

Abstract
It is generally accepted that a distinguishing property of stem cells (SCs), as compared to their more differentiated progenitors, is that of infrequent division, often referred to as ‘quiescence’. As regards hematopoietic stem cells (HSC), their resistance to antiproliferative drugs supports this notion. Maintenance of quiescence is thought to be critical for the preservation of HSCs' function.
Keywords: cancer, quiescence, stem cell
Indeed, hyper proliferation results in the functional exhaustion of HSCs, as shown by their failure to reconstitute hematopoiesis after serial transplantations from one mouse to another (usually after 5–6 passages for the wild-type HSCs; much less for HSCs with null-mutations of cell-cycle inhibitory or DNA-repair genes). This might be due to accumulation of genomic damage during DNA replication. Mutations arising from such damage would then result in progressive loss of typical SC functions, in particular their capacity to replicate (self-renewal). By limiting the accumulation of mutations, quiescence could have the additional function of preventing transformation of HSCs. However, this still requires experimental confirmation.
But what exactly is the SC quiescence? That most SCs are non-cyclic under steady-state conditions is well established. It is not clear though if quiescence is simply the interval between one cell division and the next (however long it may be) or a defined state of ‘dormancy’. This is not just a semantic issue. In the former case, SCs should be regarded as a homogeneous population of cells that divide infrequently (and asynchronically). In the latter, SCs would have the capacity of actively entering and exiting a ‘functional’ state of dormancy. Available experimental evidence is conflicting. Recent in vivo studies in mice have demonstrated that the fraction of slowly proliferating HSCs is small (∼5%) and not so ‘dormant’ (turning over every ∼2 months) (Cheshier et al, 1999; Kiel et al, 2007), while other studies have documented features of ‘dormancy’ such as reduced metabolism and ribosomal biogenesis, in a non-cycling fraction of the adult HSCs.
»What exactly is the SC quiescence?«
Much of the debate concerning this issue arises from technological challenges: (i) HSCs are extremely rare (<0.01% in the bone marrow) and their purification—based on surface markers—is difficult (no more than ∼20 and ∼50% in the Lin-Sca+Kit+CD34- and Lin-Sca+Kit+CD150+CD48- cell populations, respectively (Kiel et al, 2005; Osawa et al, 1996); and (ii) most importantly, it is difficult to purify HSCs based on their cell-cycle properties. The method most frequently used to analyse in vivo dormant HSCs is the 5′-bromo-2′-deoxyuridine (BrdU)-label retaining assay, where animals are first treated with BrdU for long intervals of time, usually 10 or more days (pulse), and then analysed months after suspending the BrdU intake (chase). In this assay, cells maintaining the BrdU-positivity (label retaining cell: LRC) are considered ‘quiescent’ or slowly proliferating.
Two recent studies by Andreas Trumpp's group (Essers et al, 2009; Wilson et al, 2008) address some of these issues, while opening, as often happens, new ones. In the first work, Wilson et al. demonstrate the existence of a conspicuous HSC population that spends most of its time outside the cell cycle, in a state of apparent ‘dormancy’. The experimental approach that has allowed unambiguous definition of this population is based on a new pulse–chase system, optimized by the authors, which allows purification of LRCs. The authors used transgenic mice that express, in an inducible manner, a histone H2B-green fluorescent protein (H2B-GFP) specifically in HSC/progenitors. The animals are first induced to express the H2B-GFP in HSCs (pulse) and, subsequently, chased and analysed after several months. This method allows the identification of ‘quiescent’ or slowly proliferating SCs (as the BrdU assay) and LRC (GFP+) HSCs can be purified as living cells, thus allowing functional analysis of HSCs with different proliferative histories (GFP intensity inversely correlates with the number of undergone divisions).
Wilson et al. (2008) demonstrated that <2% of the HSCs are actively cycling (S+G2/M phases) and, using either the BrdU or H2B-GPF pulse–chase system, that 20–30% of HSCs are LRC between 70 and 200 days of chase. Then the authors separated LRC (GFP+) from non-LRC (GFP−) HSCs, analysed their functions and showed that: (i) slowly proliferating LRC-HSCs have a better multilineage bone-marrow reconstitution capacity than highly-proliferating non-LRC-HSCs (by competitive bone marrow transplantation); and (ii) most notably, self-renewal potential is markedly higher in LRC-HSCs compared to non-LRC-HSCs (by serial bone marrow transplantation). These results provide a direct demonstration that the self-renewal potential of HSCs inversely correlates with the HSC mitotic division number.
This correlation, however, is not linear in time. Since GFP or BrdU-pulsed cells lose labelling after 4–5 cellular divisions, HSCs should be able to maintain self-renewal capacity intact for up to 5 cellular divisions, and, afterwards, progressively lose it. However, the mathematical modelling of the temporal dynamics of the BrdU or H2B-GFP decay in LRC-HSCs revealed the existence of two distinct sub-populations: a subset (∼15%) of ‘dormant’ HSCs (dHSCs) dividing approximately every 145 days, and a subset (85%) of ‘activated’ HSCs (aHSCs) dividing approximately every 36 days. This difference in doubling time is such that from 180 days of chasing, the LRC (GFP+) and non-LRC (GFP−) HSCs coincide, respectively, with dHSC and aHSC. Notably, these two HSC populations have different expression profiles, including a marked down-regulation in the dormant HSCs of almost all genes involved in the assembly and activation of the pre-replicative complex.
