

Abstract
Mutations of the mitochondrial PTEN (phosphatase and tensin homologue)-induced kinase1 (PINK1) are important causes of recessive Parkinson disease (PD). Studies on loss of function and overexpression implicate PINK1 in apoptosis, abnormal mitochondrial morphology, impaired dopamine release and motor deficits. However, the fundamental mechanism underlying these various phenotypes remains to be clarified. Using fruit fly and mouse models we show that PINK1 deficiency or clinical mutations impact on the function of Complex I of the mitochondrial respiratory chain, resulting in mitochondrial depolarization and increased sensitivity to apoptotic stress in mammalian cells and tissues. In Drosophila neurons, PINK1 deficiency affects synaptic function, as the reserve pool of synaptic vesicles is not mobilized during rapid stimulation. The fundamental importance of PINK1 for energy maintenance under increased demand is further corroborated as this deficit can be rescued by adding ATP to the synapse. The clinical relevance of our observations is demonstrated by the fact that human wild type PINK1, but not PINK1 containing clinical mutations, can rescue Complex 1 deficiency. Our work suggests that Complex I deficiency underlies, at least partially, the pathogenesis of this hereditary form of PD. As Complex I dysfunction is also implicated in sporadic PD, a convergence of genetic and environmental causes of PD on a similar mitochondrial molecular mechanism appears to emerge.
Keywords: Complex I, mitochondrial dysfunction, Parkinson's disease, reserve pool deficit
INTRODUCTION
PD is a common progressive neurodegenerative movement disorder characterized by postural instability, rigidity, tremor and bradykinesia (Dawson & Dawson, 2003; Thomas & Beal, 2007). The pathological hallmark of PD is loss of dopaminergic neurons in the substantia nigra and the presence of eosinophilic cytoplasmic inclusions of misfolded proteins termed Lewy bodies in affected brain areas. The etiology of PD remains unknown, although clinical and experimental evidence implicates the involvement of abnormal protein aggregation, oxidative stress and mitochondrial dysfunction (Beal, 2003; Jenner & Olanow, 1996; Kosel et al, 1999; Zhang et al, 2000).
Over the years, the case for mitochondrial impairment in at least part of the pathogenesis of PD has become quite persuasive (reviewed in Mandemakers et al, 2007). For instance, several mitochondrial toxins that block Complex I cause Parkinsonism in humans and laboratory animals and are widely used to create PD animal models (Panov et al, 2005). Complex I (NADH:ubiquinone oxidoreductase, EC 1.6.5.3; NADH, nicotinamide adenine dinucleotide) is the first and the largest (∼980 kDa) complex of the electron transport chain (ETC). In mammals, it consists of 46 subunits, seven of which are encoded by mitochondrial DNA (Vogel et al, 2007). Complex I transfers electrons from NADH to ubiquinone while pumping protons across the inner mitochondrial membrane contributing to the electrochemical proton gradient used by F1F0 ATP synthase (Complex V) to produce ATP from ADP and inorganic phosphate. In addition, Complex I may also leak reactive oxygen species (ROS), resulting in cellular damage (Smeitink et al, 2004). Interestingly, Complex I deficiency and oxidative damage are frequently observed in affected neurons of PD patients (Parker & Swerdlow, 1998).
The involvement of mitochondria in PD is also deduced from studies demonstrating how other nuclear genes associated with the familial forms of PD can directly or indirectly affect mitochondrial function. α-Synuclein, the major component of Lewy bodies, appears to contain an amino-terminal mitochondrial targeting sequence (Devi et al, 2008) and acidification of the cytosol or overexpression of α-synuclein itself can cause localization of the protein to mitochondria (Cole et al, 2008; Shavali et al, 2008). Additionally, α-synuclein-null mice demonstrate increased resistance to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Dauer & Przedborski, 2003), whereas transgenic α-synuclein animal models treated with MPTP present swollen, morphologically abnormal mitochondria (Song et al, 2004). Parkin, an E3 ubiquitin ligase, is another gene implicated in autosomal recessive PD (Shimura et al, 2000). Parkin-null mice and Parkin Drosophila mutants develop apoptotic muscle degeneration with mitochondrial pathology and decreased resistance to oxidative stress (Greene et al, 2003; Palacino et al, 2004; Pesah et al, 2004). Furthermore, in a recent study it was shown that Parkin is involved in autophagy of dysfunctional mitochondria (Narendra et al, 2008), which might provide a mechanistic explanation for the genetic interaction between Parkin and Pink1 in Drosophila (Clark et al, 2006; Poole et al, 2008; Yang et al, 2006). Another PD-related gene, DJ-1 appears to function as an oxidative stress sensor as it is translocated under those conditions from the nucleus to the mitochondrial matrix and intramembrane space (Zhang et al, 2005). Finally, mutations in the leucine-rich repeat serine/threonine protein kinase 2 (LRRK2) are the most common known cause of familial and, interestingly, also sporadic PD (Gandhi et al, 2008). Subcellular fractionation indicates that approximately 10% of the total LRRK2 protein is localized to the mitochondrial outer membrane (Biskup et al, 2006; West et al, 2005), and LRRK2-mediated cell toxicity might involve cytochrome c release and caspase 3 activation (Iaccarino et al, 2007).
