

Abstract
Loud sound exposure is a significant cause of hearing loss worldwide. We asked whether a lack of vezatin, an ubiquitous adherens junction protein, could result in noise-induced hearing loss. Conditional mutant mice bearing non-functional vezatin alleles only in the sensory cells of the inner ear (hair cells) indeed exhibited irreversible hearing loss after only one minute exposure to a 105 dB broadband sound. In addition, mutant mice spontaneously underwent late onset progressive hearing loss and vestibular dysfunction related to substantial hair cell death. We establish that vezatin is an integral membrane protein with two adjacent transmembrane domains, and cytoplasmic N- and C-terminal regions. Late recruitment of vezatin at junctions between MDCKII cells indicates that the protein does not play a role in the formation of junctions, but rather participates in their stability. Moreover, we show that vezatin directly interacts with radixin in its actin-binding conformation. Accordingly, we provide evidence that vezatin associates with actin filaments at cell–cell junctions. Our results emphasize the overlooked role of the junctions between hair cells and their supporting cells in the auditory epithelium resilience to sound trauma.
Keywords: adherens junction, mouse model, noise-induced hearing loss, organ of Corti, vezatin
INTRODUCTION
Noise-induced hearing loss (NIHL) is a major clinical concern in modern societies. This has driven efforts to understand the underlying cellular and molecular mechanisms. These mechanisms are currently viewed as mainly originating in the auditory sensory epithelium, the organ of Corti. One of them consists of excessive glutamate release from the auditory sensory cells, leading to dendritic swelling of the afferent auditory neurons and ultimately, neural degeneration (Kujawa & Liberman, 2006). Lesions of the hair bundle, the mechano-electrical transduction organelle located at the apical part of the hair cells (HCs), have also been reported (Canlon et al, 1987; Erulkar et al, 1996; Fowler et al, 1995; Mulroy & Whaley, 1984; Saunders et al, 1986). A possible contribution of cell–cell junction defects to NIHL, however, has not yet been investigated.
The organ of Corti contains two types of HCs, the inner HCs (IHCs) that are the genuine sensory cells, organized in a single row, and the outer HCs (OHCs), organized in three rows. Both cell types transduce sound-evoked mechanical stimulation into electrical signal, i.e. changes in membrane potential. As a result, IHCs release glutamate neurotransmitter, and thereby transfer acoustic information to the auditory neurons, whereas OHCs supply forces for local amplification of the sound-induced mechanical stimulation of the organ of Corti. This amplification process is thought to involve alternating changes of the length and stiffness of OHC lateral wall in response to membrane potential fluctuations, a process known as electromotility (Ashmore, 1987; Brownell et al, 1985; Dallos et al, 2008; He & Dallos, 1999; Liberman et al, 2002; Russell et al, 2007). The reticular lamina, the surface layer of the organ of Corti formed by the tightly joined apical parts of HCs and their supporting cells (SCs) (Gulley & Reese, 1976), not only contributes to the ion barrier between the two cochlear fluids, endolymph and perilymph (Wangemann, 2006), but also withstands mechanical stress imposed by the sound-evoked vibration of the organ of Corti (Chan & Hudspeth, 2005; Fridberger et al, 2002; Karavitaki & Mountain, 2007; Tomo et al, 2007). In most epithelia, the sealing between cells is operated by the apical junctional complex (AJC), which is comprised of a tight junction (TJ) that serves as a solute permeability barrier, and an adherens junction (AJ) that maintains tissue integrity under mechanical stress (Aijaz et al, 2006; Bershadsky, 2004; Ivanov, 2008). TJs and AJs most often have distinct spatial distributions and consist of mostly non-overlapping sets of proteins (Farquhar & Palade, 1963; Perez-Moreno et al, 2003). In the organ of Corti, HCs form junctions with their SCs only. Until recently, these junctions were thought to be composed of several alternating TJ and AJ specializations along their apico–basal length (Raphael & Altschuler, 2003). Freeze-fracture electron microscopy and immunolabelling studies, however, have shown that the junctions between OHCs and their SCs, the Deiters cells (DCs), are single large (3–5 µm in lengh) occluding junctions with AJ features, referred to as tight AJs (TAJs) (Nunes et al, 2006). Indeed, the intercellular space at the TAJ is typical of TJs, i.e. less than 14 nm in width (Gulley & Reese, 1976; Jahnke, 1975). In addition, TJ proteins, including claudins and ZO1, are present all along the TAJ, whereas α-catenin, β-catenin, p120-catenin and the electron-dense cytoplasmic plaque, i.e. canonical AJ elements, extend along the basal half of the TAJ (Nunes et al, 2006).
How cell–cell junctions of the organ of Corti cope with their constant exposure to sound-induced mechanical stress is still not understood. Vezatin, an ubiquitous AJ protein of unknown topology, is especially abundant at the junctions between HCs and their SCs, which suggests its possible involvement in the resistance of these junctions to mechanical stress. We therefore studied whether vezatin deprivation in cochlear HCs alters their ability to resist loud sound exposure in vivo. We especially focused on the OHC–DC junction that undergoes strong mechanical stress, due not only to the organ of Corti passive oscillation produced by the sound waves, but also to the local mechanical forces exerted by OHC electromotility on this oscillation (see Ashmore, 2008).
RESULTS
Vezatin is an integral membrane protein with two adjacent transmembrane domains
Two main mouse vezatin isoforms, long and short (accession nos. AAX12551 and AAX12552), with estimated molecular weights of 88 kDa and 71 kDa, respectively, have been detected in the mouse pre-implantation embryo. According to vezatin transcript analysis, these isoforms are likely to differ only in their C-terminal regions (Hyenne et al, 2005). It is still unclear whether these isoforms are submembrane or integral membrane proteins. Four membrane-topology prediction programs were used to analyse the vezatin sequences, and gave different results. Two programs, MEMSAT (Jones et al, 1994) and SOSUI (Mitaku et al, 1998), predict two TM domains (aa 134–158 and 166–188) and both amino-terminal and carboxy-terminal cytoplasmic domains (Nin–Cin). The third program (TMHMM) (Krogh et al, 2001; Moller et al, 2001) predicts a single TM domain (aa 149–182), cytoplasmic N-terminus and extracellular C-terminus (Nin–Cout), whereas the last program (HMMTOP) (Tusnady & Simon, 2001) predicts a single TM domain (aa 158–182) with the opposite configuration (Nout–Cin). Therefore, we undertook a topological study of vezatin experimentally.
We first tested whether the N- or the C-terminus of vezatin required plasma membrane permeabilization to be immunostained. Transfected MDCKII cells producing a vezatin fusion protein with a Myc tag either at the N- or at the C-terminus could be immunostained by an anti-Myc antibody only upon Triton X-100 cell membrane permeabilization. Likewise, the endogenous vezatin located at the junctions between non-transfected MDCKII cells was immunolabelled by antibodies directed against N-terminal (aa 1–123) and C-terminal (aa 460–607) fragments of the protein (see Section 4) only after Triton X-100 treatment of the cells (Fig 1A and data not shown). This suggests that both N- and C-terminal ends of vezatin are intracellular.
Figure 1. Vezatin topology.
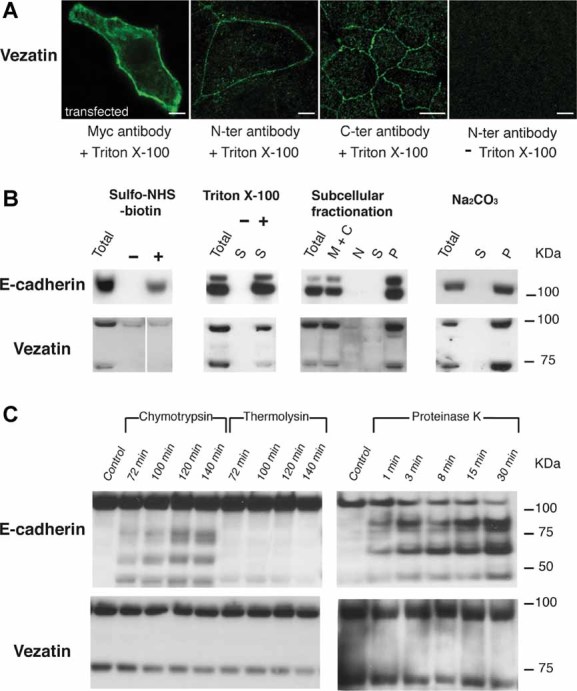
- Vezatin staining in confluent MDCKII cells. Left panel: transfected cells producing the entire vezatin fused to an N-terminal Myc tag, immunostained with an anti-Myc antibody after cell permeabilization with 1% Triton X-100. Middle panels: untransfected cells immunostained, after permeabilization, with polyclonal antibodies directed against either an N- or a C-terminal vezatin fragment to detect the endogenous protein. In all three conditions, the bulk of the protein is detected at the plasma membrane. Right panel: untransfected cells incubated with a polyclonal antibody directed against the vezatin N-terminal fragment in the absence of Triton X-100. No immunostaining is observed. Scale bars = 5 µm.
- Biochemical assays. In all immunoblots, ‘Total’ refers to MDCKII homogenates. The two vezatin isoforms detected on Western blots using the antibody directed against the C-terminal region of the protein (see Section 4) have apparent molecular weights of approximately 90 and 70 kDa, similar to the expected ones (88 and 71 kDa). Left panel: biotin ‘pull-down’ experiment. E-cadherin, but not vezatin, is biotinylated in the presence (+) of sulpho-NHS-biotin, and retained by a streptavidin resin. Dividing line has been used to separate lanes that were not contiguous in the original gel. Middle left panel: Protein extraction. + and − denote the presence and absence of Triton X-100, respectively. Both E-cadherin and vezatin are present in the supernatant fraction (S) only in the presence of Triton X-100. Middle right panel: subcellular fractionation by centrifugation. Lanes: M + C, membrane and cytosolic fraction; N, nuclear fraction; S, supernatant; P, pellet (see Section 4). Vezatin and E-cadherin are present in membrane-enriched (M + C and P) fractions. Right panel: cell lysis experiment in the presence of sodium carbonate (Na2CO3). (S) and (P) denote supernatant and pellet fractions, respectively. Neither E-cadherin nor vezatin are extracted by Na2CO3, and both are recovered in the pellet fraction.