A critical question is whether these two HSC populations are hierarchically linked. Although preliminary, Wilson's data suggest that aHSCs originate from dHSCs and, subsequently, are able to re-enter a state of dormancy: (i) nearly all the LRC-dHSCs enter the cell cycle (thus becoming non-LRC-aHSCs) after treatment with the chemotherapeutic 5′-fluorouracil (5-FU), and (ii) 5-FU-activated and BrdU-pulsed HSCs are able to reconstitute a steady state pool of LRC-dHSCs. But what are the physiological stimuli regulating the progression of one dHSC into an aHSC? Essers et al. (2009) demonstrated that dHSCs efficiently exit quiescence and enter an active cell cycle after treatment with the hematopoietic growth factor G-CSF or with interferon (INFα), a cytokine that is produced by mononuclear phagocytes in response to viral infections. G-CSF and interferons are implicated in triggering tissue regeneration following injury, thus suggesting that they might represent specific mediators of SC hyper-proliferation after tissue injury.
»What are the physiological stimuli regulating the progression of one dHSC into an aHSC?«
Altogether, the data presented in the above cited papers are in line with the hypothesis that under homeostatic conditions, HSCs belong to two separate cellular pools: one consisting of dHSCs, rarely proliferating (every 150 days, ∼5 divisions per mouse lifetime) and which maintain a near-intact self-renewal potential, and one consisting of aHSCs which instead proliferate frequently (every 30 days) but with decrease in self-renewal potential. dHSCs might serve as a reserve pool of HSCs and can be robustly activated to proliferate in response to injury. After the lost tissue or cells have been replaced, the dormant HSC pool is then restored by aHSCs and homeostasis re-established.
But how is the dHSC pool maintained intact, when, in answer to injury or specific stimuli, it is induced to massively proliferate? In particular, after 5-FU, G-CSF or INFα treatments, most of the LRC-dHSC become non-LRC, and, consequently, should have their self-renewal ability strongly reduced. They maintain, instead, an intact self-renewal potential, as shown by the transplantation experiments using bone marrow from mice treated three consecutive times with INFα. Thus, dHSCs can proliferate much more than five times without losing the self-renewal potential, in apparent contradiction with the previous results. One possibility is that a non-LRC-aHSC can re-enter a state of dormancy, thus replenishing the pool of dHSCs, similarly to what happens in the Drosophila ovary, for example, where progenitor cells can re-achieve stem cell properties when germline stem cells are eliminated (Xie & Spradling, 2000).
Alternatively, only a fraction of the dHSC pool may be induced to proliferate after 5-FU, G-CSF or INFα, and, therefore, the dHSC pool is not drained by these treatments. The presence of a fraction, even a considerable fraction, of non-proliferating dHSCs after tissue injury could remain undetected due to its dilution with hyper proliferating aHSCs. Indeed, the total number of HSCs expands dramatically after tissue injury (Passegue et al, 2005; Randall & Weissman, 1997). According to this hypothesis, at each division, dHSCs might generate two daughter SCs: one will re-enter dormancy, thus replenishing the pool of dHSCs, while the other will actively proliferate, thus supporting tissue regeneration (Fig. 1). Implicit in this model is the fact that, at each new treatment with G-CSF, INFα or 5-FU, the dHSCs will lose some of their self-renewal potential, until functional exhaustion. This is indeed what happens after eight consecutive treatments with INFα (Essers et al, 2009), or after 4–5 consecutive treatments with 5-FU (Cheng et al, 2000). By pushing this model further, one could hypothesize that similar mechanisms might also be active under steady-state conditions, when dHSCs are prompted to cycle in order to replenish exhausting aHSCs and maintain tissue homeostasis. This might happen very infrequently at any given time, yet continuously during a life span, leading to the progressive exhaustion of the HSC pool. The number of self-renewing dHSCs would thereby reflect the age of an individual. Notably, in aged mice, HSCs are more in number, yet are functionally exhausted (Rossi et al, 2007).
Figure 1. Hierarchical model of regulation of dormant and activated HSCs.