Although available data on α-synuclein, LRRK2 and Parkin already make a strong case for a role of mitochondria in PD, the most convincing evidence for the involvement of mitochondrial dysfunction is the presence of mutations in the mitochondrial serine/threonine kinase PINK1 that cause recessive hereditary PD in several families (Beilina et al, 2005; Silvestri et al, 2005). PINK1 is involved in the phosphorylation of the mitochondrial proteins TRAP1 (TNF receptor-associated protein 1), Htr2a (5-hydroxytryptamine (serotonin) receptor 2a)/OMI (Plun-Favreau et al, 2007; Pridgeon et al, 2007) and also Parkin (Kim et al, 2008), and associates with Hsp90 (heat shock protein 90)/Cdc37 (cell division cycle 37) chaperones (Moriwaki et al, 2008; Weihofen et al, 2007). Pink1 deficient mice reveal impaired dopamine release (Kitada et al, 2007), deficient mitochondrial respiration and increased sensitivity to oxidative stress (Gautier et al, 2008), while overexpression and loss of function studies suggest an anti-apoptotic activity for PINK1 (Deng et al, 2005; Lin & Kang, 2008; Petit et al, 2005). Additionally, studies in Drosophila and in HeLa cells indicate that Pink1 deficiency leads to altered mitochondrial morphology (Clark et al, 2006; Exner et al, 2007; Park et al, 2006; Poole et al, 2008; Yang et al, 2006). Evidence that pink1 genetically interacts with the fission/fusion machinery of mitochondria has supported the hypothesis that Pink1 deficiency affects various mitochondrial functions via interference with mitochondrial dynamics (Deng et al, 2008; Yang et al, 2008). It should be noted however that genetic interactions between pink1 and drp1 (dynamin related protein) or fis1 (fission 1), genes implicated in mitochondrial fission (Yoon et al, 2003), do not imply necessarily that pink1 is in the same molecular pathway as these proteins. Indeed studies performed on fibroblasts from patients carrying the homozygous W437X (Trp437 → Xxx) nonsense PINK1 mutation revealed a normal mitochondrial tubular network (Piccoli et al, 2008).
The interpretation of the various phenotypes caused by Pink1 deficiency or mutation thus remains somewhat controversial and the question of the fundamental mechanism underlying the various reported phenotypes needs to be further tested. We set out in the current work to investigate systematically the impact of Pink1 deficiency on fundamental mitochondrial functions and demonstrate that the earliest and most consistent defect lies in Complex I. We discuss how this deficit can explain the various phenotypes described previously in Pink1 deficient flies and mice.