- Limited proteolysis experiments on confluent MDCKII cells. Time lengths for enzyme digestion are indicated on top of each lane. E-cadherin, but not vezatin, undergoes proteolysis by chymotrypsin, thermolysin or proteinase K.
We then searched for evidence of an extracellular region in vezatin. MDCKII cells were treated with the membrane impermeant reagent sulfo-NHS-biotin that covalently couples biotin to free amino groups of cell surface proteins (see Section 4). Solubilized membrane proteins were then incubated with streptavidin beads and bound proteins were analysed by immunoblotting using anti-vezatin antibodies. None of the two vezatin isoforms present in the soluble fraction did bind to streptavidin (Fig 1B, left panel). We next examined the sensitivity of vezatin present at AJs to limited proteolysis, and found that digestion by thermolysin, chymotrypsin or proteinase K had no effect on the vezatin isoforms in non-permeabilized MDCKII cells (Fig 1C and Fig S1A of Supporting Information). These results indicated that vezatin does not have a large extracellular region. To determine if vezatin is a cytoplasmic or a plasma membrane protein (either integral or membrane-associated), we carried out whole-cell protein extraction with or without Triton X-100, and a subcellular fractionation experiment by means of density gradient centrifugation. In the protein extraction experiment, E-cadherin and vezatin were extracted only in the presence of Triton X-100 (Fig 1B, middle left panel). In the fractionation experiment, vezatin was not found in appreciable amounts in the soluble fraction, and the bulk of the protein (95% of the staining intensity on immunoblot) was present in the membrane fraction, together with E-cad (Fig 1B, middle right panel). Moreover, in confluent MDCKII cells mechanically disrupted in iodixanol (OptiPrep) solution and analysed by ultracentrifugation (see Section 4) (Li et al, 2004; Li & Donowitz, 2008), the bulk of vezatin was recovered in the plasma membrane fraction with E-cad (Fig S1B of Supporting Information and data not shown). Therefore, vezatin is not a soluble protein, but a plasma membrane-associated or integral membrane protein. To discriminate between these two possibilities, extraction of MDCKII cell proteins was carried out with 0.1 M sodium carbonate (pH 11.3), that releases peripheral membrane proteins in the soluble fraction (Fujiki et al, 1982). Vezatin was collected in the membrane pellet, together with E-cad (Fig 1B, right panel). We conclude that vezatin is an integral membrane protein with two transmembrane domains, and cytoplasmic N- and C-terminal regions. Based on Sosui program analysis (Hirokawa et al, 1998), the two transmembrane domains are located at positions 134–158 and 166–188 of the amino acid sequence, that is, only seven residues apart from one another (Fig S2 of Supporting Information).
Mutant mice defective for vezatin in cochlear hair cells (HCs) have increased susceptibility to noise exposure
The vezatin immunoreactivity at the junctions between HCs and their SCs was found to increase from postnatal day 4 (P4) to P16, the latter corresponding to a fully mature organ of Corti (Fig 2). To study the role of vezatin at the junctions between cochlear HCs and their SCs, we produced a biallelic conditional deletion of Vezt in the HCs by crossing Veztfl/fl mice (Hyenne et al, 2007) with transgenic mice expressing the Cre-recombinase under the control of the Prestin promoter (Tian et al, 2004). The expected Vezt deleted transcript could be detected in the cochlea of P7 homozygous recombinant mice (thereafter referred to as Veztfl/fl:PrestinCre mice) by RT-PCR analysis (Fig S3 of Supporting Information).
Figure 2. Vezatin staining of the organ of Corti at 4, 8 and 16 postnatal days.
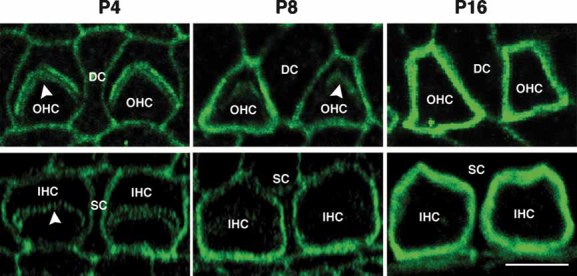
Vezatin labelling intensity increases both at OHC–DC and IHC–SC junctions between P4 and P16. Vezatin is also transiently detected at the base of the V-shaped (OHCs) or U-shaped (IHCs) hair bundles (arrowheads) at P4 and P8, but not at P16, as previously reported (Küssel-Andermann et al, 2000; Michalski et al, 2007). Scale bar = 4 µm.
Hearing thresholds were determined by recording auditory brainstem responses (ABR) to tone bursts at 5, 10, 15, 20, 32 and 40 kHz in 7-week old Veztfl/fl and Veztfl/fl:PrestinCre mice. At this age, thresholds were not significantly different between Veztfl/fl:PrestinCre mice and their Veztfl/fl littermates (Fig 3A). Accordingly, morphological analysis of the Veztfl/fl:PrestinCre cochlear sensory epithelium, both by immunofluorescence confocal microscopy and scanning electron microscopy (SEM), did not show abnormal features at this age (data not shown).
Figure 3. Hearing thresholds and cochlear hair cell morphology before and after noise exposure in 7-week old vezatin-mutant mice.
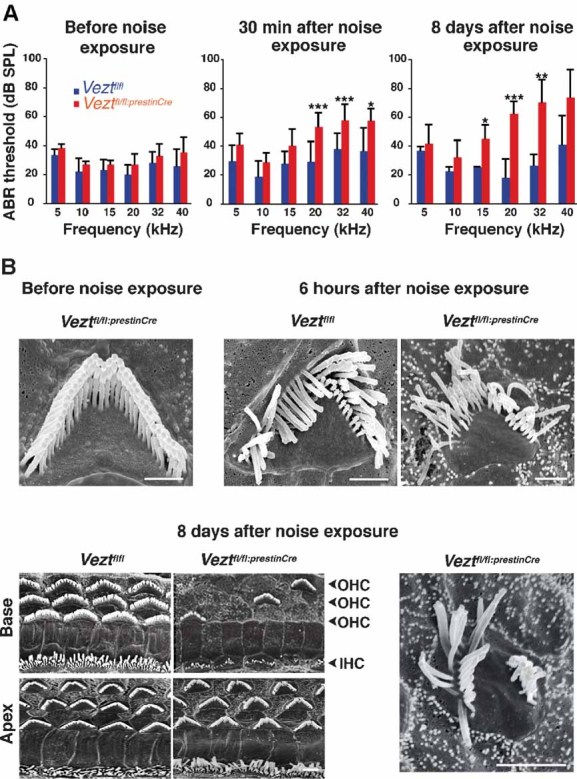
- ABR thresholds. Left panel: before noise exposure. The ABR thresholds (mean ± standard error of mean (s.e.m.)) are not significantly different between Veztfl/fl:PrestinCre (red histograms) and Veztfl/fl (blue histograms) mice (Student's t-test, p > 0.05 for all sound frequencies). Middle panel: 30 min after noise exposure (105 dB for 1 min). Both Veztfl/fl and Veztfl/fl:PrestinCre mice show ABR threshold elevation at intermediate and high sound frequencies (15–40 kHz), but threshold increase is larger in Veztfl/fl:PrestinCre mice on average. The mean threshold differences between Veztfl/fl:PrestinCre and Veztfl/fl mice at 5, 10, 15, 20, 32 and 40 kHz are +7 dB (p = 0.1), +9 dB (p = 0.08), +10 dB (p = 0.1), +21 dB (p = 0.0006), +17 dB (p = 0.0004) and +18 dB (p = 0.03), respectively. Right panel: 8 days after noise exposure, Veztfl/fl mice, but not Veztfl/fl:PrestinCre mice, have recovered their initial ABR thresholds, except at 40 kHz. At intermediate and high sound frequencies (15–40 kHz), Veztfl/fl:PrestinCre mice have increased ABR thresholds compared to those recorded 30 min after noise exposure. The mean threshold differences between Veztfl/fl:PrestinCre and Veztfl/fl mice at 5, 10, 15, 20, 32 and 40 kHz are +5 dB (p = 0.5), +10 dB (p = 0.1), +19 dB (p = 0.004), +44 dB (p = 0.0006), +43 dB (p = 0.002) and +32 dB (p = 0.05), respectively. *, ** and *** indicate statistically significant differences (Student's t-test with p < 0.05, p < 0.01 and p < 0.001, respectively).
- Scanning electron micrographs of the cochlear sensory epithelium before noise exposure, 6 hours after, and 8 days after noise exposure. Top left panel: before noise exposure. No hair bundle disorganization or loss is observed in Veztfl/fl:PrestinCre mice. Top middle and right panels: 6 hours after noise exposure. Some disarrayed hair bundles are seen in Veztfl/fl and Veztfl/fl:PrestinCre mice. Lower panels: 8 days after noise exposure. A substantial loss of OHC bundles is observed only at the base of the cochlea in Veztfl/fl:PrestinCre but not Veztfl/fl mice (left panels). The bottom right panel shows an example of disorganization of an OHC hair bundle in a Veztfl/fl:PrestinCre mouse. Scale bars = 1 µm (upper panels) or 2 µm (lower panels).
We then compared the effect of loud sound exposure in 7-week old Veztfl/fl:PrestinCre and Veztfl/fl mice. Mice were exposed during 1 min to a 105 dB SPL continuous broadband noise (flat spectrum in the 2 kHz–50 kHz range), and ABR thresholds were determined 30 min and 8 days later. At 30 min post-exposure, both Veztfl/fl and Veztfl/fl:PrestinCre mice had increased hearing thresholds, predominantly on high frequencies. In Veztfl/fl mice, the shifts were only 7 dB at 15 kHz, and 12 dB at 20, 32 and 40 kHz, on average. The shifts, however, were significantly larger in the mutants at all sound frequencies studied, with mean differences between Veztfl/fl:PrestinCre and Veztfl/fl mice of 7–10 dB at 5, 10 and 15 kHz frequencies, and 17–21 dB at 20, 32 and 40 kHz. Eight days later, the hearing loss had progressed substantially in the Veztfl/fl:PrestinCre mice across the entire range of frequencies above 10 kHz (reaching 44 dB at 20 kHz), whereas Veztfl/fl mice had recovered normal thresholds, except at 40 kHz, the highest frequency tested (Fig 3A). Disarrayed hair bundles were observed 6 h after the sound exposure both in Veztfl/fl:PrestinCre and Veztfl/fl mice, by SEM (Fig 3B, upper panels). Eight days later, however, hair bundle anomalies, including fused and missing stereocilia, were observed in Veztfl/fl:PrestinCre mice only (Fig 3B, bottom right panel). Moreover, at that time, mutant mice showed a substantial loss of HCs (66%) at the cochlear base, mainly affecting OHCs (80%), whereas HCs from the cochlear apex were almost unaffected (Fig 3B, bottom left panels).
