In addition, tumour cells close to a blood vessel can experience hypoxia due to disruptions in blood flow, a common characteristic of malformed tumour vasculature (Brown & Wilson, 2004). The transcription factor HIF is activated under hypoxia and triggers the transcription of a large number of genes that promote various adaptive cellular responses ranging from anaerobic metabolism, erythropoiesis and angiogenesis to cell survival. HIF-induced genes are known to drive oncogenesis and as a result, HIF overexpression is frequently associated with increased phenotypic aggressiveness and poor prognosis in numerous tumour types including brain, breast, lung, colon, skin, prostate and kidney cancers (Kim & Kaelin, 2004; Semenza, 2003).
HIF is a heterodimeric transcription factor composed of two subunits, HIF-α and aryl hydrocarbon receptor nuclear translocator (ARNT). HIF-α and ARNT are members of the basic-helix–loop–helix (bHLH) Per/ARNT/Sim (PAS) family of transcription factors. The basic domain is essential for DNA binding, whereas the HLH and PAS domains are necessary for heterodimerization and DNA binding. HIF-α contains two transactivation domains (NAD and CAD, located in the amino (N) and carboxy (C)termini, respectively), whereas ARNT contains one transactivation domain (TAD) in the C-terminus. Under low oxygen tension, HIF-α recruits transcriptional co-activators p300/CBP and binds ARNT. The active HIF complex binds to hypoxia-responsive elements (HREs) in the promoters/enhancers of the numerous HIF-target genes such as vascular endothelial growth factor (VEGF), glucose transporter-1 (GLUT1), transforming growth factor-α (TGF-α) and erythropoietin (EPO) to initiate their transcription (Kim & Kaelin, 2004; Semenza, 1999). Under normal oxygen tension, HIF-α is hydroxylated on conserved proline residues in the oxygen-dependent degradation domain (ODD) by prolyl hydroxylase domain-containing family of enzymes (PHD1-3) (Bruick & McKnight, 2001; Epstein et al, 2001; Ivan et al, 2001; Jaakkola et al, 2001; Masson et al, 2001). This oxygen-dependent modification of HIF-α permits recognition by the VHL tumour suppressor protein, which functions as the substrate-recognition component of an E3 ubiquitin ligase ECV (Elongins BC/Cul2/VHL) that polyubiquitylates HIF-α for subsequent proteasomal degradation (Cockman et al, 2000; Kamura et al, 2000; Maxwell et al, 1999; Ohh et al, 2000; Tanimoto et al, 2000). Unlike HIF-α, ARNT is constitutively expressed and stable, and thus the regulation of HIF is at the level of HIF-α stability (Kim & Kaelin, 2004).
VHL protein has two major functional domains: the α domain is required for the nucleation of the Elongins BC/Cul 2/VHL complex and the β domain is required for prolyl-hydroxylated HIF-α recognition (Stebbins et al, 1999). Furthermore, functional inactivation of the VHL protein is the cause of hereditary VHL cancer syndrome—characterized by the development of hypervascular tumours in multiple organs including the brain, spine, retina and kidney, and is also associated with the development of the vast majority of sporadic clear-cell renal cell carcinoma (CCRCC), which is the most common form of kidney cancer (Kim & Kaelin, 2004). Notably, all CCRCC-causing VHL mutants tested-to-date have shown a failure in either assembling into an ECV complex or binding to HIF-α (Kim & Kaelin, 2004). Concordantly, tumour cells including CCRCC devoid of functional VHL protein have enhanced expression of HIF-target genes irrespective of oxygen tension. In addition, growth factors binding to their cognate receptor tyrosine kinases (RTKs) and ensuing activation of the phosphoinositide 3-kinase and MAPK signalling pathways can regulate HIF-1α protein levels in an oxygen-independent fashion (Maynard & Ohh, 2007; Semenza, 2003). Both pathways can activate mTOR-mediated cap-dependent translation of HIF-1α mRNA, and PI3K can also increase translation of HIF-1α mRNA through an internal ribosomal entry site (IRES)-dependent mechanism (Maynard & Ohh, 2007). Thus, tumour-associated mutations impinging on the PI3K or MAPK pathways including gain-of-function mutations in RTKs and Ras or loss-of-function mutations in phosphatase and tensin homolog and tumour suppressor complex 1/2 tumour suppressor genes, increase HIF-1α synthesis. In addition to mutations in various oncogenes and tumour suppressor genes, the most common mechanism of HIF-α stabilization in cancer arguably involves the general oxygen-sensing pathway in regions of tumour hypoxia, in which, for example, a functional VHL protein would be rendered ineffectual in negatively regulating HIF-α stability.
Overexpression of HIF-1α or HIF-2α has been strongly associated with tumour progression and resistance to therapy, implicating HIF-1α and HIF-2α as compelling therapeautic targets for anti-cancer therapy (Kondo et al, 2002; Semenza, 2003). Currently, there are a number of compounds either in clinical development or approved by the Food and Drug Administration that have been shown to block HIF-1α activity. For example, gefitinib and erlotinib, trastuzumab, and everolimus and temsirolimus have been shown to inhibit HIF-1α synthesis by blocking upstream oncogenic epidermal growth factor receptor, human epidermal growth factor receptor 2/Neu and mammalian target of rapamycin signalling pathways, respectively (Melillo, 2007). A topoisomerase I inhibitor topotecan and a microtubule polymerization inhibitor 2ME2 have also been found to interfere with HIF-1α mRNA translation. DNA-binding molecule echinomycin interrupts the DNA binding of HIF-1α, whereas HIF-1α-mediated transcription is reduced by the proteasome inhibitor velcade and the antifungal agent amphotericin B. In addition, inhibition of the chaperone Hsp90 by 17-AAG and 17-DMAG, as well as inhibition of HDAC by LAQ824, has been found to induce HIF-1α protein degradation (Melillo, 2007; Semenza, 2007). However, none of the above agents directly targets HIF-1α and each drug has multiple functions other than blocking HIF-1α. Moreover, the inhibitory effect of these agents on HIF-2α is largely unknown, despite an increasing evidence supporting an important role of HIF-2α in tumourigenesis. For example, HIF2 has recently been shown to transactivate Oct-4, a transcription factor essential for maintaining stem cell pluripotency, and angiopoietin-1 receptor, Tie-2 and VEGFR2 have been established as HIF2-target gene products (Covello et al, 2006; Duan et al, 2005; Elvert et al, 2003; Tian et al, 1997). HIF2 also has a higher transactivation activity than HIF1 on the promoters of VEGF, TGF-α and EPO (Gunaratnam et al, 2003; Warnecke et al, 2004; Wiesener et al, 1998). In addition, several lines of evidence have shown the stabilization of HIF-2α, but not HIF-1α, to be the critical oncogenic event upon the loss of VHL protein in CCRCC (Kondo et al, 2002, 2003).
In the present study, we demonstrate that a bioengineered VHL protein can engage and degrade HIF-1α and HIF-2α irrespective of oxygen tension, eliminating the necessity for prolyl-hydroxylation of HIF-α for degradation. We further show that adenovirus-mediated delivery of a bioengineered VHL protein dramatically inhibits angiogenesis and regresses CCRCC xenografts in vivo. This is the first report illustrating the feasibility of an E3 ubiquitin ligase designed to remove the oxygen constraint as an alternative mode to directly and constitutively target and destroy HIF-α for rational anti-cancer therapy.
RESULTS
Unlike binding to VHL protein, prolyl-hydroxylation of HIF-α is not required for binding ARNT since heterodimerization occurs under hypoxic conditions. Thus, we sought to generate a VHL–ARNT chimaera containing the minimal region of ARNT required for binding HIF-α fused to the α domain of VHL known to bind elongin C, which bridges the VHL protein to the rest of the ECV complex (Fig 1A). A prediction is that the VHL–ARNT chimaera would bind HIF-α irrespective of oxygen to promote its degradation.