RESULTS
Drosophila pink1 mutants fail to maintain normal synaptic transmission and show defects in reserve pool function
We used previously characterized pink1 mutant flies (Park et al, 2006), which display motor disturbances and structural changes of mitochondria. Loss of mitochondria at synapses has been implicated in specific defects in neurotransmission at Drosophila neuromuscular junction (NMJ) (Verstreken et al, 2005). We therefore measured neurotransmission using current clamp recordings at third instar larval NMJs in 2 or 0.5 mM Ca2+. As shown in Fig 1A–D, pink1B9 null mutants did not show deficits in neurotransmitter release or in amplitude or frequency of the spontaneous vesicle fusion measured in 0.5 mM Ca2+ and tetrodotoxin (TTX) (Fig 1E–H) implying that basal release characteristics, such as synaptic vesicle size, neurotransmitter filling and post-synaptic receptor clustering, are not significantly affected in the mutants. However, when pink1 mutants were stimulated at a higher and more intense frequency of 10 Hz, a small but significant progressive decline in synaptic transmission (Fig 1I) was observed. This might be caused by reduced endocytosis and inefficient replenishment of the cycling vesicle pool (ECP) or, alternatively, by defects in the utilization of the reserve pool (RP) that becomes mobilized only during high frequency stimulation (Kuromi & Kidokoro, 2002). FM1–43 dye reversibly binds membranes and is internalized in synaptic vesicles. Using specific stimulation paradigms (Kuromi & Kidokoro, 2002) this dye differentially labels both the vesicle pools. The vesicle endocytosis and labelling of the ECP in pink1B9 is not affected (Fig 2A and B); however, the RP cannot be efficiently labelled in pink1B9 mutants (Fig 2C, D). This was rescued by expressing human PINK1 in the affected neurons (Fig 2C and D). These specific defects at pink1 deficient synapses are very reminiscent, albeit less severe, to those observed in Drosophila drp1 mutants where most of the mitochondria fail to localize to synapses (Verstreken et al, 2005).
Figure 1. Drosophila pink1 mutants fail to maintain normal synaptic transmission during intense activity.
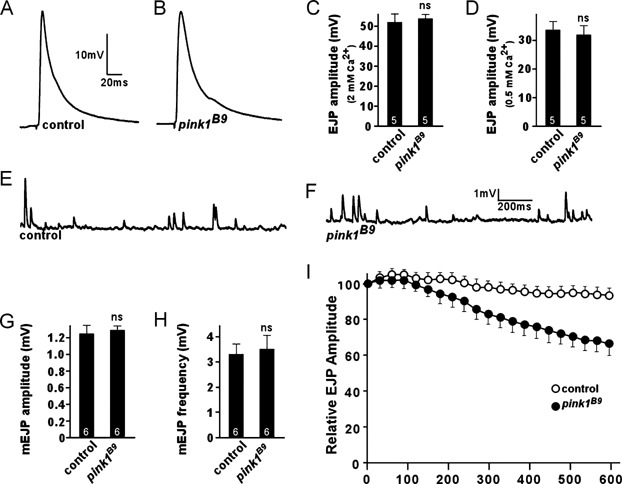
A–D. Neurotransmitter release in controls (w pink1REV) and Drosophila pink1 mutants (w pink1B9) in 2 mM (C) and 0.5 mM Ca2+ (A, B, D). Excitatory Junctional Potential traces (EJPs) (A, B) and quantification of EJP amplitudes (C, D) do not show a significant difference, indicating that basal neurotransmitter release is not affected in pink1 mutants.
E–H. Spontaneous vesicle fusion (mEJPs) recorded in 0.5 mM Ca2+ in the presence of TTX in controls (E) and pink1 mutants (F). Quantification of mEJP amplitude (G) and mEJP frequency (H) did not show a significant difference between control and pink1. We did not observe a statistical difference between the two genotypes.
I. Relative EJP amplitudes measured in 2 mM Ca2+ during 10 min of 10 Hz stimulation in controls and pink1 mutants. EJP amplitudes were binned per 30 s and normalized to the average amplitude of the first 10 EJPs. EJPs in pink1 mutants gradually declined to a level lower than that in controls. n = 4; ns = non-significant. The number of animals tested is indicated in the bar graphs and error bars indicate SEM.
Figure 2. Drosophila pink1 mutants show defects in reserve pool function that can be rescued by ATP.