Veztfl/fl:PrestinCre mice undergo spontaneous late onset progressive hearing loss and vestibular dysfunction related to hair cell (HC) death
Evidence from several mouse mutants supports the implication of common molecular mechanisms in noise-induced and age-related hearing losses (see Ohlemiller, 2006 for review). We thus studied hearing thresholds in Veztfl/fl:PrestinCre mice after 7 weeks of age. Between 3 and 6 months of age, spontaneous progressive hearing loss was found, with hearing thresholds at 24 weeks being over 110 dB SPL for all sound frequencies analysed (Fig 4A). In addition, 12-week and 24-week old, but not 7-week old Veztfl/fl:PrestinCre mice, displayed vestibular dysfunction, which manifested as a spontaneous circling behaviour, and abnormal elevated-platform and swimming tests (data not shown).
Figure 4. Hearing thresholds and DPOAEs in vezatin-mutant mice at different ages.
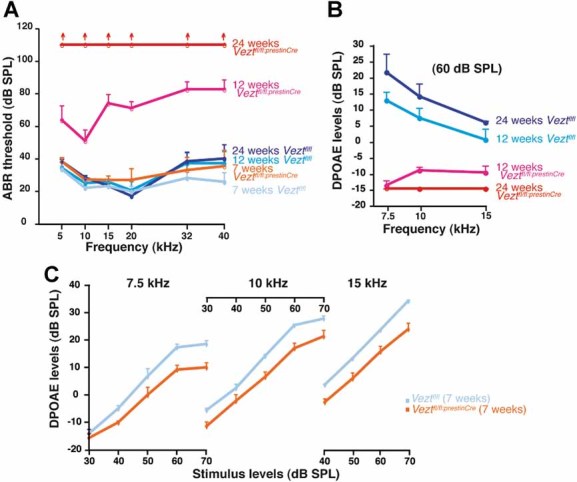
- ABR thresholds at 7, 12 and 24 weeks. Red and blue curves (means ± s.e.m.) correspond to Veztfl/fl:PrestinCre and Veztfl/fl mice, respectively. At 7 weeks, there is no significant difference between Veztfl/fl:PrestinCre and Veztfl/fl mice at all sound frequencies tested (ANOVA one-way, p > 0.05). At 12 weeks, there is a significant threshold increase in Veztfl/fl:PrestinCre mice compared to Veztfl/fl mice at all frequencies (ANOVA one-way, p < 0.0001). Note that threshold increases are larger for intermediate and high frequencies (15–40 kHz) than for low frequencies (5–10 kHz). In 24-week old Veztfl/fl:PrestinCre mice, ABR thresholds are beyond 110 dB SPL for all frequencies.
- DPOAE levels (means ± s.e.m.) for sound stimuli at 60 dB SPL. In 12-week old mice, a significant difference is detected between Veztfl/fl and Veztfl/fl:PrestinCre mice (test of Mann–Whitney, p < 0.001). In 24-week old mutant mice, DPOAEs are at the average noise floor (−15 dB SPL).
- DPOAE growth functions in 7-week old mice. Mean values ± s.e.m. have been plotted. There is a significant difference between Veztfl/fl and Veztfl/fl:PrestinCre mice at 7.5 and 10 kHz (test of Mann–Whitney, p < 0.05) and at 15 kHz (test of Mann–Whitney, p < 0.01) for all stimulus levels tested, except at 7.5 kHz, 30 and 50 dB SPL, and at 10 kHz, 40 dB SPL (p > 0.05 in each case).
Hearing sensitivity relies on the amplification of sound stimuli by the OHCs. These cells also produce conspicuous sound distortion that shows up in the otoacoustic emissions, sounds emitted by the cochlea that can be recorded in the auditory canal. In response to bitonal stimuli (frequencies f1, f2), OHCs produce distortion product otoacoustic emissions (DPOAEs) at various intermodulation frequencies. The predominant DPOAE has a frequency 2f1–f2 (Kim et al, 1980; Robles et al, 1997). Notably, 2f1–f2 DPOAE already showed a 10 dB threshold shift in 7-week old Veztfl/fl:PrestinCre mice, that is, prior to any detectable loss of hearing sensitivity (Fig 4C). In 12-week old Veztfl/fl:PrestinCre mice, only a small islet of DPOAEs at 10 and 15 kHz was still recorded, and DPOAEs had completely disappeared at 24 weeks of age in the mutant mice (Fig 4B).
Morphological analysis of the cochlear and vestibular sensory epithelia in 12-week and 24-week old Veztfl/fl:PrestinCre mice showed a loss of HCs (Fig 5A,B and Table I). In the cochlea, OHCs disappeared before IHCs. In 12-week old Veztfl/fl:PrestinCre mice, the loss of OHCs was predominant at the base of the cochlea, where HCs are high frequency-tuned. This was in agreement with the hearing loss being more pronounced for high frequency sounds (40 dB mean threshold elevation) than for low frequency sounds (20 dB mean threshold elevation) (see Fig 4A). In contrast, IHCs were preserved throughout the cochlea. In 24-week old Veztfl/fl:PrestinCre mice, most OHCs were missing throughout the cochlea, and many IHCs were absent in the cochlear basal region. Likewise, many vestibular HCs were lost in 12-week (data not shown) and 24-week old (Fig 5B) mutant mice. In addition, various hair bundle anomalies (loss, fusion and shortening of stereocilia) were observed in the 12-week and 24-week old mutant mice, both in OHCs, IHCs (data not shown) and vestibular HCs (Fig 5B).
Figure 5. Hair cell morphology in vezatin-mutant mice at different ages.
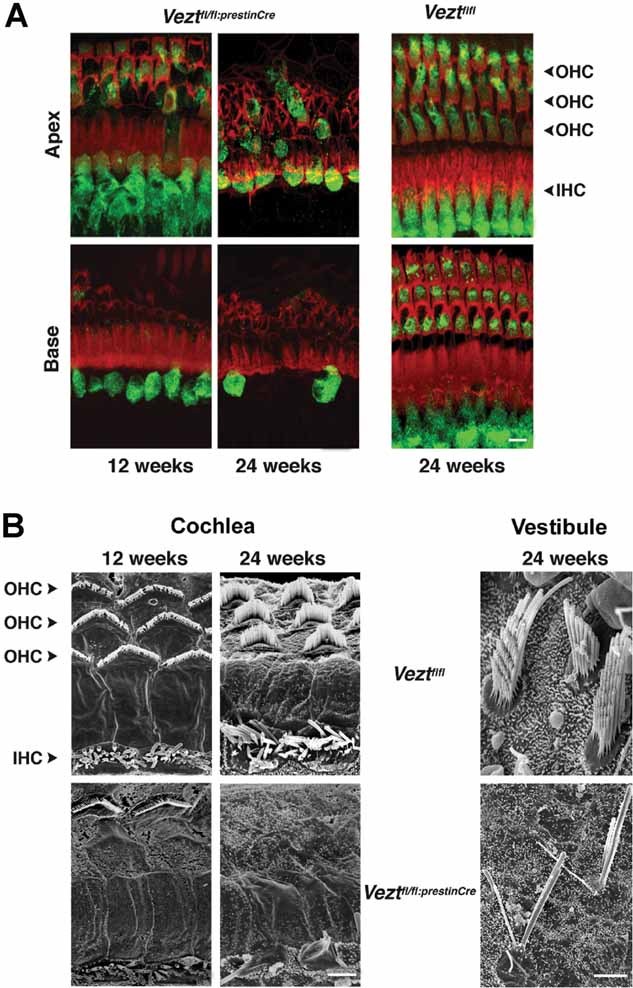
- Confocal images of the organ of Corti from the basal and apical regions of the cochlea at 12 and 24 weeks. HCs are stained for actin (red) and myosin VI (green), a marker of their cell bodies. In the 12-week old Veztfl/fl:PrestinCre mouse (left panels), almost all OHCs, but no IHCs, are missing at the base (lower panel), whereas only a few OHCs and no IHCs are absent at the apex (upper panel). In the 24-week old Veztfl/fl:PrestinCre mouse (middle panels), all OHCs are missing and only a few IHCs are still present at the base of the cochlea (lower panel), whereas most of the OHCs are absent and IHCs persist at the cochlear apex (upper panel). Images from a 24-week old Veztfl/fl mouse (right panels) are shown for comparison. Scale bars = 10 µm.
- Scanning electron micrographs of the organ of Corti apical surface at the base of the cochlea at 12 and 24 weeks, and of a vestibular sensory epithelium (utricle) at 24 weeks. In the 12-week old Veztfl/fl:PrestinCre mouse (bottom left panel), numerous OHC hair bundles are missing, but IHC hair bundles are preserved. In the 24-week old Veztfl/fl:PrestinCre mouse (bottom middle panel), all OHC hair bundles are missing, and the IHC hair bundles either have disappeared or are completely disorganized. Micrographs from 12 and 24-week old Veztfl/fl mice (top left and middle panels) are shown for comparison. In 24-week old Veztfl/fl:PrestinCre mouse (bottom right panel), the hair bundles of vestibular HCs from the utricle either have disappeared or are completely disorganized. A microphotograph from a 24-week old Veztfl/fl mouse is shown for comparison (top right panel). Scale bars = 1 µm.
Table I.