Figure 1. VHL–ARNT fusion proteins bind HIF-1α in vitro.
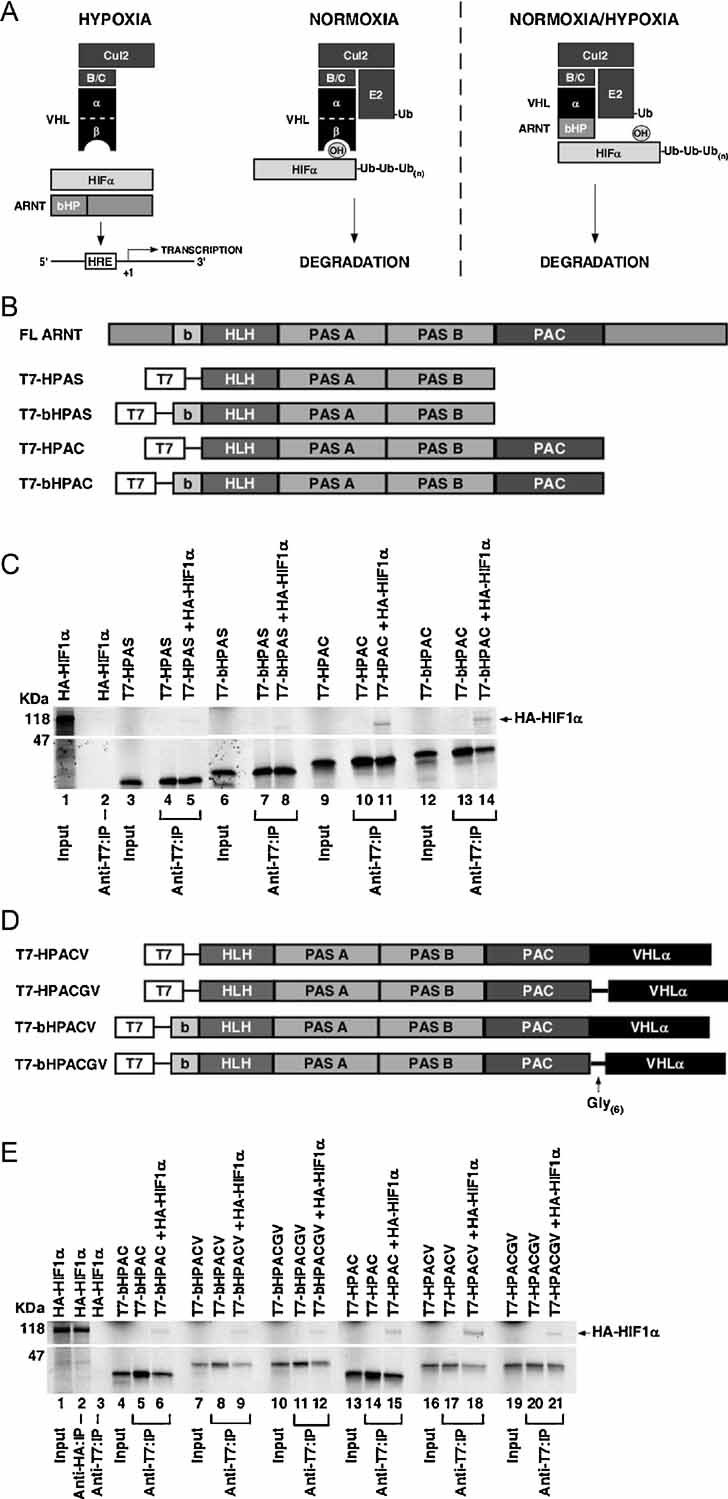
- Schematic diagram of a model depicting VHL–ARNT binding HIF-α independent of its prolyl-hydroxylation status and promoting HIF-α polyubiquitylation via ECV (see text for details). B/C, Elongins BC; bHP, basic-helix–loop–helix and PAS; HRE, hypoxia-responsive element; Ub, ubiquitin.
- Schematic diagram of the various T7-tagged ARNT truncation mutants generated for defining minimal regions required for binding HIF-1α. FL ARNT, full-length ARNT; b, basic; HLH, helix–loop–helix; PAS, Per-ARNT-Sim; PAC, PAS-associated C-terminal domain.
- 35S-labelled in vitro translated HA-HIF-1α was mixed with the indicated 35S-labelled in vitro translated T7-ARNT truncation mutants. Sample mixtures were immunoprecipitated with anti-T7 antibody, resolved by SDS–PAGE and visualized by autoradiography. The image shown was generated from one autoradiograph.
- Schematic diagram of the VHL α domain fused to the indicated ARNT truncation mutants with or without the flexible Gly(6)linker.
- 35S-labelled in vitro translated HA-HIF-1α was mixed with the indicated 35S-labelled in vitro translated T7-tagged VHL–ARNT fusion proteins. Sample mixtures were immunoprecipitated with anti-T7 or anti-HA antibodies. Bound proteins were resolved by SDS–PAGE and visualized by autoradiography. The image shown was generated from one autoradiograph. IP, immunoprecipitation.
VHL–ARNT fusion proteins bind HIF-α and form an ECV complex
The bHLH, PAS A and PAS B domains of ARNT are required for dimerization with HIF-1α and HIF-2α. C-terminal to the PAS B domain is the PAS C-terminal domain (PAC) that is less well-described, but has been proposed to play a similar role in heterodimerization (Maynard & Ohh, 2004). To define the minimal region of ARNT required for strong dimerization with HIF-α, we generated T7-tagged truncation plasmids encoding the following domains: HLH, PAS A, PAS B (T7-HPAS, residues 103–419); bHLH, PAS A, PAS B (T7-bHPAS, residues 90–419); HLH, PAS A, PAS B, PAC (T7-HPAC, residues 103–467); and bHLH, PAS A, PAS B, PAC (T7-bHPAC, residues 90–467) (Fig 1B). An in vitro binding assay was performed with 35S-labelled in vitro translated HA-HIF-1α mixed with 35S-labelled in vitro translated T7-tagged ARNT truncation mutants. The reaction mixtures were immunoprecipitated with an anti-T7 antibody, resolved by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and visualized by autoradiography (Fig 1C). The ARNT truncation mutants containing the PAC domain showed increased binding to HIF-1α (Fig 1C, compare lanes 5 and 8 with lanes 11 and 14). Based on these findings, T7-HPAC and T7-bHPAC were used to generate the VHL–ARNT chimaera.
VHL–ARNT chimaeras were generated by fusing the VHL α domain (residues 151–194) C-terminal to T7-HPAC and T7-bHPAC with or without a 6-Glycine flexible linker between the two heterologous protein fragments, giving rise to the following constructs: T7-HPACV, T7-HPACGV, T7-bHPACV and T7-bHPACGV (Fig 1D). We next tested the ability of the fusion proteins to bind HIF-1α by performing an analogous in vitro binding assay (Fig 1E). The addition of VHL α domain did not diminish the ability of ARNT truncation mutants to bind HIF-1α, and T7-HPACV and T7-HPACGV chimaeras displayed stronger interaction with HIF-1α than T7-bHPACV or T7-bHPACGV containing the basic DNA binding sequences (Fig 1E, compare lanes 9 and 12 with lanes 18 and 21).
