

Abstract
For many years research in Parkinson's disease (PD) has linked mitochondrial dysfunction with the characteristic loss of dopaminergic neurons of the substantia nigra, accumulation of cytoplasmic inclusions termed Lewy bodies, and motor dysfunction (Henchcliffe & Beal, 2008). The most compelling connection is that Parkinsonism can be observed in both humans and animals following exposure to inhibitors of complex I of the electron transport chain (Betarbet et al, 2002).
Keywords: complex I, PINK1, mitochondrial respiration
An understanding of how mitochondrial dysfunction arises in the tissue of a person afflicted with the disease has been elusive. The discovery of seemingly unrelated mutant genes responsible for familial forms of PD, including α synuclein (PARK1/PARK4), LRRK2 (PARK8), parkin (PARK2), DJ-1 (PARK6), ATP13A2 (PARK9), initially seemed to confound rather than solve the mystery, until the discovery of PD-associated mutations in a bona fide mitochondrial protein, PINK1 (PTEN-induced kinase 1 or PARK6) (Valente et al, 2004). Since then, a flurry of studies have detailed how this serine threonine kinase affects mitochondrial function and dynamics, and have put forward different hypotheses to explain the role(s) of mutant PINK1 in Parkinson's disease.
In this issue of EMBO Molecular Medicine, Morais et al make a significant contribution to our understanding of the PINK1-mediated mitochondrial protection. Using Drosophila and mouse models, the researchers assert that an early effect of PINK1 deficiency is the disruption of Complex I function. This results in decreased mitochondrial membrane potential and compromised transmission at neuromuscular junctions in Drosophila, that can be rescued by supplementing ATP in the synaptic terminal. Thus, the work lends insight into functional consequences of the mitochondrial defects in neuromuscular junctions that could account for defects in normal motor control in the disease. The authors also found that complex I activity and synaptic function could be replenished by expression of human wild type but not by PINK1 PD clinical mutants. Their findings highlight the pivotal role of PINK1 in maintaining respiration and mitochondrial ATP production and its relevance in PD.
»This study pinpoints the respiratory defect specifically at the level of complex I activity.«
The consequences reported so far regarding the genetic manipulation of PINK1 on mitochondria are multifaceted, and discrepancies exist between model systems and between laboratories even when measuring similar endpoints (Table I). There is, however, a general agreement of a drop in mitochondrial membrane potential as a result of PINK1 deficiency, and evidence that mitochondria are hydrolyzing glycolytically produced ATP to generate the membrane potential (Morais et al, this issue; Gandhi et al, 2009). Morais et al propose that defects in the mitochondrial respiratory chain lie upstream of these alterations. Deficiencies in several respiratory complexes have been reported in PINK1 deficient cells (Gautier et al, 2008; Gegg et al, 2009; Hoepken et al, 2007; Piccoli et al, 2008); however, this study pinpoints the respiratory defect specifically at the level of complex I activity. This is an important distinction, as a generalized loss of respiratory competence may signal a different underlying mechanism than specific complex I inhibition. Another recent publication ascribes similar importance of PINK1 in the maintenance of mitochondrial bioenergetic function, but with a slightly different twist (Gandhi et al, 2009). Instead of complex I as the primary dysfunction resulting from PINK1 deficiency, these authors suggest a key effect on the Na+/Ca2+ exchanger at the inner mitochondrial membrane that is responsible for Ca2+ efflux from mitochondria in electrically excitable cells. Uptake and efflux of Ca2+ from mitochondria are important aspects of normal neurotransmitter responses; however, excessive matrix Ca2+ is known to significantly compromise mitochondrial function by multiple mechanisms. Each of these patterns of inhibition would predict increased reactive oxygen species (ROS) production, as reported in multiple studies.
Table I.