Quantitative analysis of IHC and OHC loss in vezatin-mutant mice
| IHC loss % (number of IHCs analysed) | OHC loss % (number of OHCs analysed) | ||||
|---|---|---|---|---|---|
| Base | Apex | Base | Apex | ||
| 12 weeks | Veztfl/fl:prestinCre | 2 (25) | 1 (40) | 64 (68) | 53 (118) |
| Veztfl/fl | 1 (26) | 2 (62) | 2 (58) | 4 (156) | |
| 24 weeks | Veztfl/fl:prestinCre | 72 (30) | 40 (50) | 94 (70) | 77 (110) |
| Veztfl/fl | 2 (32) | 1 (38) | 1 (44) | 3 (80) | |
The quantitative analysis was carried out on whole-mount organs of Corti from 12 and 24-week old mice, labelled for actin and myosin VI (marker of cell bodies) by using confocal microscopy. Three Veztfl/fl and three Veztfl/fl:prestinCre mice were studied at each age. In the 12 and 24-week old Veztfl/fl:PrestinCre mice, a large proportion of the OHCs are missing, both at the base and at the apex of the cochlea. In addition, many IHCs are missing in the 24-week old, but not the 12-week old mutant mice.
Vezatin is recruited at mature AJs in MDCKII cells
As a first attempt to get an insight into the function of vezatin at the subcellular level, we studied how vezatin is recruited at MDCKII cell–cell contacts in comparison with E-cadherin, that is present at AJs from the earliest stages of their formation onwards (Nelson, 2008). In growing MDCKII cells, E-cadherin was detected at nascent cell–cell contacts, and subsequently, at mature AJs. In contrast, vezatin was not detected at nascent cell–cell contacts. In confluent cells, however, vezatin and E-cadherin labellings, both formed discrete spots or short continuous lines at cell–cell junctions, with some overlapping dot-like staining (Fig 6A).
Figure 6. Comparison of E-cadherin and vezatin recruitments at AJs between MDCKII cells during the formation or reformation of cell–cell contacts.
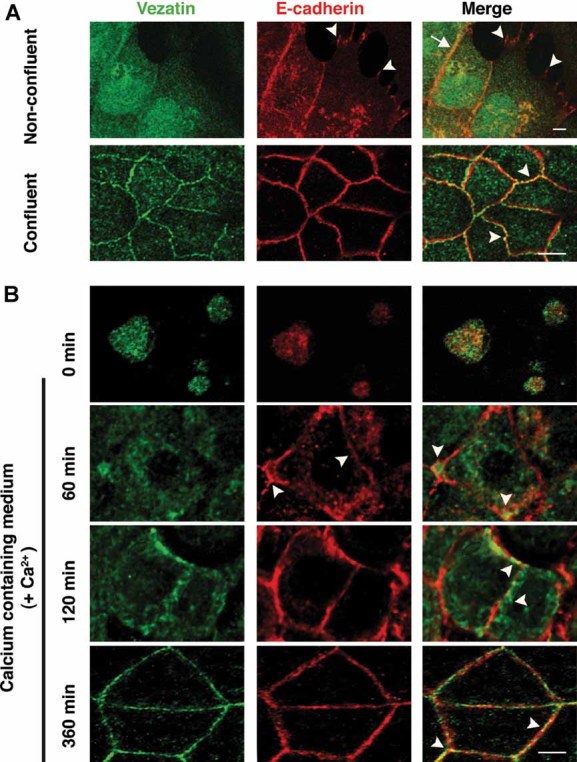
- Non-confluent and confluent cell cultures. In non-confluent MDCKII cells (upper panels), only E-cadherin but not vezatin can be detected at nascent cell–cell contacts (arrowheads), whereas the proteins colocalize at stabilized cell–cell contacts (arrow). In contrast, in confluent MDCKII cells (lower panels), vezatin immunostaining is seen as spots at cell–cell junctions, where it partly colocalizes with E-cadherin (arrowheads). Note also the punctate vezatin staining in the cytoplasm of confluent cells.
- Calcium switch experiment. Confluent MDCKII cells are incubated in a calcium-free culture medium for 6 h to disrupt all cell–cell junctions. Cell–cell junctions are then allowed to reform in a medium containing normal calcium concentration. The distributions of vezatin and E-cadherin are studied at various times between t = 0 and t = 360 min. At t = 0 min, the E-cadherin staining is cytoplasmic. At t = 60 min, the E-cadherin staining is present at cell–cell contacts in nearly all the cells, either as dots or as a more continuous linear staining (arrowheads in middle panel). Vezatin immunostaining is diffuse in the cytoplasm, with dot-like aggregates preferentially located at the proximity of tricellular contacts (arrowheads in right panel). At t = 120 min, the E-cadherin staining at cell–cell contacts is stronger, and forms more continuous ribbons between adjacent cells. Vezatin is detected mainly as patches beneath the junctions and occasionally at putative junctions (arrowheads). At t = 360 min, junctional punctate E-cadherin and vezatin stainings can be seen throughout the MDCKII monolayer and partly overlap (arrowheads). Scale bars 10 µm in A and 5 µm in B, C.
We then carried out calcium switch experiments to monitor the recruitment of vezatin during the reassembling of AJs promoted by the restoration of the calcium concentration in the culture medium after calcium deprivation. Confluent MDCKII cell cultures were first incubated in calcium-free medium for 6 h, then switched back to normal medium and processed for immunofluorescence to detect E-cadherin and vezatin at different times. Accumulation of E-cadherin, but not vezatin, at sites of cell–cell contacts could be seen as a punctate labelling at the plasma membrane as soon as 30 min after returning cells to the normal culture medium (data not shown). At 60 min, E-cadherin junctional staining had become more intense, whereas the bulk of vezatin labelling was seen in the cytoplasm. At 120 min, intense vezatin labelling underneath AJs was observed. At 360 min, vezatin-immunoreactive junctional spots were eventually observed, many of which did not clearly overlap with the E-cadherin staining (Fig 6B). We conclude that vezatin recruitment at AJs in MDCKII cells is markedly delayed compared to that of E-cadherin.
Vezatin and radixin directly interact at cell–cell junctions in the organ of Corti
Vezatin has been shown to bind to the C-terminal FERM domain of the myosin VIIa tail (Küssel-Andermann et al, 2000). Radixin, another actin-binding protein of the ezrin–radixin–moesin (ERM) family which is involved in human deafness (Khan et al, 2007), is abundant in the HCs' apical region (Kitajiri et al, 2004; Pataky et al, 2004; Shin et al, 2007). We thus asked whether vezatin can also interact with this protein via its FERM domain. Confocal microscopy analysis (0.25 µm steps along the z-axis) of double labelling experiments on the mature organ of Corti showed a colocalization of vezatin and radixin at the same emplacement as β-catenin and p120-catenin along the OHC–DC TAJs. Vezatin and radixin were also colocalized at DC-DC and IHC-SC AJs (Fig 7 and data not shown). Colocalization of these proteins was observed in MDCKII cells too (see Fig 9A). Moreover, vezatin and radixin could be coimmunoprecipitated in protein extracts from MDCKII cells or mouse cochlear sensory epithelium by using an antibody directed against vezatin C-terminal region (Fig 8A). Finally, the purified C-terminal cytosolic region (aa 188–607) of vezatin was able to bind to the radixin FERM domain (aa 1–310), but neither to the radixin C-terminal region (aa 476–583) nor to radixin carrying the Thr564Ala substitution that maintains the protein in the closed, i.e. non-actin-binding conformation (Fig 8B). We conclude that vezatin is likely to interact with radixin in its actin-binding conformation at AJs and TAJs. Supporting the in vivo relevance of this interaction, we observed a substantial decrease in the radixin immunolabelling at OHC–DC junctions in Veztfl/fl:PrestinCre mice, whereas ZO1 and β-catenin labellings were unchanged (Fig 8C and data not shown).
Figure 7. Distributions of vezatin, ZO1, β-catenin, p120-catenin and radixin in the cochlear sensory epithelium.
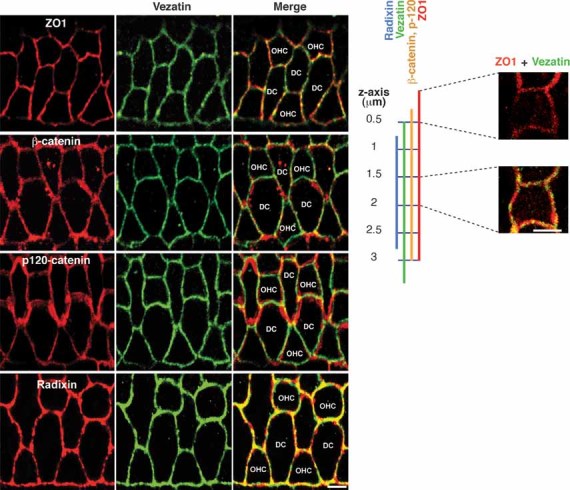
Confocal images of the mature organ of Corti, stained for vezatin (green) and ZO1, β-catenin, p120-catenin or radixin (red). Vezatin, ZO1, β-catenin and p120-catenin immunostainings are superimposed at several emplacements along the DC–DC and OHC–DC junctions, especially at tricellular junctions. A schematic representation of the distributions of ZO1, β-catenin, p120-catenin, vezatin and radixin at the OHC–DC TAJ is shown on the right, from a series of x–y planes along the z-axis (0.25 µm steps), analysed by confocal microscopy. Vezatin is excluded from the most apical part of the TAJ that is ZO1 immunoreactive (right upper panel), but colocalizes with ZO1 along the rest of the TAJ (right lower panel). Scale bars = 5 µm.
Figure 9. Effects of calcium depletion and F-actin disrupting drugs on vezatin and radixin distributions in MDCKII cells.