We explored whether the VHL–ARNT fusion proteins bound HIF-1α in vivo. HEK293A cells were transiently co-transfected with the mammalian expression plasmids encoding HA-HIF-1α and empty plasmid (MOCK) or T7-HPACV, T7-HPACGV, T7-bHPACV or T7-bHPACGV. Cells were treated with the proteasome inhibitor MG132 to stabilize the oxygen-labile HIF-1α. Cells were then lysed, immunoprecipitated with an anti-HA antibody, bound proteins resolved by SDS–PAGE and immunoblotted with anti-HA and anti-T7 antibodies. All VHL–ARNT chimaeras co-precipitated HA-HIF-1α (Fig 2A). Moreover, VHL–ARNT chimaeras likewise bound HA-HIF-2α (data not shown). We next examined whether the VHL–ARNT chimaeras formed an ECV complex. HEK293A cells were transiently transfected with an empty plasmid, T7-VHL, T7-bHPAC, T7-HPACV, T7-HPACGV, T7-bHPACV or T7-bHPACGV. Cells were then lysed, immunoprecipitated with anti-T7 antibody, bound proteins resolved by SDS–PAGE and immunoblotted with anti-Cul2 and anti-T7 antibodies (Fig 2B). T7-VHL served as a positive control showing co-precipitation of the scaffold component Cul2 (Fig 2B, lane 3), while the ARNT truncation mutant T7-bHPAC lacking the VHL α domain served as a negative control showing a failure in co-precipitating Cul2 (Fig 2B, lane 4). VHL–ARNT chimaeras, when normalized for expression, exhibited Cul2 binding with efficiency comparable to that of wild-type VHL protein, indicating their ability to form an ECV complex.
Figure 2. VHL–ARNT fusion proteins bind HIF-1α and form ECV complexes in vivo.
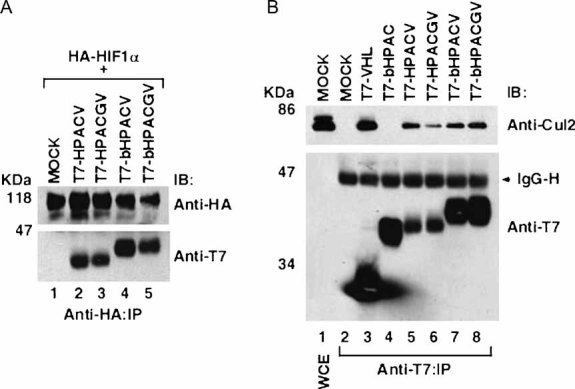
- HEK293A cells transfected with the indicated plasmids were treated with the proteasome inhibitor MG132, lysed, immunoprecipitated with anti-HA antibody and visualized by immunoblotting.
- HEK293A cells transfected with the indicated plasmids were lysed, immunoprecipitated with anti-T7 antibody, resolved by SDS–PAGE and immunoblotted with anti-Cul2 and anti-T7 antibodies. WCE, whole cell extract; IP, immunoprecipitation; IB, immunoblot; IgG-H, immunoglobulin heavy chain.
VHL–ARNT chimaeras promote HIF-α degradation and inhibit HRE-mediated transcription under hypoxia
We asked whether T7-HPACV or T7-HPACGV could degrade HIF-1α under hypoxia. HEK293A cells were transiently co-transfected with plasmids encoding HA-HIF-1α and empty plasmid or T7-VHL, T7-HPACV or T7-HPACGV. Prior to lysis, cells were treated with or without proteasome inhibitor MG132 and maintained under hypoxia (1% oxygen). Cells were lysed and equal amounts of the whole cell lysates were resolved by SDS–PAGE and immunoblotted with an anti-HIF-1α antibody (Fig 3A). As expected, co-transfection of T7-VHL had negligible effect on HIF-1α protein levels, evidenced by the similar amounts of HIF-1α detected with or without MG132 (Fig 3A, compare lanes 3 and 4). Notably, the endogenous VHL protein in HEK293A cells had likewise no discernable effect on HA-HIF-1α expression under hypoxia (Fig 3A, lane 2). In contrast, the expression of either T7-HPACV or T7-HPACGV caused dramatic attenuation of HIF-1α levels in the absence of MG132 (Fig 3A, lanes 6 and 8). These results indicate that both T7-HPACV and T7-HPACGV effectively promote HIF-1α for proteasome-dependent degradation under hypoxia. Consistent with this notion, non-hydroxylated HIF-1α(Pro564Ala) (Ivan et al, 2001; Jaakkola et al, 2001), which has been shown to be stable in the presence of wild-type VHL protein under normoxia, was effectively degraded by T7-HPACGV in a proteasome-dependent manner (Fig S1 of Supporting Information).
Figure 3. HPACV and HPACGV bind and degrade HIF-1α under hypoxia and decrease HRE-mediated transcription.
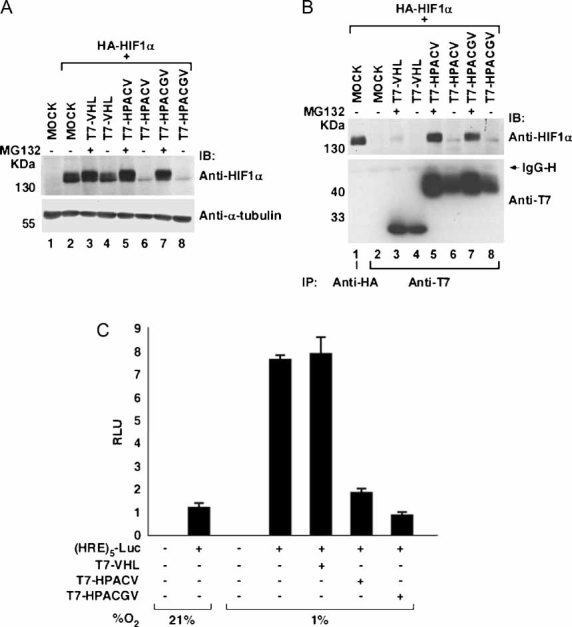
A, B. HEK293A cells transfected with the indicated plasmids were treated with (+) or without (−) MG132 for 4 h and maintained at 1% oxygen. Cells were lysed and whole cell extracts (A) or anti-HA and anti-T7 immunoprecipitated proteins (B) were resolved by SDS–PAGE and immunoblotted with the indicated antibodies.
C. Dual-luciferase assay was performed in HEK293A cells transfected with (HRE)5-Luc in combination with the indicated plasmids. SV40-driven renilla luciferase was transfected as internal control. Cells were maintained at 21 or 1% oxygen for 16 h prior to lysis and dual-luciferase assay was performed. Experiments and transfections were performed in triplicates with one representative experiment shown. Error bars represent standard deviations. RLU, relative light units.
To assess the binding of VHL–ARNT chimaeras to HIF-1α under hypoxia, an analogous experiment in HEK293A cells was performed with anti-HA and anti-T7 immunoprecipitations of the whole cell lysates. Bound proteins were resolved by SDS–PAGE and immunoblotted with anti-HIF-1α and anti-T7 antibodies (Fig 3B). As expected, T7-VHL failed to co-precipitate HA-HIF-1α under hypoxia even in the presence of MG132 (Fig 3B, lane 3). In contrast, both T7-HPACV and T7-HPACGV co-precipitated HA-HIF-1α in the presence of MG132 (Fig 3B, lanes 5 and 7). These results strongly suggest that VHL–ARNT chimaeras bind HIF-1α under hypoxia to promote its proteasome-dependent degradation. AhRryl Hydrocarbon Receptor (AhR) is another well-characterized ARNT binding partner ubiquitously expressed and involved in endo- and xenobiotic metabolism (Kewley et al, 2004). AhR as expected bound ARNT, but failed to bind HPACGV in an in vitro binding assay (Fig S2A and S2B of Supporting Information). Accordingly, HPACGV failed to downregulate the expression of AhR in vitro or in vivo (Fig S2C and D of Supporting Information). Thus HPACGV, which was generated and optimized for binding and degrading HIF-α, does not interact with arguably the next best-characterized ANRT-binding partner AhR.