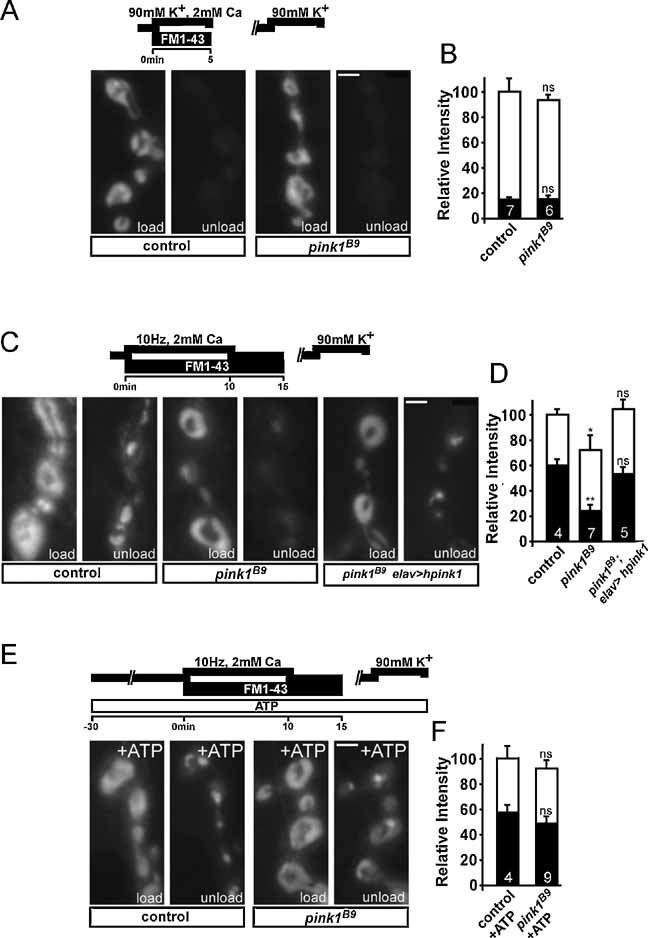
A, B. Exo–endo cycling vesicle pool (ECP) labelling in controls and pink1 mutants. Third instar larval fillets incubated for 5 min in HL-3 with 90 mM KCl, 2 mM Ca2+ and 4 µM FM1-43 to label the exo–endo cycling pool were washed in Ca2+ free HL-3 and imaged (A, ‘Load’) (Nikon FN-1 with DS-2MBWc digital camera, 40× water objective, NA 0.8). ECP vesicles were subsequently re-mobilized by incubating preparations for 5 min in 90 mM KCl, 2 mM Ca2+ and after washing in Ca2+ free HL-3, the same synapses imaged after loading were imaged again (A, ‘unload’). Fluorescence intensity of loading and unloading, normalized to loading intensity of controls, was quantified (B), but did not show a statistically significant difference. Scale bar = 0.2 µm.
C, D. Reserve pool (RP) labelling in controls, pink1 mutants (pink1B9) and pink1 mutants neuronally expressing human PINK1 (hpink1). Both the ECP and RP were labelled by electrically stimulating motor neurons of third instar fillets in HL-3 with 2 mM Ca2+ for 10 min and then leaving the dye with the preparation to rest for 5 min following stimulation (C, ‘load’). ECP vesicles, but not RP vesicles, were subsequently re-mobilized by incubating preparations for 5 min in 90 mM KCl, 2 mM Ca2+ and after washing in Ca2+ free HL-3, the same synapses imaged after loading were imaged again (C, ‘unload’). Fluorescence intensity of loading and unloading, normalized to loading intensity of controls, was quantified (D). Note the much weaker RP labelling in pink1 mutants compared to controls. Scale bar = 0.2 µm.
E, F. Reserve pool (RP) labelling with forward-filling ATP in controls and pink1 mutants. Loading of FM1-43 after forward-filling control and pink1B9 neurons with 1 mM ATP was performed as described above. Both ECP and RP vesicles are labelled after loading (E, ‘load’) and subsequently the ECP vesicles, but not RP vesicles, were re-mobilized by incubating preparations for 5 min in 90 mM KCl, 2 mM Ca2+ (E, ‘unload’). Fluorescence intensity of loading and unloading, normalized to loading intensity of controls, was quantified (F). Statistical analysis was performed using the unpaired Student's t-test, where *p < 0.05; **p < 0.01 when compared to control. ns = non-significant. The number of animals tested is indicated in the bar graphs and error bars indicate SEM. White bars: load; black bars: unload. Scale bar = 0.2 µm.
The pink1B9 flies harbour lower overall ATP levels (Clark et al, 2006). Thus, we forward filled control and pink1B9 motor neurons with ATP. As shown in Fig 2E and F, pink1B9 mutants and controls internalized similar amounts of FM1–43 dye in the presence of ATP. Subsequent unloading of ECP vesicles showed an increase in FM1–43 retention in the ATP-treated pink1B9 mutants compared to the non-ATP treated mutants (compare pink1B9 ‘unload’ in Fig 2C with Fig 2E; as well as pink1B9 black bars in Fig 2D with Fig 2F). Thus, impaired RP mobilization in pink1B9 mutants is, at least in part, caused by synaptic ATP depletion, similar to the defects observed in the mitochondria depleted drp1 mutant synapses (Verstreken et al, 2005).