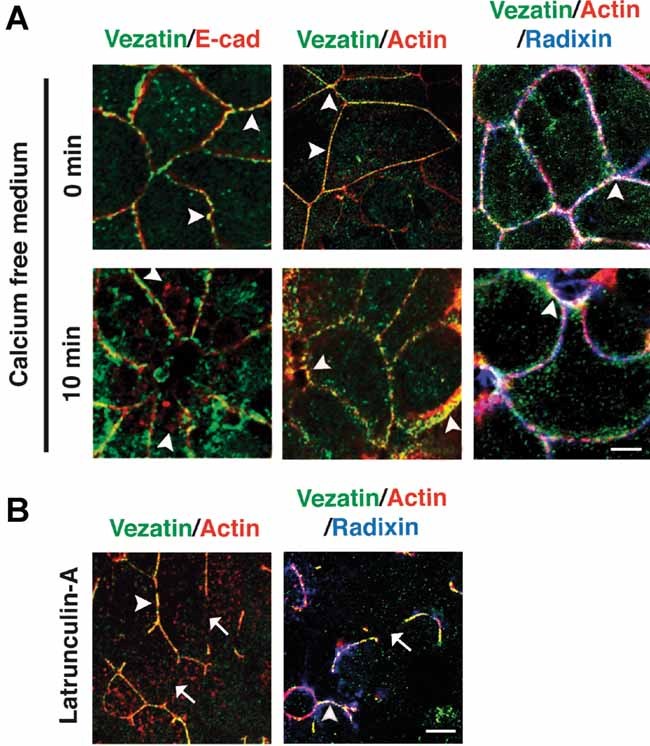
- Calcium depletion experiment. Fully polarized MDCKII cells are switched from a calcium-containing culture medium to a medium devoid of calcium to induce junctional complex disassembly. Cells are processed for immunofluorescence at different times after the switch to follow the distributions of E-cadherin, vezatin and actin during junctional complex disassembly. At t = 0 min (upper panels) E-cad, vezatin, radixin and actin are colocalized at cell–cell contacts (arrowheads). At t = 10 min after the switch to calcium-free medium (lower panels), some disruption of E-cad, vezatin, radixin and actin junctional stainings is observed. Spots of E-cad staining are visible in the cytoplasm (lower left panel), but are not colocalized with vezatin staining (arrowheads). Most of the vezatin staining, however, colocalizes with radixin and actin at the membrane (lower middle and right panels; arrowheads).
- F-actin disruption experiments. In untreated MDCKII cells, vezatin, radixin and actin are colocalized at cell–cell contacts (see upper panels in (A)). Treatment of the cells by latrunculin-A causes disruption of F-actin bundles, as well as vezatin and radixin at AJs. F-actin, radixin and vezatin labellings become discontinuous at AJs (arrows). The residual vezatin, radixin and actin junctional labellings at the plasma membrane, however, still colocalize (arrowhead). In addition, spots of radixin staining are visible in the cytoplasm and do not overlap with either vezatin or actin stainings. Scale bars = 5 µm.
Figure 8. Vezatin–radixin interaction in the cochlear sensory epithelium.
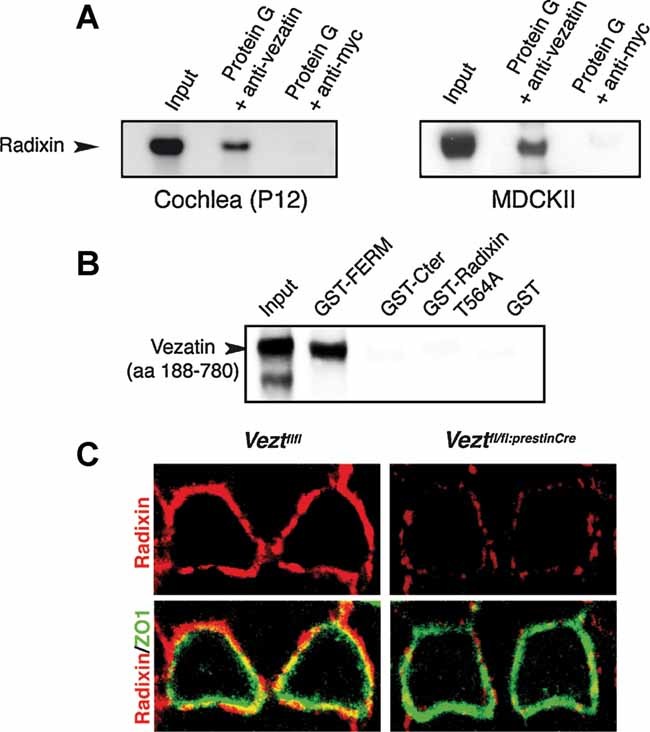
- Co-immunoprecipitation experiments. Protein extracts from P12 mouse cochleas (left panel) or MDCKII cells (right panel) were used. The endogenous radixin is immunoprecipitated in the presence of the anti-vezatin N-terminal fragment antibody, but not in the presence of the anti-Myc antibody.
- In vitro binding assays. GST and different GST–Radixin fusion proteins, namely GST–FERM (aa 1–325), GST–Cter (aa 476–583) or GST–radixinThr564Ala (entire protein carrying the T564A substitution) were immobilized on glutathione-Sepharose. A vezatin fragment encompassing aa 188–780 of the protein interacts only with the GST–FERM construct.
- Radixin distribution at DC–OHC junctions in Veztfl/fl and Veztfl/fl:PrestinCre mice. In both Veztfl/fl and Veztfl/fl:PrestinCre organs of Corti, radixin (red) can be detected along the OHC–DC junctions, but the labelling is much less intense in the vezatin-deficient mouse. In contrast, the intensity of the ZO1 labelling (green) is unchanged.
A large body of evidence has established the close association between AJs and the cortical actin cytoskeleton (Bershadsky, 2004; Hartsock & Nelson, 2008; Pokutta & Weis, 2007). Firstly, we studied how calcium depletion that results in disassembly of AJs and reorganization of the perijunctional actin cytoskeleton, affects vezatin and radixin localization in MDCKII cells. Fully polarized MDCKII cell monolayers were switched from a medium containing calcium to a medium devoid of calcium to induce junctional complex disassembly (Ivanov et al, 2004 and see Section 4). This treatment perturbed the localization of F-actin, E-cadherin, radixin and vezatin at AJs. E-cadherin was partly internalized as early as after 10 min, and formed large aggregates under the membrane 20 min after calcium depletion. Vezatin and E-cadherin labellings were not colocalized at either time. In contrast, vezatin and radixin colocalized with F-actin at the disassembled junctions (Fig 9A and data not shown). We then probed the effect of F-actin perturbation by cytochalasin-D (7 µM) and latrunculin-A (7 µM) on the distribution of vezatin at mature junctions (Wakatsuki et al, 2001). Cytochalasin-D shortens microfilaments by inhibiting the addition of new monomers to their barbed ends, whereas latrunculin-A, which binds to actin monomers, prevents their incorporation into filaments, thus preferentially affecting the formation of the most dynamic microfilaments (Hyman et al, 2006). Under both experimental conditions, the loss of junctional F-actin was accompanied by a loss of vezatin and radixin immunolabellings at AJs (data not shown and Fig 9B). In latrunculin-treated cells, vezatin and radixin stainings were colocalized with intracytoplasmic F-actin bundles. Under the same conditions, the E-cadherin immunostaining persisted at AJs, even though it became diffuse (data not shown and Fig S4 of Supporting Information). Together, these results suggest the existence of an interaction between vezatin and the cortical actin cytoskeleton through radixin at AJs.
DISCUSSION
Here, we show that in the absence of vezatin in the HCs, the resistance of the organ of Corti to mechanical stress is severely compromised. Indeed, a single short exposure to a loud broadband sound was sufficient to induce irreversible cochlear HC damage in mutant mice deprived of vezatin in these cells. The progressive hearing impairment that spontaneously occurs in these mice together with a substantial HC loss is also consistent with a cumulative deleterious effect of the less intense sounds of their breeding environment. Finally, the fact that OHCs were affected before IHCs is in agreement with both the large forces applied to TAJs by OHC electromotility (around 100 pN/mV) (Iwasa & Adachi, 1997 and see Ashmore, 2008), and the forces associated with sound-induced passive deformation of the cochlear partition that are stronger at OHC than IHC level (Tomo et al, 2007). In addition, the more pronounced damage to basal cochlear HCs that are high frequency-tuned, compared to apical HCs that are low frequency-tuned, is also consistent with mechanically driven alterations of the junctions between HCs and their SCs in Veztfl/fl:PrestinCre mice (see Ashmore, 2008). Two different missense mutations in VEZT, c.275C>G (p.A92G) and c.1747A>T (p.D503V), which affect amino acid residues located in the first transmembrane segment and the C-terminal region, respectively, have recently been found in heterozygous state in two families affected by progressive hearing loss predominating on high frequency sounds (C Zadro, D Licastro and P Gasparini, unpublished results, 2008). The effects of these mutations on protein activity remain to be determined.
Notably, a decreased DPOAE response was found in Veztfl/fl:PrestinCre mice before their hearing thresholds, hence the OHC-driven amplification of the sound stimuli, were affected. Properly recorded DPOAEs are thought to be as sensitive to auditory threshold shifts due to OHC damage as routine pure-tone audiometry (Sutton et al, 1994). Such an example of decreased DPOAEs anticipating auditory threshold shifts by several weeks in Veztfl/fl:PrestinCre mice is unusual. Because vezatin is located at TAJs, the audiometric situation in 7-week old vezatin-deficient mice suggests that the mechanical distortion of sound waves operated by normal OHC hair bundles involves the connection of the TAJ to the actin rootlets of OHCs' peripheral stereocilia (Furness et al, 2008). As a corollary, this connection would be defective in the mutant mice already at 7-weeks of age, despite the absence of detectable hair bundle morphological anomalies.