Under hypoxia, HIF-α dimerizes with ARNT to form an active transcription factor HIF, which engages HREs in the promoters of a myriad of hypoxia-inducible genes to initiate their transcription. We sought to determine the effect of T7-HPACV and T7-HPACGV on HRE-driven transcription by performing a dual-luciferase assay using a firefly luciferase reporter driven by five contiguous HRE elements from the phosphoglycerate kinase-1 promoter (Fig 3C). HEK293A cells were transiently co-transfected with plasmids encoding (HRE)5-Luc and T7-VHL, T7-HPACV or T7-HPACGV. Cells were maintained at either normoxia (21% oxygen) or hypoxia (1% oxygen) for 16 h prior to lysis. The transactivation activity from the HRE promoter was markedly higher under hypoxia than normoxia, as expected. Also, the ectopic expression of T7-VHL did not influence HRE-driven transactivation under hypoxia, since a VHL protein is ineffective in targeting HIF-α for destruction under hypoxia. In contrast, T7-HPACV and T7-HPACGV significantly reduced the transactivation from the HRE promoter under hypoxia (Fig 3C), indicating a marked loss of endogenous HIF function. Notably, T7-HPACGV was reproducibly more potent in attenuating HIF-mediated transcription than T7-HPACV (Fig 3C), and thus the T7-HPACGV chimaera was selected for subsequent experimentation. Furthermore, HPAC in the absence of a VHL protein diminished HIF-driven transcription under hypoxia by forming an inactive transcriptional complex, whereas HPACGV in comparison, dramatically attenuated HIF-driven transcription (Fig S3 of Supporting Information), suggesting that the potency of HPACGV is due to the rapid E3 enzymatic activity causing the degradation of HIF-α upon binding.
Adenoviral delivery of T7-HPACGV attenuates HIF-α and HIF-target gene expression independent of oxygen tension
A cardinal feature of CCRCC is the overexpression of hypoxia-inducible genes. This is principally due to the inactivation of VHL protein that is observed in the vast majority of CCRCC. Interestingly, CCRCC harbouring wild-type VHL protein also displays strong hypoxic signatures and several lines of evidence suggest the stabilization of HIF-2α to be a critical oncogenic event in the pathogenesis of CCRCC (Kondo et al, 2002). We generated recombinant adenoviruses expressing enhanced green fluorescence protein (EGFP) alone or in combination with T7-VHL (Ad-EGFP-T7-VHL) or T7-HPACGV (Ad-EGFP-T7-HPACGV) and tested their ability to form an ECV complex in the CCRCC cell line 786-O (VHL−/−; HIF-1α−/−), a widely used cell system for CCRCC with constitutive activation of HIF-2α. Upon high-efficiency infection of 786-O cells with the indicated adenoviruses (>95%, as determined by EGFP fluorescence; data not shown), cells were lysed and immunoprecipitated with anti-T7 antibody. Bound proteins were resolved by SDS–PAGE and immunoblotted with anti-T7, anti-Cul2 and anti-Elongin B antibodies (Fig 4A). Both Ad-EGFP-T7-VHL and Ad-EGFP-T7-HPACGV, but not Ad-EGFP, co-precipitated Cul2 and Elongin B to comparable levels, suggesting equal ability of ECV complex formation (Fig 4A).
Figure 4. Ad-EGFP-T7-HPACGV forms an ECV complex and attenuates HIF-α levels and HIF-target gene expression independent of oxygen tension.

- 786-O (VHL−/−, HIF-1α−/−) cells infected with the indicated recombinant adenoviruses were lysed 48 h post-infection, immunoprecipitated with anti-T7 antibody and visualized by immunoblotting.
- 786-O cells uninfected (MOCK) or infected with the indicated adenoviruses were maintained for 48 h at 21 or 1% oxygen and whole cell extracts were analysed by immunoblotting. Asterisk represents proteins non-specifically recognized by T7 antibody.
- Luciferase assay was performed on 786-HRE-Luc cells infected with the indicated adenoviruses and maintained at 21 or 1% oxygen for 48 h. Experiments were performed in triplicate with one representative experiment shown. Error bars represent standard deviations. RLU, relative light units.
- 786-O cells were infected as above, maintained at 21 or 1% oxygen for 48 h. Whole cell extracts were then resolved by SDS–PAGE and immunoblotted with the indicated antibodies. Relative GLUT1 intensity was determined by densitometry and presented as percentage values in relation to GLUT1 signal in MOCK infected cell lysates (below top panel).
- 786-O cells were infected and maintained at 21 or 1% oxygen as above. Expression levels of GLUT1, VEGF and BNIP3L mRNAs were measured by quantitative real-time PCR and normalized to TCF8 mRNA level. Transcript level of the indicated HIF-target genes was arbitrarily set to 1.0 in cells infected with Ad-EGFP and represented as the mean value ± standard deviation of three independent experiments performed in triplicate.
We next determined the effect of Ad-EGFP-T7-HPACGV on HIF-2α protein levels under normoxia and hypoxia. 786-O cells were uninfected (MOCK) or infected with Ad-EGFP, Ad-EGFP-T7-VHL or Ad-EGFP-T7-HPACGV and maintained at 21 or 1% oxygen for 48 h. Cells were then lysed, resolved by SDS–PAGE and immunoblotted with anti-HIF-2α and anti-T7 antibodies (Fig 4B). Cells infected with Ad-EGFP-T7-VHL and Ad-EGFP-T7-HPACGV, but not Ad-EGFP, showed near-complete loss of HIF-2α expression under normoxia (Fig 4B, left panel). Importantly, while HIF-2α protein level was unaffected by Ad-EGFP-T7-VHL or Ad-EGFP infection, Ad-EGFP-T7-HPACGV dramatically attenuated the expression of HIF-2α under hypoxia (Fig 4B, right panel). These results demonstrate that, unlike Ad-EGFP-T7-VHL, which is only capable of degrading HIF-2α under normoxia, Ad-EGFP-T7-HPACGV degrades HIF-2α irrespective of oxygen tension.
We next generated 786-O cells stably expressing a firefly luciferase reporter gene driven by five HRE elements from the VEGF promoter (786-HRE-Luc). Infection of 786-HRE-Luc cells with Ad-EGFP-T7-VHL and Ad-EGFP-T7-HPACGV, but not Ad-EGFP, significantly decreased HIF-dependent HRE-driven luciferase activity under normoxia (Fig 4C). Under hypoxia, only Ad-EGFP-T7-HPACGV infected cells showed marked loss of luciferase activity (Fig 4C). Intriguingly, Ad-EGFP-T7-HPACGV had noticeably greater effect on the attenuation of HIF transactivation activity than Ad-EGFP-T7-VHL even under normoxia. This is likely a reflection of the restricted binding of VHL protein to prolyl-hydroxylated HIF-α, which is critically dependent on the Fe2+ ion, 2-oxoglutarate and oxygen- dependent activity of prolyl hydroxylase domain (PHD) enzymes compared to the unrestricted capacity of VHL–ARNT fusion protein to bind any and all unmodified or modified HIF-α.
We next assessed the endogenous HIF transcriptional activity in 786-O cells infected with Ad-EGFP, Ad-EGFP-T7-VHL or Ad-EGFP-T7-HPACGV. Consistent with the aforementioned biochemical analyses, Ad-EGFP-T7-VHL and Ad-EGFP-T7-HPACGV, but not Ad-EGFP, significantly decreased the protein level of GLUT1 under normoxia (Fig 4D, left panel). Under hypoxia, only Ad-EGFP-T7-HPACGV resulted in the marked reduction in GLUT1 (Fig 4D, right panel). Moreover, HIF-target gene transcripts such as GLUT1, VEGF and BNIP3L were reduced upon Ad-EGFP-T7-VHL and Ad-EGFP-T7-HPACGV infection under normoxia (Fig 4E). In contrast, only Ad-EGFP-T7-HPACGV was effective at decreasing GLUT1, VEGF and BNIP3L mRNA levels under hypoxia (Fig 4E).