Recent data on the effects of PINK1 knockout or knockdown on mitochondria and related functions
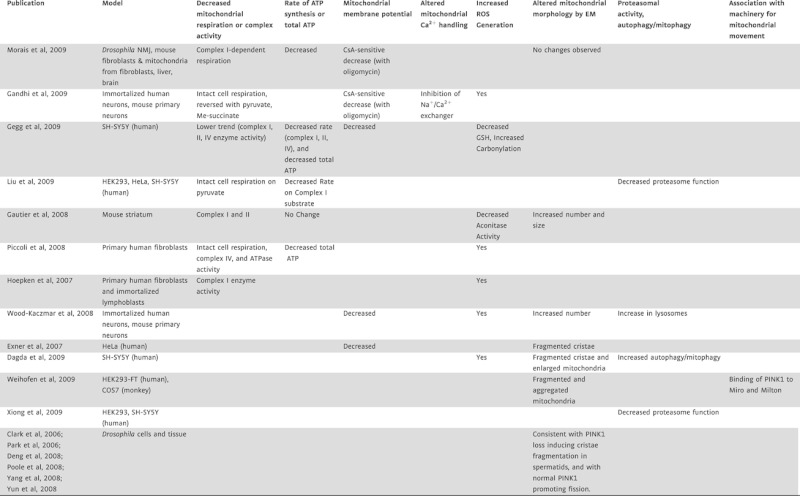 |
A clear link between the loss of PINK1 expression and alterations in mitochondrial morphology has been established (see Table I). The observations of both morphological and bioenergetic compromise raise the quintessential ‘chicken and egg’ question, as these two aspects of mitochondrial function are closely intertwined (see Twig et al, 2008). Genetic studies in Drosophila (Deng et al, 2008; Poole et al, 2008; Yang et al, 2009) suggest that PINK1 normally plays a role in promoting fission, although Morais et al have not observed defects in the morphology or number of mitochondria at the Drosophila neuromuscular junction (NMJ) of Pink1 mutants. The data available for mammalian cells and tissues are also not clear; Morais et al, did not observe changes in mitochondrial morphology of Pink1 mouse mutant neurons while others have described cristae and mitochondrial fragmentation as well as an increased size (Table I). If mitochondrial respiratory dysfunction or enhanced Ca2+ retention is indeed the primary consequence of PINK1 deficiency, one might expect mitochondrial fission rather than fusion to result, as mitochondrial depolarization and Ca2+ sequestration have generally been associated with fission events (Saotome et al, 2008; Twig et al, 2008). Clearly, the link between mitochondrial bioenergetics and morphology is complex, and much remains to be revealed on the topic.
»A direction in which the PINK flurry should now converge.«
What can explain the divergent observations of effects of PINK1 deficiency? The simplest is that PINK1 may be a multifunctional protein with numerous binding partners and kinase targets that are differentially expressed in the many models that have been created. The reported effectors–substrates of PINK1 include the mitochondrial molecular chaperone TRAP1 (Pridgeon et al, 2007), the matrix serine protease HtrA2/Omi (Plun-Favreau et al, 2007), and the ubiquitin E3 ligase parkin (Kim et al, 2008). It is currently unclear how these fit into pathways controlling bioenergetic function, although a theoretical argument can easily be made that chaperone and protease function could have important effects on protein import and respiratory complex assembly. The identification of the targets of PINK1's kinase activity and/or binding partners will help to further unravel the role of PINK1 in the regulation of mitochondrial function and PD. Morais et al provide us with a direction in which the PINK flurry should now converge—we can narrow the search considerably by focusing on substrates involved in Complex I activity and/or regulation, and hope that this approach will soon bring us novel insights and additional PD therapeautic targets.
The author declares that she has no conflict of interest.
References
- Betarbet R, Sherer TB, Di Monte DA, Greenamyre JT. Mechanistic approaches to Parkinson's disease pathogenesis. Brain Pathol. 2002;12:499–510. doi: 10.1111/j.1750-3639.2002.tb00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Dagda RK, Cherra S,J, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of pink1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009 doi: 10.1074/jbc.M808515200. Mar 10. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exner N, Treske B, Paquet D, Holmström K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci USA. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Schapira AH, Taanman JW. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE. 2009;4:e4756. doi: 10.1371/journal.pone.0004756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- Hoepken HH, Gispert S, Morales B, Wingerter O, Del Turco D, Mülsch A, Nussbaum RL, Müller K, Dröse S, Brandt U, et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol Dis. 2007;25:401–411. doi: 10.1016/j.nbd.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- Liu W, Vives-Bauza C, Acín-Peréz- R, Yamamoto A, Tan Y, Li Y, Magrané J, Stavarache MA, Shaffer S, Chang S, et al. PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson's disease. PLoS ONE. 2009;4:e4597. doi: 10.1371/journal.pone.0004597. Epub 2009 Feb 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais VA, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M, Haddad D, Frezza C, Mandemakers W, Vogt-Weisenhorn D, et al. Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med. 2009;1 doi: 10.1002/emmm.200900006. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Piccoli C, Sardanelli A, Scrima R, Ripoli M, Quarato G, D'Aprile A, Bellomo F, Scacco S, De Michele G, Filla A, et al. Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem Res. 2008;33:2565–2574. doi: 10.1007/s11064-008-9729-2. [DOI] [PubMed] [Google Scholar]
- Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, et al. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 Protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry. 2009:20. doi: 10.1021/bi8019178. Jan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AY, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE. 2008;3:e2455. doi: 10.1371/journal.pone.0002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, Xia K, Jiang W, Ronai Z, Zhuang X, et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009;119:650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YX, Wood NW, Latchman DS. Molecular basis of Parkinson's disease. NeuroReport. 2009;20:150–156. doi: 10.1097/WNR.0b013e32831c50df. [DOI] [PubMed] [Google Scholar]
- Yun J, Cao JH, Dodson MW, Clark IE, Kapahi P, Chowdhury RB, Guo M. Loss-of-function analysis suggests that Omi/HtrA2 is not an essential component of the PINK1/PARKIN pathway in vivo. J Neurosci. 2008;28:14500–14510. doi: 10.1523/JNEUROSCI.5141-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]