What could be the role of vezatin at the HC–SC junctions? The delayed vezatin recruitment at the junctions with respect to their formation in MDCKII cells is inconsistent with a role of this protein in the establishment of the first cell–cell contacts (‘puncta’ structures) or in the following step of junction formation, that is the transformation of ‘puncta’ in a continuous adhesive belt (Vasioukhin et al, 2000). In contrast, the subsequent junctional integration of vezatin in these cells suggests that it is involved in maturation steps or in the maintenance of junction integrity. Vezatin and F-actin similar behaviours upon either disassembly of the junctions by Ca2+ depletion or treatment of the cells by cytochalasin-D or latrunculin-A argue in favour of an association of vezatin with F-actin at mature junctions. Two distinct populations of F-actin have been identified at AJs, a highly dynamic one and a more stable one, based on their turnover and sensitivity to latrunculin treatment of the cells (Cavey et al, 2008). The partial mislocalization of both F-actin and vezatin in MDCKII cells following latrunculin treatment suggests an association between vezatin and both the dynamic and stable pools of junctional F-actin. Therefore, we cannot exclude the involvement of vezatin in homo-E-cadherin stability related to the stable pool of actin, which would be consistent with the previously reported coimmunoprecipitation of these two junctional proteins (Küssel-Andermann et al, 2000). Along this line, the ERM protein moesin has recently been shown to participate in F-actin reorganization at cell–cell junctions, which in turn stabilizes E-cadherin (Pilot et al, 2006). Moesin is not present at cell–cell junctions in the organ of Corti. Ezrin, another ERM protein, is detected until P14, but is no longer present at HC junctions in the mature organ of Corti (A Bahloul, unpublished results). Based on our findings in vivo and in vitro, we suggest that still another ERM protein, radixin, upon its phosphorylation and conformation changes to an actin-binding conformation (Fievet et al, 2004; Yonemura et al, 2002) – an activation process that requires interaction with the membrane phosphatidylinositol 4,5-bisphosphate (PIP2) – bridges vezatin to F-actin at the junctions between cochlear HCs and their SCs. Activation of phosphatidylinositol-4-phosphate 5-kinase as a result of the homophilic interaction between E-cadherin monomers (Akiyama et al, 2005) may produce the PIP2 locally required for radixin activation. This provides a possible mechanism by which vezatin participates in the strengthening of mature cell–cell junctions. In addition, the C- and N-terminal cytoplasmic regions of vezatin are likely to bind to other, as yet unidentified, proteins of the junctional complex. Based on the in vitro binding of vezatin to the FERM domains of radixin (this study) and myosin VIIa (Küssel-Andermann et al, 2000), vezatin binding partners at the junctions between HCs and their SCs may include not only radixin and myosin VIIa, but also other unconventional myosins with FERM tail domains (myosins VIIb, X, XVa) (Oliver et al, 1999) that could be present at these junctions.
MATERIALS AND METHODS
Antibodies
The following primary antibodies were used: mouse monoclonal antibodies against β-catenin, E-cadherin, p120-catenin (BD Transduction Laboratories), ZO1 (Zymed Laboratories) and radixin (ABNOVA), rabbit anti-myosin VI (gift from Dr S Safieddine, Institut Pasteur, Paris, France) and anti-vezatin (Küssel-Andermann et al, 2000) polyclonal antibodies, anti-Myc mono- and polyclonal antibodies (clone 9E10 and SC 789 from Santa Cruz Biotechnology, Inc.). For vezatin topology and distribution studies in HCs, rabbit anti-sera were produced against two bacterially expressed peptides derived from the N-terminal (aa 1–123) and C-terminal regions (aa 460–607) of mouse vezatin sequence (accession no. AAX12551). Antibodies were affinity-purified from immune sera using the recombinant antigen coupled to a NHS-column (GE Healthcare). The specificity of each antibody was tested in transfected MDCKII cells producing vezatin, by using Western blot and immunoprecipitation. Primary antibodies were detected using Alexa488- or Cy3-conjugated secondary antibodies (Molecular Probes). F-actin was visualized with TRITC-conjugated phalloidin (Sigma).
Recombinant expression vectors
The cDNAs encoding mouse, the long (AAX12551) and short (AAX12552) isoforms of vezatin, were subcloned either in the pCMV expression vector, which enabled the production of the N-terminal Myc-tagged vezatin protein, or in the pCS2 modified vector (kindly provided by Dr FJ del Castillo, Ramon y Cajal Hospital, Madrid, Spain), which produced a C-terminal Myc-tagged vezatin protein. For in vitro binding experiments, a cDNA encoding a vezatin fragment (aa 188–607) was subcloned in pFast Bac vector. In addition, cDNAs encoding the FERM domain (residues 1–310) and C-terminal domain (residues 476–583) of mouse radixin (AK162204) in fusion with a glutathione sulpho-transferase (GST) tag were subcloned into the prokaryotic expression vector pGST//1. The T564A missense mutation in radixin was engineered by using the QuickChange mutagenesis kit (Stratagene).
Cell surface protein biotinylation
Cell surface proteins were biotinylated by incubating the cells with 1.5 mg/ml sulpho-NHS-SS-biotin (Pierce Chemical) for 1 h at 4°C, and free biotin was quenched with a blocking solution (50 mM NH4Cl in phosphate buffered saline (PBS) containing 1 mM MgCl2 and 0.1 mM CaCl2). Cells were then directly extracted in a radio-immunoprecipitation assay (RIPA) buffer (1% NP-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulphate (SDS), 150 mM sodium chloride, 50 mM Tris-HCl (pH 7.4), containing a cocktail of protease inhibitors. Cell extracts were centrifuged and incubated with streptavidin magnetic beads (Dynal) to collect biotinylated proteins. Samples were then analysed by SDS-PAGE and immunoblotting was performed using antibodies against E-cad and vezatin.
Cell lysis and immunoblotting
Cell lysates extracted in RIPA buffer were resolved by 3–8% SDS-PAGE. Proteins were transferred to nitrocellulose (Millipore), immunoprobed and detected by enhanced chemiluminescence (PerkinElmer).
Proteolysis experiments
Enzymatic digestions were carried out at 4°C on MDCKII cell monolayers (Transwell filters) at one protease concentration for different lengths of time at 4°C. Chymotrypsin, thermolysin and proteinase K were used at a 1:500 dilution in each well (mg enzyme/mg total protein; one well was assayed for protein amount by Bradford quantification, with the assumption that the amount of cells in each well was equivalent). Proteolysis was stopped by heat inactivation of the enzyme (15 min at 95°C).
Subcellular fractionation
Confluent MDCKII cells were washed three times in fresh PBS, and harvested by centrifugation at 1000 g for 10 min. Cells were resuspended twice in 10 ml of 250 mM sucrose, 10 mM triethanolamine–10 mM acetic acid, pH 7.8, in the presence of protease inhibitors and harvested again. Cells were incubated for 10 min on ice and homogenized by 150 passes in a 2 ml Dounce homogeneizer. Cell disruption was monitored under the microscope. The samples were immediately centrifuged at 800 g for 10 min to isolate the nuclear fraction. The cytosol and membrane fractions were then centrifuged at 100,000 × g for 60 min. For OptiPrep gradient (Li, 2008 #19), 200 µl of lysate were mixed with 200 µl of a 50% OptiPrep solution (Axis-Shield) (25% final concentration). An OptiPrep gradient was prepared by layering 6 ml of 25% OptiPrep in lysis buffer and 6 ml of buffer alone on top. Gradients were centrifuged at 200,000 × g for 3 h at 4°C, using a swinging rotor. Twenty-four 500 µl fractions were recovered from the gradient.
For carbonate extraction of peripheral membrane proteins, MDCKII cells were resuspended in 0.1 M Na2CO3, pH 11.5, with care to ensure that the final protein concentration was between 0.5 and 1 mg/ml (Fujiki, 1982 #20). The samples were incubated for 30 min at 4°C, and spun at 200,000 × g for 30 min to separate the membranes from the soluble material. The pellet was resuspended in a volume equal to that of the supernatant.
Animal handling
Experiments on mice were carried out according to INSERM and Pasteur Institute welfare guidelines.
Noise exposure, ABR and DPOAE recordings, and vestibular testing
Seven-week-old mice were anaesthetized and exposed to a broadband continuous noise (flat spectrum in the 2–50 kHz range) at an intensity of 105 dB SPL (sound pressure level) for 1 min. Auditory function and HC morphology were then assessed 30 min, 6 h and 1 week after noise exposure. To control for any possible effect of one week ageing in the mice, a small group (n = 6) of 8-week old Veztfl/fl:PrestinCre mice not exposed to the noise were tested for auditory function.
ABRs and DPOAEs were recorded and analysed as described in Le Calvez et al (1998). The DPOAE at frequency 2f1 − f2 was recorded in response to two equal level primary tones, f1 and f2, with f2/f1 = 1.20. Cubic DPOAEs were elicited by a CubeDis system (Mimosa Acoustics, v2.43). Frequency f2 was swept at one-tenth-octave steps from 4 to 20 kHz (levels increased stepwise from 30 to 70 dB SPL). Input–output curves, representing DPOAE levels at every f2 tested against stimulus intensity, were plotted, averaged across Veztfl/fl and Veztfl/fl:PrestinCre mice and compared using the non-parametric test of Mann–Whitney, due to the widely different variances among samples forbidding the use of Student's t-test.
Analysis of variance (one-way ANOVA) was used to compare ABR thresholds between experimental groups. Term of group included same genotype, age and time after noise exposure. A criterion of p < 0.05 was chosen to define statistically significant differences. Student's t-test was used to compare ABR thresholds for a given sound frequency, in Veztfl/fl and Veztfl/fl:PrestinCre mice.
Mice were evaluated for vestibular function using the elevated-platform test, and swimming tests (Steel & Harvisty, 1996).
Immunoprecipitation and in vitro binding experiments
For co-immunoprecipitation experiments, cochleas from postnatal day 12 (P12) mice were homogenized in 500 µl lysis buffer (20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 1 mM MgCl2, 0.2% Triton X-100, 0.01% SDS, 1% glycerol, and protease inhibitor cocktail (GE Healthcare)). After centrifugation at 100,000 × g for 10 min at 4°C, the soluble fraction was incubated for 2 h at 4°C with 40 µl protein G-agarose (Pierce Biotechnology, Inc.) preincubated either with 3 µg of purified anti-vezatin polyclonal antibody or with anti-myc antibody.
To test for vezatin molecular interactions, the C-terminal fragment (aa 188–607) of the protein was produced in a baculovirus system and purified on a Ni2+-agarose column. The purified protein was incubated with glutathione beads coupled to GST fusion proteins in RIPA buffer containing 50 mM Hepes (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1.5 mM MgCl2, 10% glycerol, 0.1% SDS, 1% Triton, 0.5% sodium deoxycholate, 1 mM sodium orthovanadate and protease inhibitor cocktail (GE Healthcare) for 2 h at 4°C.