Treatment of CCRCC xenografts with Adeno-EGFP-T7-HPACGV results in decreased angiogenesis and significant tumour necrosis
Optical coherence tomography (OCT) is a non-invasive near-infrared imaging technique that provides depth-resolved microstructural information in biological tissue at near-histology resolution (1–10 µm) (Huang et al, 1991). Depending on tissue optical properties, optical coherence tomography imaging depth ranges from 2 mm in highly scattering samples such as solid tumors to >20 mm in the eye. Recent advances in spectral domain OCT made three-dimensional in vivo imaging feasible. We developed a method of mapping normal microvasculature with high sensitivity and tracking photodynamic therapy-induced microvascular changes within the window chamber using an interframe speckle variance (SV) technique (Mariampillai et al, 2008). We apply, for the first time, the speckle variance technique to monitor tumour angiogenesis in response to the various recombinant adenoviral treatments within the skin-fold window chamber.
On day 2, 3 and 8, post-implantation of 786-O cells stably expressing red fluorescent protein DsRed2 (786-dsRed) in the dorsal skin-fold window chambers on severe combined immunodeficiency mice (see Fig S4 of Supporting Information), intratumoural injections of Ad-EGFP, Ad-EGFP-T7-VHL or Ad-EGFP-T7-HPACGV were administered at 2 × 108 ifu. The adenovirus infections were monitored by the area and intensity of EGFP expression as measured by green fluorescence imaging, and tumour xenografts were visualized using red fluorescence microscopy, while angiogenesis was assessed using white-light microscopy and sv–OCT (Fig. 5). Adenoviruses infected the tumours with high efficiency and importantly, there was a complete lack of EGFP signal in the normal mouse tissues surrounding the xenografts. This allowed the observed effects to be directly attributed to specific adenoviral infection of the tumour (Fig. 5, second rows). Ad-EGFP treatment had negligible effect on the growth of tumours, which showed extensive angiogenesis within the tumour mass (Fig 5A). Ad-EGFP-T7-VHL treatment had a modest negative effect on tumour growth with some angiogenesis occurring within the tumour as compared to the Ad-EGFP treated group (Fig 5B). In contrast, Ad-EGFP-T7-HPACGV treatment dramatically inhibited tumour angiogenesis and showed significant tumour regression, especially in the central core region by day 8 post-implantation and most noticeably by day 10, the final day of the assay (Fig 5C).
Figure 5. Ad-EGFP-T7-HPACGV treatment inhibits human CCRCC tumour xenograft angiogenesis in a dorsal skin-fold window chamber model.
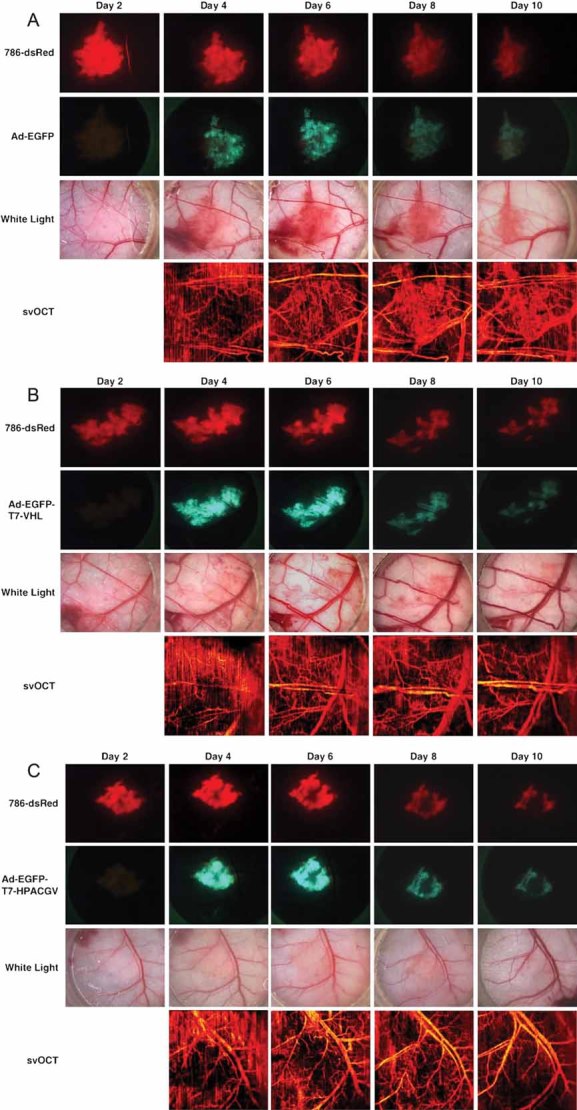
786-dsRed cells were implanted into dorsal skin-fold window chambers in SCID mice. Tumours were intratumourally injected with
- Ad-EGFP on day 2 post-implantation;
- Ad-EGFP-T7-VHL on day 3 post-implantation;
- Ad-EGFP-T7-HPACGV on day 8 post-implantation.
Tumours were visualized by red fluorescence microscopy and positivity of adenoviral infectionwas monitored by green fluorescence microscopy.Tumour angiogenesis was visualized by white-lightmicroscopy and sv–OCT. Four mice received treatments per recombinant adenovirus. Representative images are shown from each treatment group.
To further examine the regression in Ad-EGFP-T7-HPACGV-infected tumours, we performed analogous experiments in which the tumours were resected 7 days post-implantation, corresponding to 4 days post-first adenoviral treatment, for immunohistochemical analysis (Fig 6). Immediately prior to sacrificing the mice, fluorescent, white-light and svOCT images were collected for analysis. Consistently, Ad-EGFP-infected tumours were highly angiogenic, while Ad-EGFP-T7-HPACGV-infected tumours exhibited markedly lower levels of neovascularization in the tumour core (Fig 6A). Green fluorescence microscopy and anti-GFP immunohistochemical analysis of the resected tumours revealed positive GFP expression throughout Ad-EGFP-infected specimens (Fig 6A). However, while green fluorescence microscopy showed similar GFP expression in the Ad-EGFP-T7-HPACGV-infected tumours, anti-GFP immunohistochemical staining from numerous Z-stacked sections of the tumour revealed striking absence of GFP staining in the tumour core (Fig 6A). Consistent with this observation, hematoxylin and eosin (H&E) staining showed viable tumour cells throughout the Ad-EGFP-treated tumour mass, while Ad-EGFP-T7-HPACGV-treated tumours displayed an interface of viable to necrotic tumour cells in which the tumour periphery contained mostly viable cells with admixed early necrotic changes at the viable–necrotic interface to a largely necrotic tumour core with infiltrating inflammatory cells (Fig 6A and B). These results show that adenovirus-mediated expression of the VHL–ARNT fusion protein dramatically attenuates tumour angiogenesis and growth in a mouse xenograft model.
Figure 6. Adenoviral delivery of T7-HPACGV causes tumour cell death by necrosis.

- Analogous experiments were performed as in Fig 5 using Ad-EGFP (left panel) and Ad-EGFP-T7-HPACGV (right panel). Images were taken from day 7 post-implantation, corresponding to 4 days post-first adenoviral treatment. Tumours were visualized by red fluorescence microscopy and positivity of adenoviral infection was monitored by green fluorescence microscopy. Tumour angiogenesis was visualized by white-light microscopy and sv–OCT. Tumours were then resected, and H&E and anti-GFP immunohistochemistry were performed. Dashed line, viable/necrotic interface; V, viable cells; N, necrotic cells; N + I, necrotic and inflammatory cells.