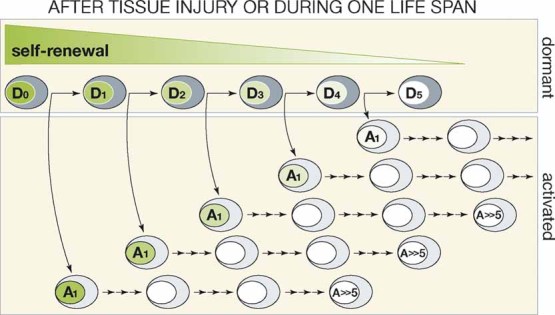
A dormant HSC (D) undergoes asymmetric division and generates one daughter cell which re-enters into dormancy and one daughter cell which actively proliferates (A1-A ≫ 5). Both dormant and activated HSCs progressively lose the self-renewal potential during proliferation (loss of self-renewal occurs after five divisions as experimentally determined). While entry into cell cycle is assumed to occur infrequently under steady-state condition, it occurs simultaneously for almost all cells after tissue injury or cumulatively during life-span.
The existence of dormant SCs that can be induced to proliferate with specific factors bears important implications for cancer treatment. Emerging evidence supports the hypothesis that individual tumours are biologically heterogeneous and contain a minor population of cells (cancer SCs) that share the self-renewal feature of normal SCs, are responsible for sustaining the tumour and give rise to proliferating and progressively differentiating cells. An obvious consequence is that cancer cure is unlikely to occur if the rare cancer SCs are not targeted. This might constitute one reason for the frequent failure of current anticancer therapies, since they have been developed to decrease the bulk of the tumour mass, thus not necessarily involving targeting of cancer SCs.
»Cancer cure is unlikely to occur if the rare cancer SCs are not targeted.«
A number of recent reports suggest that a sizeable fraction of cancer SCs are quiescent (Ishikawa et al, 2007; Viale et al, 2009), implying that cell-cycle restriction, like in the wild-type SCs, might serve in cancer SCs to prevent self-renewal exhaustion. Notably, it has been recently reported that expression of leukaemia-associated oncogenes in HSCs induces DNA damage and activates a cellular response that, in turn, imposes cell-cycle restriction and favours repair of damaged DNA, thus protecting leukaemia SCs from accumulation of excess genomic damage and physiological exhaustion (Viale et al, 2009). Together, these data imply that cell-cycle restricted leukaemia SCs are critical for the maintenance of the leukaemic clone, suggesting that their targeting might be critical to disease eradication.
One potential approach to target cell-cycle restricted cancer SCs is suggested by the two papers discussed here: a concomitant treatment with agents that boost proliferation of dormant cancer SCs and drugs that kill the proliferating cells (cell-cycle specific). In principle, this treatment program might render dormant cancer SCs sensitive to chemotherapy. The idea of using growth factors or cytokines to make cancer cells more sensitive to chemotherapy is not a new one. Results, however, have not been satisfactory. In a randomized clinical trial involving a large cohort of leukaemia patients, the combination of G-CSF and chemotherapy reduced the relapse rate without, however, improving the rate of complete responses or overall survival (Lowenberg et al, 2003). Should we then conclude that this novel approach is already over? In the clinical trial, G-CSF was administered chronically, before and during the first two courses of chemotherapy, while it was given as bolus injection in the mouse studies discussed here, a difference that might explain the modest effects on leukaemias of this treatment modality. Alternatively, quiescent leukaemia SCs might be partially resistant to G-CSF priming. Quiescence of leukaemic SCs is imposed by increased expression of the cell-cycle inhibitor p21 in response to oncogene-induced DNA damage (Viale et al, 2009), a condition that might reduce sensitivity to G-CSF. Do we need more potent signals (other growth factors or combinations) to push the dormant leukaemia SCs into the cell cycle? Should we target instead the p21-pathway? Notably, deletion of p21 in mouse models induces hyper-proliferation of leukaemia SCs and inhibits leukaemia outgrowth (Viale et al, 2009). In summary, this new biological and genetic information clearly indicates that awaking leukaemia stem cells from dormancy is a potentially powerful strategy for leukaemia eradication. We look forward to further developments in this exciting and promising new field.
Acknowledgments
We thank Saverio Minucci and Luisa Lanfrancone for a critical reading of this manuscript and Paola Dalton for her scientific editing.
The authors declare that they have no conflict of interest.
References
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl Acad. Sci. USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009 doi: 10.1038/nature07815. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007;25:1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, He S, Ashkenazi R, Gentry SN, Teta M, Kushner JA, Jackson TL, Morrison SJ. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature. 2007;449:238–242. doi: 10.1038/nature06115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Lowenberg B, van Putten W, Theobald M, Gmur J, Verdonck L, Sonneveld P, Fey M, Schouten H, de Greef G, Ferrant A, et al. Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. New Engl J Med. 2003;349:743–752. doi: 10.1056/NEJMoa025406. [DOI] [PubMed] [Google Scholar]
- Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall TD, Weissman IL. Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood. 1997;89:3596–3606. [PubMed] [Google Scholar]
- Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, Ronchini C, Ronzoni S, Muradore I, Monestiroli S, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukemia stem cells. Nature. 2009;457:51–56. doi: 10.1038/nature07618. [DOI] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Xie T, Spradling AC. A niche maintaining germ line stem cells in the Drosophila ovary. Science. 2000;290:328–330. doi: 10.1126/science.290.5490.328. [DOI] [PubMed] [Google Scholar]