The paper explained
PROBLEM:
Loud sound exposure is a major cause of hearing loss in modern societies. Lesions and malfunctioning of the auditory sensory cells that occur after noise exposure have been characterized. The junctions between these cells and their supporting cells undergo mechanical stress imposed by the sound-evoked vibration of the sensory epithelium. However, contribution of these junctions to the noise-induced hearing loss has remained unexplored.
RESULTS:
In this study, a mouse model lacking the cell–cell junction integral membrane protein vezatin in the sensory cells of the inner ear was engineered. The mutant mice undergo irreversible hearing loss and sensory cell death after a short exposure to a loud broadband sound. Molecular analyses also show that the physical connection between the plasma membrane and underlying actin cytoskeleton at the junctions between auditory sensory cells and their supporting cells involves a direct interaction between vezatin and radixin, an actin-binding protein. As a corollary, the lack of vezatin in the sensory cells is likely to affect mechanical resistance of these junctions.
IMPACT:
The results point to cell–cell junctions of the cochlear sensory epithelium as a plausible primary target for sound trauma in humans, especially in genetically predisposed individuals who may carry deleterious mutations in genes encoding proteins involved in the strengthening of these junctions.
Cell culture and immunocytofluorescence analysis
MDCKII cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS, Invitrogen) at 37°C (5% CO2). For transient transfection experiments, cells were transfected with the DNA of interest (1 µg) using Jet-PEi, plated at a density of 4 × 105 cells per well, and grown for 5 days.
For immunocytofluorescence experiments, cell samples were fixed using −20°C methanol for 5 min (or PFA for 15 min for phalloidin staining and for topology studies), incubated for 1 h in PBS containing 10% goat serum, stained with primary antibody for 1 h at 20°C, washed in PBS three times for 5 min at 20°C, stained with secondary antibody for 1 h at 20°C, washed as above and mounted using Fluorosave (Calbiochem).
Latrunculin-A and cytochalasin-d treatments
MDCKII cell monolayers were washed and equilibrated in Hepes-buffered saline plus glucose (HBSG; 10 mM Hepes, pH 7.4, 5.4 mM KCl, 137 mM NaCl, 1.3 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose) for 30 min at 37°C. Cells were then incubated in HBSG with either 7 µM cytochalasin-D (Sigma) or 7 µM latrunculin-A (Invitrogen) for 1 h at 37°C.
Calcium switch experiments
To study the disassembly of AJs, confluent MDCKII cell monolayers were incubated in a low-calcium medium (DMEM-without calcium (Gibco), supplemented with 10% FCS, 2 mM glutamine, 1 mM sodium pyruvate and 4 mM ethylene glycol tetra-acetic acid (EGTA) for two different lengths of time (10 and 20 min), at 37°C (5% CO2). To induce reassembly of the AJs after a 6 h incubation in calcium-free culture medium, the cells were returned to normal medium for different lengths of time (from 30 to 360 min) at 37°C.
Cochlear dissection and immunostaining
Whole-mount preparations of the organ of Corti of mouse were obtained as follows. Animals were killed by exposure to CO2 followed by decapitation, and the inner ears were removed and placed in PBS. The organ of Corti was exposed by removing the stria vascularis and the tissues were fixed by immersion in methanol at −20°C for 5 min. The samples were washed three times in PBS, incubated for 1 h at 20°C in PBS containing 20% goat serum and stained overnight with the affinity-purified anti-vezatin and anti-β-catenin antibodies diluted in PBS containing 1% bovine serum albumin (BSA). The samples were washed three times with PBS, and incubated with either Alexa 488-conjugated or Cy3-conjugated goat anti-rabbit Fab'2 antibodies diluted in PBS containing 20% goat serum, for 1 h at 20°C. After three washes with PBS, the tectorial membrane was carefully removed, and the pieces were mounted using Fluorosave. Samples were analysed by means of a Zeiss LSM510 Meta confocal microscope.
Genotyping and RT-PCR
For PCR genotyping of newborn or adult animals, genomic DNA from tail clipping was used as a template. Tail DNA extraction and PCR amplification were realized according to the manufacturer's recommendation (REDExtract-N-Amp™ Tissue PCR Kit, Sigma). Primers used to amplify the various allelic combinations are described in Hyenne (2007 #15).
For RT-PCR analysis of vezatin transcripts in the cochlear sensory epithelium of Veztfl/fl:PrestinCre and Veztfl/fl P7 mice, the sequences of forward and reverse primers were 5′-TTTTGAGAATTCTCCACTTTACCAGTACC-3′ and 5′-AGGCAGAGTGAGCATGAGGTAGACC-3′, respectively. In the Veztfl/fl mice, the detected amplification product is 1136 bp in length. In the Veztfl/fl:PrestinCre mice, an additional product of 860 bp lacking exon 5 is also detected.
Scanning electron microscopy (SEM)
Samples were processed as described in Delmaghani (2006 #34). Processed specimens were mounted on aluminium stubs with colloidal silver adhesive and sputter-coated with gold palladium before imaging in a JSM-6700 F Jeol scanning electron microscope.9.
Author contributions
AB designed and performed the biochemistry and cell biology experiments. MCS produced the Veztfl/fl mice (Hyenne et al, 2007); requests for these mice should be addressed to her. VM performed confocal microscopy analyses of the organ of Corti. ML carried out the SEM experiments. IP contributed to the cell biology studies. IR contributed to the DNA constructs, with help from SN. DW carried out the RT-PCR analysis. JZ produced the transgenic mice expressing Cre under the control of the Prestin promoter (Tian et al, 2004). PA performed the auditory tests in the mice. CZ, DL and PG collected DNA from hearing impaired individuals and searched for mutations in VEZT. AB, JPH and CP wrote the article. CP supervised the whole project.
Acknowledgments
The authors wish to thank Dr Monique Arpin for fruitful discussions, Dr Jacqueline Levilliers for her help in the preparation of the manuscript, Emilie Bizard and Olinda Alegria-Prevot for technical help. AB benefited from a Roux fellowship (Institut Pasteur). This work was supported by grants from European Commission FP6 Integrated Project EuroHear LSHG-CT-2004-512063, Région Ile-de-France project Sésame 2002-E1666 (confocal microscope), Pasteur–Weizmann Joint Research Program (2006–2007), French National Research Agency (ANR-Maladies Rares, ANR-05-MRAR-015-01) and the French National Research Agency (ANR-Maladies Neurologiques et Psychiatriques).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
For more information
Hereditary Hearing loss Homepage: http://webh01.ua.ac.be/hhh/
Eurohear, European research consortium (EC-FP6): http://www.eurohear.org/
Christine Petit laboratory: http://www.pasteur.fr/ip/easysite/go/03b-000023-001/genetics-and-physiology-of-hearing
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Aijaz S, Balda MS, Matter K. Tight junctions: molecular architecture and function. Int Rev Cytol. 2006;248:261–298. doi: 10.1016/S0074-7696(06)48005-0. [DOI] [PubMed] [Google Scholar]
- Akiyama C, Shinozaki-Narikawa N, Kitazawa T, Hamakubo T, Kodama T, Shibasaki Y. Phosphatidylinositol-4-phosphate 5-kinase gamma is associated with cell-cell junction in A431 epithelial cells. Cell Biol Int. 2005;29:514–520. doi: 10.1016/j.cellbi.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Ashmore J. Cochlear outer hair cell motility. Physiol Rev. 2008;88:173–210. doi: 10.1152/physrev.00044.2006. [DOI] [PubMed] [Google Scholar]
- Ashmore JF. A fast motile response in guinea-pig outer hair cells: the cellular basis of the cochlear amplifier. J Physiol. 1987;388:323–347. doi: 10.1113/jphysiol.1987.sp016617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershadsky A. Magic touch: how does cell-cell adhesion trigger actin assembly? Trends Cell Biol. 2004;14:589–593. doi: 10.1016/j.tcb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Brownell WE, Bader CR, Bertrand D, de Ribaupierre Y. Evoked mechanical responses of isolated cochlear outer hair cells. Science. 1985;227:194–196. doi: 10.1126/science.3966153. [DOI] [PubMed] [Google Scholar]
- Canlon B, Miller J, Flock A, Borg E. Pure tone overstimulation changes the micromechanical properties of the inner hair cell stereocilia. Hear Res. 1987;30:65–72. doi: 10.1016/0378-5955(87)90184-5. [DOI] [PubMed] [Google Scholar]
- Cavey M, Rauzi M, Lenne PF, Lecuit T. A two-tiered mechanism for stabilization and immobilization of E-cadherin. Nature. 2008;453:751–756. doi: 10.1038/nature06953. [DOI] [PubMed] [Google Scholar]
- Chan DK, Hudspeth AJ. Mechanical responses of the organ of corti to acoustic and electrical stimulation in vitro. Biophys J. 2005;89:4382–4395. doi: 10.1529/biophysj.105.070474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallos P, Wu X, Cheatham MA, Gao J, Zheng J, Anderson CT, Jia S, Wang X, Cheng WH, Sengupta S, et al. Prestin-based outer hair cell motility is necessary for mammalian cochlear amplification. Neuron. 2008;58:333–339. doi: 10.1016/j.neuron.2008.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erulkar JS, O'Brien DA, Saunders JC. Hair bundle morphology on surviving hair cells of the chick basilar papilla exposed to intense sound. Scanning Microsc. 1996;10:1127–1140. discussion 1140-1122. [PubMed] [Google Scholar]
- Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D, Arpin M. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol. 2004;164:653–659. doi: 10.1083/jcb.200307032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler T, Canlon B, Dolan D, Miller JM. The effect of noise trauma following training exposures in the mouse. Hear Res. 1995;88:1–13. doi: 10.1016/0378-5955(95)00062-9. [DOI] [PubMed] [Google Scholar]