- Higher magnifications of the H&E images from (A).
DISCUSSION
Accumulation of HIF-1/2α due to tumour hypoxia promotes progression and aggressiveness of cancer, and is associated with resistance to conventional cancer therapies. In addition, cancer-causing mutations on a growing list of oncogenes and tumour suppressor genes have been identified to enhance the expression of HIF-α, underscoring the significance of HIF in oncogenesis (Roberts & Ohh, 2008). Perhaps, the most direct and convincing association between tumour-causing mutations and HIF activation is the tumour suppressor VHL protein, which functions as a substrate-binding component of an E3 that polyubiquitylates prolyl-hydroxylated HIF-α for immediate destruction via the 26S proteasome (Kim & Kaelin, 2004). Moreover, CCRCC that frequently harbours mutations that inactivate the VHL protein or cause its loss, overexpresses HIF-2α and is one of the most resistant tumours to radiation or chemotherapies (Cohen & McGovern, 2005). Thus, the molecular understanding of the VHL–HIF oxygen-sensing pathway has been invaluable for conceptualizing new and targeted anti-cancer therapeutics.
Surgery by radical or partial nephrectomy is the most effective treatment option for localized renal cancer. However, in one-third of patients, tumours recur post-operatively as distant metastases, and only 4–6% of these tumours respond to chemotherapy. The standard non-surgical treatment for advanced CCRCC has been the administration of interleukin-2 (IL-2) or IFN-α. However, the response rates were generally low with considerable toxicity. For example, high-dose IL-2 regiment has a response rate of only 21% and causes significant toxicities (Cohen & McGovern, 2005). Recently, clinical trials of RTK inhibitors, such as VEGFR2 and PDGFR-β inhibitors sorafenib and sunitinib have yielded promising results by prolonging progression-free survival in approximately 70% of patients with metastatic CCRCC. However, neither drug has had a significant effect on overall patient survival (Brugarolas, 2007; Oudard et al, 2007). Sorafenib and sunitinib as well as bevacizumab (a humanized monoclonal anti-VEGF antibody) are anti-angiogenic agents that curtail tumour neovascularization (Brugarolas, 2007; Oudard et al, 2007). However, while angiogenesis is one, albeit a major cellular response initiated by HIF, there are other important HIF-regulated functions, including anaerobic metabolism and cell survival, unlikely to be affected by these drugs. Thus, some of the strategies are aimed at targeting HIF-α itself rather than the downstream targets of HIF. For example, the most recent FDA approval was granted to temsirolimus, an injectable mTOR inhibitor as a first-line therapy for advanced CCRCC (Costa & Drabkin, 2007). The anti-tumour effects of temsirolimus are presumed to be through the inhibition of cap-dependent translation of HIF-1α. However, mTOR-mediated translation decreases under hypoxia as an energy-conserving measure. Under these circumstances, HIF-1α mRNA translation is ensured through IRES-dependent mechanisms (van den Beucken et al, 2006). This alternative method for HIF-1α synthesis under hypoxia questions the activity of mTOR inhibitors in hypoxic tumours. Moreover, numerous other small molecule inhibitors such as gefitinib, trastuzumab or topotecan in comparison to HDAC inhibitor LAQ824 have been shown to reduce the expression of HIF-1α (Melillo, 2007; Semenza, 2007). However, none of these agents directly targets HIF-1α with each drug having additional functions other than blocking HIF-1α. Thus, new strategies to inactivate HIF-1α, as well as HIF-2α, exclusively and directly would constitute a major conceptual advancement in anti-cancer therapeutics.
In the present study, we describe the re-engineering of an E3 ligase ECV containing the VHL–ARNT chimaeric F-box component to remove the oxygen constraint in the recruitment of HIF-α to promote its rapid degradation under any oxygen tension. The expression of VHL–ARNT fusion protein in hypoxic tumour cells with constitutive stabilization of HIF-1α and/or HIF-2α leads to a dramatic and highly efficient downregulation of HIF activity, ultimately suppressing the growth of CCRCC xenografts. Notably, the strategy of chimaeric F-box proteins in the context of SCF (Skp1/Cul1/F-box protein) E3 ligase has been described for targeted proteolysis of cellular proteins including Rb (Zhou et al, 2000), β-catenin (Cong et al, 2003; Liu et al, 2004; Su et al, 2003), c-myc (Cohen et al, 2004) and cyclin A–Cdk2 complex (Chen et al, 2004). For example, Cong et al showed that an SCF complex containing a chimaeric protein with the β-catenin binding domain of E-cadherin fused to the F-box protein βTrCP targets the stable β-catenin mutant for ubiquitin-mediated destruction (Cong et al, 2003). Hence, the replacement of the substrate-binding interface of any F-box protein with the HIF-α-binding region of ARNT (i.e., HPAC) may have been sufficient to promote oxygen-independent degradation of HIF-α. However, emerging evidence suggests that HIF-α triggers its own degradation by initiating the conjugation of ubiquitin-like NEDD8 onto Cul2, which enhances the E3 activity of ECV (Sufan & Ohh, 2006). SCF was also enriched in neddylated Cul1 upon SCF-specific substrate recruitment (Read et al, 2000), suggesting coordination between substrate-SCF binding and neddylation. The precise mechanism(s) governing substrate-dependent triggering of E3 function remains incompletely understood, but suggests molecular interplay between substrates and substrate-specific E3s. While we have presented evidence of feasibility in promoting human CCRCC xenograft regression and cell death using the bioengineered VHL–ARNT chimaera in an animal model system, it remains an outstanding question whether such a treatment would lead to the growth suppression of other tumour types exhibiting strong hypoxic signatures with overexpression of HIF-α.
MATERIALS AND METHODS
Cells
786-O (VHL−/−; HIF-1α−/−) CCRCC cells and human embryonic kidney cells (HEK293A) were obtained from American Type Culture Collection (Rockville, MD, USA). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (Sigma, St. Louis, MO, USA) at 37°C in a humidified 5% CO2 atmosphere. 786-O cells stably expressing DsRed2 (786-dsRed) were generated by nucleofecting (Amaxa, Gaithersburg, MD, USA) the mammalian expression plasmid encoding DsRed2 (Clontech, Palo Alto, CA, USA). Clones were selected with 500 µg/ml G418 (Sigma–Aldrich, Oakville, ON, Canada) and red fluorescence was confirmed using a fluorescence microscope (Nikon Eclipse TE200). Similarly, 786-HRE-Luc cells were generated by nucleofecting 5xHRE-Luciferase mammalian expression plasmid (kindly provided by Dr R. P. Hill) in combination with an empty mammalian expression plasmid pcDNA3.1 encoding the neomycin resistance gene. Positive clones were selected with 500 µg/ml G418 and luciferase expression was confirmed by luciferase assay.
Antibodies
Monoclonal anti-T7, anti-HA (12CA5), anti-α-tubulin, anti-vinculin, anti-GFP and anti-AhR antibodies were obtained from Novagen (Madison, WI, USA), Boehringer Ingelheim (Laval, QC, Canada), Sigma–Aldrich (Oakville, ON, Canada), Millipore (Billerica, MA, USA), Covalence (Berkeley, CA, USA) and Abcam (Cambridge, MA, USA), respectively. Monoclonal anti-HIF-1α and polyclonal anti-HIF-2α were obtained from Novus Biologicals (Littleton, CO, USA). Anti-Cul2, anti-Elongin B and anti-GLUT1 polyclonal antibodies were obtained from Invitrogen (Burlington, ON, Canada), Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Alpha Diagnostics International (San Antonio, TX, USA), respectively. MG132 proteasome inhibitor was obtained from Boston Biochem (Cambridge, MA, USA).