Absence of PINK1 does not lead to dramatic changes in mitochondrial fragmentation
Absence of Pink1 and also deficiencies in Drp1, have been associated with alterations in mitochondrial morphology. To visualize mitochondria we used a mitochondrially targeted EGFP (Verstreken et al, 2005), but no alterations at pink1B9 mutant NMJs were observed, nor did we observe a difference in synaptic mitochondrial localization (Fig 3A and B) (Li et al, 2004; Verstreken et al, 2005). Also in fibroblasts obtained from Pink1 mouse mutants expressing mitochondrially targeted yellow fluorescent protein (YFP), no significant alterations in mitochondrial morphology were observed (Fig 3C and D). These findings were corroborated using high voltage transmission electron microscopy to image mouse brain and heart muscle mitochondria (Fig 3E). Hence, it appears that under basal conditions, and in multiple tissues, under the conditions tested, Pink1 deficiency did not affect mitochondrial localization or morphology. The synaptic dysfunction on the other hand clearly demonstrates that a Pink1 functional deficit is already present when morphology of the mitochondria is not yet affected, strongly arguing that this defect is upstream of morphological alterations observed by others (Clark et al, 2006; Exner et al, 2007; Park et al, 2006; Poole et al, 2008; Yang et al, 2006).
Figure 3. Morphology of mitochondria is not altered in the absence of PINK1.
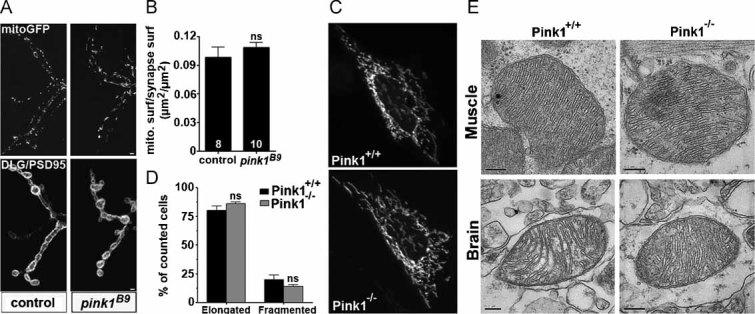
A, B. Confocal images of mitochondrial green fluorescent protein (mitoGFP) labelling (top panel) in control (w pink1REV) and pink1 mutant (w pink1B9). Drosophila third instar larval NMJs were also labelled with discs large (DLG/PSD95) (bottom panel) marking pre- and post-synaptic areas of synaptic boutons (A). Mitochondria at the synapse were quantified (B) by measuring mitochondrial surface to synaptic surface in maximum intensity projections (adapted from Verstreken et al (2005)). We did not observe a statistical difference between the two genotypes; ns = non-significant. The number of animals tested is indicated in the bar graphs (eight synapses from w pink1REV and 10 synapses from w pink1B9) and error bars indicate SEM. Scale bar = 0.2 µm.
C, D. Mitochondrial morphology of wild-type (Pink1+/+) and Pink1 knock-out (Pink1−/−) fibroblast cells transfected with a mitochondrially targeted YFP (mtYFP). Twenty-four hours post-transfection, cells were imaged on an inverted Olympus IMT-2 microscope equipped with a Cell®R imaging system (C). The corresponding morphometric analysis (D) was performed as described previously in Cipolat et al (2004). We did not observe a statistical difference between the two genotypes. Data represent the average ±SEM of three independent experiments (100 images for Pink1+/+, 120 images for Pink1−/−); ns = non-significant.
E. High voltage transmission electron microscopy of Pink1+/+ and Pink1−/− mouse brain and muscle (heart) tissue does not show morphological changes in the mitochondria. Ultrathin tissue sections (70 nm) were cut with an ultramicrotome and negatively stained with uranyl acetate and lead citrate. Samples were examined in a JEOL JEM-2100. Scale bar = 0.2 µm.