- Fridberger A, Boutet de Monvel J, Ulfendahl M. Internal shearing within the hearing organ evoked by basilar membrane motion. J Neurosci. 2002;22:9850–9857. doi: 10.1523/JNEUROSCI.22-22-09850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness DN, Mahendrasingam S, Ohashi M, Fettiplace R, Hackney CM. The dimensions and composition of stereociliary rootlets in mammalian cochlear hair cells: comparison between high- and low-frequency cells and evidence for a connection to the lateral membrane. J Neurosci. 2008;28:6342–6353. doi: 10.1523/JNEUROSCI.1154-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulley RL, Reese TS. Intercellular junctions in the reticular lamina of the organ of Corti. J Neurocytol. 1976;5:479–507. doi: 10.1007/BF01181652. [DOI] [PubMed] [Google Scholar]
- Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He DZ, Dallos P. Somatic stiffness of cochlear outer hair cells is voltage-dependent. Proc Natl Acad Sci USA. 1999;96:8223–8228. doi: 10.1073/pnas.96.14.8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- Hyenne V, Louvet-Vallée S, El-Amraoui A, Petit C, Maro B, Simmler M-C. Vezatin, a protein associated to adherens junctions, is required for mouse blastocyst morphogenesis. Dev Biol. 2005;287:180–191. doi: 10.1016/j.ydbio.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Hyenne V, Souilhol C, Cohen-Tannoudji M, Cereghini S, Petit C, Langa F, Maro B, Simmler MC. Conditional knock-out reveals that zygotic vezatin-null mouse embryos die at implantation. Mech Dev. 2007;124:449–462. doi: 10.1016/j.mod.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Hyman T, Shmuel M, Altschuler Y. Actin is required for endocytosis at the apical surface of Madin-Darby canine kidney cells where ARF6 and clathrin regulate the actin cytoskeleton. Mol Biol Cell. 2006;17:427–437. doi: 10.1091/mbc.E05-05-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AI. Actin motors that drive formation and disassembly of epithelial apical junctions. Front Biosci. 2008;13:6662–6681. doi: 10.2741/3180. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Nusrat A, Parkos CA. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell. 2004;15:176–188. doi: 10.1091/mbc.E03-05-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa KH, Adachi M. Force generation in the outer hair cell of the cochlea. Biophys J. 1997;73:546–555. doi: 10.1016/S0006-3495(97)78092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnke K. The fine structure of freeze-fractured intercellular junctions in the guinea pig inner ear. Acta Otolaryngol Suppl. 1975;336:1–40. [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry. 1994;33:3038–3049. doi: 10.1021/bi00176a037. [DOI] [PubMed] [Google Scholar]
- Karavitaki KD, Mountain DC. Imaging electrically evoked micromechanical motion within the organ of corti of the excised gerbil cochlea. Biophys J. 2007;92:3294–3316. doi: 10.1529/biophysj.106.083634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SY, Ahmed ZM, Shabbir MI, Kitajiri S, Kalsoom S, Tasneem S, Shayiq S, Ramesh A, Srisailpathy S, Khan SN, et al. Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Hum Mutat. 2007;28:417–423. doi: 10.1002/humu.20469. [DOI] [PubMed] [Google Scholar]
- Kim DO, Molnar CE, Matthews JW. Cochlear mechanics: nonlinear behavior in two-tone responses as reflected in cochlear-nerve-fiber responses and in ear-canal sound pressure. J Acoust Soc Am. 1980;67:1704–1721. doi: 10.1121/1.384297. [DOI] [PubMed] [Google Scholar]
- Kitajiri S, Fukumoto K, Hata M, Sasaki H, Katsuno T, Nakagawa T, Ito J, Tsukita S, Tsukita S. Radixin deficiency causes deafness associated with progressive degeneration of cochlear stereocilia. J Cell Biol. 2004;166:559–570. doi: 10.1083/jcb.200402007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J Neurosci. 2006;26:2115–2123. doi: 10.1523/JNEUROSCI.4985-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küssel-Andermann P, El-Amraoui A, Safieddine S, Nouaille S, Perfettini I, Lecuit M, Cossart P, Wolfrum U, Petit C. Vezatin, a novel transmembrane protein, bridges myosin VIIA to the cadherin-catenins complex. EMBO J. 2000;19:6020–6029. doi: 10.1093/emboj/19.22.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Calvez S, Avan P, Gilain L, Romand R. CD1 hearing-impaired mice. I: Distortion product otoacoustic emission levels, cochlear function and morphology. Hear Res. 1998;120:37–50. doi: 10.1016/s0378-5955(98)00050-1. [DOI] [PubMed] [Google Scholar]
- Li X, Donowitz M. Fractionation of subcellular membrane vesicles of epithelial and nonepithelial cells by OptiPrep density gradient ultracentrifugation. Methods Mol Biol. 2008;440:97–110. doi: 10.1007/978-1-59745-178-9_8. [DOI] [PubMed] [Google Scholar]
- Li X, Leu S, Cheong A, Zhang H, Baibakov B, Shih C, Birnbaum MJ, Donowitz M. Akt2, phosphatidylinositol 3-kinase, and PTEN are in lipid rafts of intestinal cells: role in absorption and differentiation. Gastroenterology. 2004;126:122–135. doi: 10.1053/j.gastro.2003.10.061. [DOI] [PubMed] [Google Scholar]
- Liberman MC, Gao J, He DZ, Wu X, Jia S, Zuo J. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature. 2002;419:300–304. doi: 10.1038/nature01059. [DOI] [PubMed] [Google Scholar]
- Michalski N, Michel V, Bahloul A, Lefèvre G, Barral J, Yagi H, Chardenoux S, Weil D, Martin P, Hardelin J-P, et al. Molecular characterization of the ankle link complex in cochlear hair cells and its role in the hair bundle functioning. J Neurosci. 2007;27:6478–6488. doi: 10.1523/JNEUROSCI.0342-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitaku S, Hirokawa T, Ono M. Classification of membrane proteins by types of transmembrane helices using SOSUI system. Genome Inform. 1998;9:367–368. [Google Scholar]
- Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17:646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- Mulroy MJ, Whaley EA. Structural changes in auditory hairs during temporary deafness. Scan Electron Microsc. 1984:831–840. [PubMed] [Google Scholar]
- Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;36:149–155. doi: 10.1042/BST0360149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes FD, Lopez LN, Lin HW, Davies C, Azevedo RB, Gow A, Kachar B. Distinct subdomain organization and molecular composition of a tight junction with adherens junction features. J Cell Sci. 2006;119:4819–4827. doi: 10.1242/jcs.03233. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK. Contributions of mouse models to understanding of age- and noise-related hearing loss. Brain Res. 2006;1091:89–102. doi: 10.1016/j.brainres.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Oliver TN, Berg JS, Cheney RE. Tails of unconventional myosins. Cell Mol Life Sci. 1999;56:243–257. doi: 10.1007/s000180050426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pataky F, Pironkova R, Hudspeth AJ. Radixin is a constituent of stereocilia in hair cells. Proc Natl Acad Sci USA. 2004;101:2601–2606. doi: 10.1073/pnas.0308620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- Pilot F, Philippe JM, Lemmers C, Lecuit T. Spatial control of actin organization at adherens junctions by a synaptotagmin-like protein Btsz. Nature. 2006;442:580–584. doi: 10.1038/nature04935. [DOI] [PubMed] [Google Scholar]
- Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol. 2007;23:237–261. doi: 10.1146/annurev.cellbio.22.010305.104241. [DOI] [PubMed] [Google Scholar]
- Raphael Y, Altschuler RA. Structure and innervation of the cochlea. Brain Res Bull. 2003;60:397–422. doi: 10.1016/s0361-9230(03)00047-9. [DOI] [PubMed] [Google Scholar]
- Robles L, Ruggero MA, Rich NC. Two-tone distortion on the basilar membrane of the chinchilla cochlea. J Neurophysiol. 1997;77:2385–2399. doi: 10.1152/jn.1997.77.5.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell IJ, Legan PK, Lukashkina VA, Lukashkin AN, Goodyear RJ, Richardson GP. Sharpened cochlear tuning in a mouse with a genetically modified tectorial membrane. Nat Neurosci. 2007;10:215–223. doi: 10.1038/nn1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders JC, Canlon B, Flock A. Changes in stereocilia micromechanics following overstimulation in metabolically blocked hair cells. Hear Res. 1986;24:217–225. doi: 10.1016/0378-5955(86)90020-1. [DOI] [PubMed] [Google Scholar]
- Shin JB, Streijger F, Beynon A, Peters T, Gadzala L, McMillen D, Bystrom C, Van der Zee CE, Wallimann T, Gillespie PG. Hair bundles are specialized for ATP delivery via creatine kinase. Neuron. 2007;53:371–386. doi: 10.1016/j.neuron.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel KP, Harvisty R. What's Wrong With My Mouse? New Interplays between Mouse Genes and Behavior. Washington DC: Society for Neuroscience; 1996. Assessing hearing, vision and balance in mice; pp. 26–38. [Google Scholar]
- Sutton LA, Lonsbury-Martin BL, Martin GK, Whitehead ML. Sensitivity of distortion-product otoacoustic emissions in humans to tonal over-exposure: time course of recovery and effects of lowering L2. Hear Res. 1994;75:161–174. doi: 10.1016/0378-5955(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Tian Y, Li M, Fritzsch B, Zuo J. Creation of a transgenic mouse for hair-cell gene targeting by using a modified bacterial artificial chromosome containing Prestin. Dev Dyn. 2004;231:199–203. doi: 10.1002/dvdy.20106. [DOI] [PubMed] [Google Scholar]
- Tomo I, Boutet de Monvel J, Fridberger A. Sound-evoked radial strain in the hearing organ. Biophys J. 2007;93:3279–3284. doi: 10.1529/biophysj.107.105072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–219. doi: 10.1016/s0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- Wakatsuki T, Schwab B, Thompson NC, Elson EL. Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J Cell Sci. 2001;114:1025–1036. doi: 10.1242/jcs.114.5.1025. [DOI] [PubMed] [Google Scholar]
- Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol. 2006;576:11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S, Matsui T, Tsukita S, Tsukita S. Rho-dependent and -independent activation mechanisms of ezrin/radixin/moesin proteins: an essential role for polyphosphoinositides in vivo. J Cell Sci. 2002;115:2569–2580. doi: 10.1242/jcs.115.12.2569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.