The paper explained
PROBLEM:
Under normal oxygen tension, von Hippel–Lindau (VHL) tumour suppressor protein promotes the degradation of a key transcription factor called the hypoxia-inducible factor (HIF), which governs cellular adaptation to hypoxia or low oxygen tension. Tumour hypoxia, a common feature of solid tumours, or mutations in the VHL gene cause inappropriate accumulation of HIF, the extent of expression of which correlates with disease progression, poor prognosis and resistance to radiation and chemotherapy. Thus, various drugs designed to block the activity of HIF have been recognized as attractive strategies for cancer therapy, but have shown limited success in the clinic.
RESULTS:
In the present study, the authors describe the bioengineering of a VHL protein that can recognize HIF and promote its degradation in the absence of oxygen. The study goes further and demonstrates the potency of this engineered VHL on the growth of arguably one of the most resistant tumours to conventional cancer therapy, kidney cancer, in a mouse model system.
IMPACT:
This report introduces the concept and feasibility of ‘bio-tailored’ proteins in the treatment of tumours that overexpress HIF and highlights the potential of this novel anti-cancer strategy.
Plasmids
The ARNT truncation plasmids T7-bHPAS (residues 90-419), T7-HPAS (103-419), T7-bHPAC (90-467) and T7-HPAC (103-467) were generated using the following primers introducing 5′-BamHI and 3′-XbaI sites. ARNT basic region forward primer: 5′-GCGCGGATCCATGGCCAGGGAAAATCAC-3′, ARNT HLH forward primer: 5′-GCGCGGATCCATGAACAA GATGACAGCC-3′, ARNT PAS B reverse primer: 5′-GCGCTCTAGA-TCATTTTAATTTCACTAC-3′, ARNT PAC reverse primer: 5′-GCGCTCTAGATCAAGAGTTCTTCACATTG-3′. The polymerase chain reaction (PCR) products were digested with BamHI and XbaI and ligated into pcDNA3-T7 cut with these restriction enzymes. T7-HPACV, T7-HPACGV, T7-bHPACV and T7-bHPACGV (VHL α domain residues 151–194) were generated using the above-mentioned forward primers, a reverse PAC primer introducing a 3′-XbaI site without a Stop codon: 5′-GCGCTCTAGAAGAGTTCTTCACATTG-3′, and the appropriate combination of the following VHL primers, introducing a 5′-XbaI site and a 3′-ApaI site. VHL forward primer: 5′-GCGCTCTAGAATCACACTGCCAGTGTATAC-3′, VHL forward primer including the Gly(6)linker: 5′-GCGCTCTAGAGGTGGTGGTGGTGGTGGTATCACACTGCCAGTG-3′ and VHL reverse primer: 5′-GCGCGGG-CCCTCACACATTTGGGTGGTC-3′. The corresponding PCR products were ligated into pcDNA3-T7 cut with BamH1 and Apa1 by a three-way ligation. All plasmids were confirmed by DNA sequencing. pcDNA3- HA-HIF-1α, pcDNA3-HA-HIF-1α(P564A) pcDNA3-T7-VHL, pcDNA3.1-(HRE)5-Luc expression plasmids were described previously (Ivan et al, 2001; Maynard et al, 2007; Ohh et al, 2000). AhR was a gift from Dr C. A. Bradfield and pGL3-5x-HRE-Luc was kindly provided by Dr R. P. Hill. A plasmid encoding DsRed2 was obtained from Clontech (Palo Alto, CA, USA).
Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblotting were performed as described previously (Evans et al, 2007).
In vitro binding assays
In vitro binding assays were performed as described previously (Ivan et al, 2001; Maynard et al, 2007; Ohh et al, 2000).
Hypoxia treatment of cells
Cells were maintained at 1% O2 in a ThermoForma (Marietta, OH, USA) hypoxia chamber. Cell lysates were prepared in the chamber in a hypoxic environment before further experimentation.
Luciferase assays
The dual-luciferase assay was performed as described previously (Evans et al, 2007).
Infection of 786-O or 786-HRE-Luc cells with recombinant adenovirus
Ad-EGFP (where EGFP stands for enhanced green fluorescence protein), Ad-EGFP-T7-VHL and Ad-EGFP-T7-HPACGV were generated by the Viral Vector Core Facility (Neuroscience Research Institute, University of Ottawa, Ottawa, ON, Canada). 786-O or 786-HRE-Luc cells were infected at 70% confluency with a final multiplicity of infection (MOI) of 300. Cells were lysed 48 h after infection for further experimentation.
Quantitative real-time PCR
Quantitative real-time PCR was performed and results were interpreted as described previously (Evans et al, 2007). The following primer sets were used: GLUT 1 5′-CACCACCTCACTCCTGTTACTT-3′ and 5′-CAAGCATTTCAAAACCATGTTTCTA-3′, VEGF 5′-CTCTCTCCCTCATCGGTGACA-3′ and 5′-GGAGGGCAGAGCTGAGTGTTAG-3′, BNIP3L 5′-CTGCACAAACTTGCACATTG-3′ and 5′-TAATTTCCACAACGGGTTCA-3′, and TCF8 5′-CCTAGTAAGCCAAATTAGGATTAAC-3′ and 5′-GGAACAAATTGGCACAAAATGTT-3′. Values were normalized to the expression of TCF8 mRNA, expressed relatively to Ad-EGFP-infected samples (arbitrarily set to 1.0), and represented as the mean value ± standard deviation of three independent experiments performed in triplicate.
Mouse dorsal skin-fold window chamber assay
All animal experiments were performed in accordance with the institutional animal care guidelines (University Health Network, ON, Canada). SCID mice were anaesthetized by intraperitoneal injection of a mixture of ketamine and xylazine (80 and 5 mg/kg, respectively). Dorsal window chambers were installed as described previously (Algire & Legallais, 1949). Briefly, a circular incision of 1 cm diameter was made in the dorsal skin and the titanium chamber was surgically implanted. 7 × 105 786-dsRed tumour cells were implanted in the dermis on the right dorsal side of the mouse using an 18G syringe. A circular glass coverslip was positioned over the incision allowing visualization of the tumour and longitudinal monitoring of the associated vasculature. Adenoviral intratumoural injections with Ad-EGFP, Ad-EGFP-T7-VHL or Ad-EGFP-T7-HPACGV were performed using 2 × 108 infectious units (ifu).
In vivo imaging
Fluorescence and white-light imaging were performed using an MZ FLIII, Leica stereomicroscope. Tumours, as identified by red fluorescence and white-light stereomicroscope images, were also imaged non-invasively using OCT (Huang et al, 1991). A 36 kHz 1.3 µm swept source OCT system was used to probe tissue microstructure and microvasculature with approximately 10 µm resolution. Three-dimensional image stacks over a 5 × 5 × 2 mm3 volume of the window chamber were acquired with 1600 × 2000 × 512 pixels. Image stacks were processed to produce two-dimensional projection maps of microvasculature within the volume using the interframe sv algorithm as described previously (Mariampillai et al, 2008). Low pass Gaussian filter was used for motion artefact removal and all images were normalized to the same false colour map. This technique allows for blood vessels with diameters as small as 25 µm to be visualized without the use of any exogenous contrast agents during longitudinal studies.
Immunohistochemical staining
Sample preparation and immunohistochemical staining were performed as described previously (Evans et al, 2007).
Acknowledgments
We thank the members of the Ohh laboratory for their helpful comments and discussions. We also thank Mark Harduar, Tim Luk, Wei Shi and Michael Leung for their assistance with the animal experiments. This work has been supported by the Canadian Institutes of Health Research (MOP77718), Canadian Cancer Society Research Institute (18460 and 16056) and the Kidney Foundation of Canada (KFOC). RIS is a recipient of the Ontario Graduate Scholarship and OR is a recipient of the Canadian Institute of Health research post-doctoral Fellowship. VXDY is a Canada Research Chair in Biophotonics. MO is a Canada Research Chair in Molecular Oncology.
Supporting Information is available at EMBO Molecular Medicine online (http://www.embomolmed.org).
The authors declare that they have no conflict of interest.
For more information
VHL Family Alliance:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Algire GH, Legallais FY. Recent developments in the transparent-chamber technique as adapted to the mouse. J Natl Cancer Inst. 1949;10:225–253. incl 228 pl. [PubMed] [Google Scholar]
- Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- Brugarolas J. Renal-cell carcinoma–molecular pathways and therapies. N Engl J Med. 2007;356:185–187. doi: 10.1056/NEJMe068263. [DOI] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- Chen W, Lee J, Cho SY, Fine HA. Proteasome-mediated destruction of the cyclin a/cyclin-dependent kinase 2 complex suppresses tumor cell growth in vitro and in vivo. Cancer Res. 2004;64:3949–3957. doi: 10.1158/0008-5472.CAN-03-3906. [DOI] [PubMed] [Google Scholar]
- Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med. 2005;353:2477–2490. doi: 10.1056/NEJMra043172. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Scott DK, Miller J, Zhang J, Zhou P, Larson JE. Transient in utero knockout (TIUKO) of C-MYC affects late lung and intestinal development in the mouse. BMC Dev Biol. 2004;4:4. doi: 10.1186/1471-213X-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong F, Zhang J, Pao W, Zhou P, Varmus H. A protein knockdown strategy to study the function of beta-catenin in tumorigenesis. BMC MolBiol. 2003;4:10. doi: 10.1186/1471-2199-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LJ, Drabkin HA. Renal cell carcinoma: new developments in molecular biology and potential for targeted therapies. Oncologist. 2007;12:1404–1415. doi: 10.1634/theoncologist.12-12-1404. [DOI] [PubMed] [Google Scholar]
- Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan LJ, Zhang-Benoit Y, Fong GH. Endothelium-intrinsic requirement for Hif-2alpha during vascular development. Circulation. 2005;111:2227–2232. doi: 10.1161/01.CIR.0000163580.98098.A3. [DOI] [PubMed] [Google Scholar]
- Elvert G, Kappel A, Heidenreich R, Englmeier U, Lanz S, Acker T, Rauter M, Plate K, Sieweke M, Breier G, et al. Cooperative interaction of hypoxia-inducible factor-2alpha (HIF-2alpha) and Ets-1 in the transcriptional activation of vascular endothelial growth factor receptor-2 (Flk-1) J Biol Chem. 2003;278:7520–7530. doi: 10.1074/jbc.M211298200. [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VW, Kim WY, Saravanan A, Maynard MA, Gervais ML, et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007;27:157–169. doi: 10.1128/MCB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaratnam L, Morley M, Franovic A, de Paulsen N, Mekhail K, Parolin DA, Nakamura E, Lorimer IA, Lee S. Hypoxia inducible factor activates the transforming growth factor-alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(−/−) renal cell carcinoma cells. J Biol Chem. 2003;278:44966–44974. doi: 10.1074/jbc.M305502200. [DOI] [PubMed] [Google Scholar]
- Huang D, Swanson EA, Lin CP, Schuman JS, Stinson WG, Chang W, Hee MR, Flotte T, Gregory K, Puliafito CA, et al. Optical coherence tomography. Science. 1991;254:1178–1181. doi: 10.1126/science.1957169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci USA. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kewley RJ, Whitelaw ML, Chapman-Smith A. The mammalian basichelix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol. 2004;36:189–204. doi: 10.1016/s1357-2725(03)00211-5. [DOI] [PubMed] [Google Scholar]
- Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–246. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Liu J, Stevens J, Matsunami N, White RL. Targeted degradation of beta-catenin by chimeric F-box fusion proteins. Biochem Biophys Res Commun. 2004;313:1023–1029. doi: 10.1016/j.bbrc.2003.12.035. [DOI] [PubMed] [Google Scholar]
- Mariampillai A, Standish BA, Moriyama EH, Khurana M, Munce NR, Leung MK, Jiang J, Cable A, Wilson BC, Vitkin IA, et al. Speckle variance detection of microvasculature using swept-source optical coherence tomography. Opt Lett. 2008;33:1530–1532. doi: 10.1364/ol.33.001530. [DOI] [PubMed] [Google Scholar]
- Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Maynard MA, Evans AJ, Shi W, Kim WY, Liu FF, Ohh M. Dominant-negative HIF-3 alpha 4 suppresses VHL-null renal cell carcinoma progression. Cell Cycle. 2007;6:2810–2816. doi: 10.4161/cc.6.22.4947. [DOI] [PubMed] [Google Scholar]
- Maynard MA, Ohh M. Von Hippel-Lindau tumor suppressor protein and hypoxia-inducible factor in kidney cancer. Am J Nephrol. 2004;24:1–13. doi: 10.1159/000075346. [DOI] [PubMed] [Google Scholar]
- Maynard MA, Ohh M. The role of hypoxia-inducible factors in cancer. Cell Mol Life Sci. 2007;64:2170–2180. doi: 10.1007/s00018-007-7082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev. 2007;26:341–352. doi: 10.1007/s10555-007-9059-x. [DOI] [PubMed] [Google Scholar]
- Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- Oudard S, George D, Medioni J, Motzer R. Treatment options in renal cell carcinoma: past, present and future. Ann Oncol. 2007;18(Suppl 10):x25–x31. doi: 10.1093/annonc/mdm411. [DOI] [PubMed] [Google Scholar]
- Read MA, Brownell JE, Gladysheva TB, Hottelet M, Parent LA, Coggins MB, Pierce JW, Podust VN, Luo RS, Chau V, et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20:2326–2333. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AM, Ohh M. Beyond the hypoxia-inducible factor-centric tumour suppressor model of von Hippel-Lindau. Curr Opin Oncol. 2008;20:83–89. doi: 10.1097/CCO.0b013e3282f310de. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12:853–859. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Kaelin WG, Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- Su Y, Ishikawa S, Kojima M, Liu B. Eradication of pathogenic beta-catenin by Skp1/Cullin/F box ubiquitination machinery. Proc Natl Acad Sci USA. 2003;100:12729–12734. doi: 10.1073/pnas.2133261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sufan RI, Ohh M. Role of the NEDD8 modification of Cul2 in the sequential activation of ECV complex. Neoplasia. 2006;8:956–963. doi: 10.1593/neo.06520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- van den Beucken T, Koritzinsky M, Wouters BG. Translational control of gene expression during hypoxia. Cancer Biol Ther. 2006;5:749–755. doi: 10.4161/cbt.5.7.2972. [DOI] [PubMed] [Google Scholar]
- Warnecke C, Zaborowska Z, Kurreck J, Erdmann VA, Frei U, Wiesener M, Eckardt KU. Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J. 2004;18:1462–1464. doi: 10.1096/fj.04-1640fje. [DOI] [PubMed] [Google Scholar]
- Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, et al. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood. 1998;92:2260–2268. [PubMed] [Google Scholar]
- Zhou P, Bogacki R, McReynolds L, Howley PM. Harnessing the ubiquitination machinery to target the degradation of specific cellular proteins. Mol Cell. 2000;6:751–756. doi: 10.1016/s1097-2765(00)00074-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.