Mitochondrial membrane potential is decreased in the absence of Pink1
We therefore focused on the mitochondrial function, testing mitochondrial membrane potential by labelling NMJ synapses with JC-1. This potentiometric dye accumulates in the mitochondrial matrix as a function of the mitochondrial membrane potential (Δψm) and forms J-aggregates which cause a green- to red-shift emission. Synaptic mitochondria of control flies showed a clear JC-1 red labelling that is completely disrupted by the mitochondrial uncoupler carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (Fig 4A). In contrast, synaptic mitochondria of pink1B9 mutants showed a less red JC-1 labelling, indicating a decreased Δψm (Fig 4A and B). Quantitative analyses indicated no difference in NMJ length and bouton number between controls and pink1 mutants (Fig 4C).
Figure 4. Absence of PINK1 causes deficits in mitochondrial membrane potential (Δψm) and increases susceptibility to apoptotic stimuli.
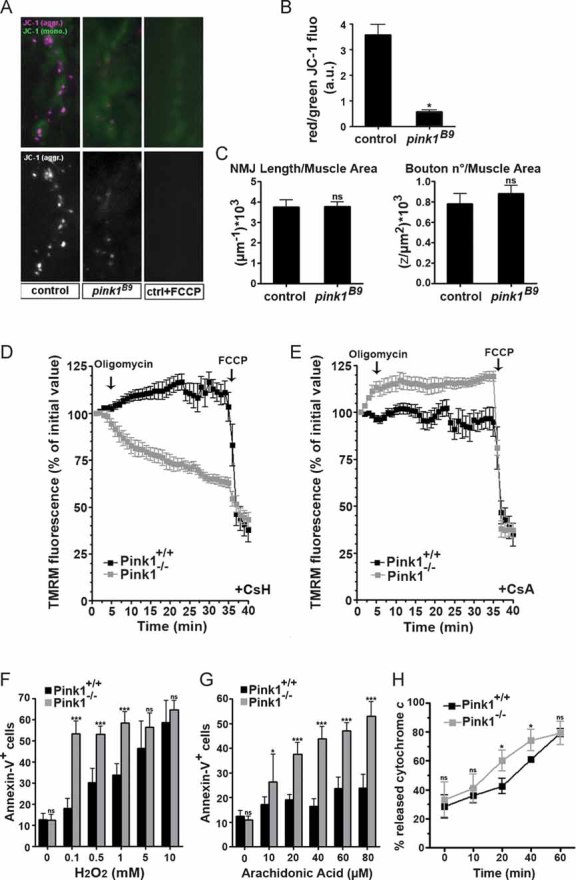
A, B. Imaging of Δψm at third instar Drosophila larval NMJs in controls (w pink1REV) and pink1 mutants (w pink1B9) using the ratiometric dye JC-1 as described previously (Verstreken et al, 2005). Pre-treatment of the preparations with 2 µM FCCP (an uncoupler) resulted in a lack of dye accumulation in mitochondria (A, third panel). Quantification of red JC-1 fluorescence emission to green emission (in the same area) showed a significant decrease in Δψm in pink1 mutants compared to controls (B).
C. Quantification of the third instar NMJ morphology by length (µm−1) and bouton number (z/µm2) both normalized to muscle surface area was similar in controls and pink1 mutants.
D, E. Imaging of Δψm in Pink1+/+ (black squares) and Pink1−/− (grey squares) fibroblast cells grown on cover slips and loaded for 30 min at 37°C with 10 nM TMRM in the presence of (D) 2 µg/ml cyclosporine H (CsH), a P-glycoprotein inhibitor, or (E) 1 µg/ml cyclosporine A (CsA), a P-glycoprotein and PTP inhibitor. Cells were then placed on the stage of an Olympus IMT-2 inverted microscope equipped with a Cell®R imaging system. Sequential images of TMRM fluorescence were acquired every 60 s over a 40 min time course. TMRM fluorescence over mitochondrial regions of interest was measured. When indicated (arrows), 2 µg/ml oligomycin (an ATP synthase inhibitor) and 2 µM FCCP (an uncoupler) were added. Data represent the average ± SEM of eight independent experiments.
F, G. Pink1+/+ and Pink1−/− fibroblasts were treated with increasing concentrations of H2O2 (F) and arachidonic acid (G) and after 2 h apoptosis was determined by the percentage of Annexin-V positive cells by flow cytometry. Data represent the average ± SEM of three independent experiments.
H. Cytochrome c release assays were performed on mitochondria isolated by differential centrifugation from Pink1+/+ and Pink1−/− mouse liver. Mitochondria were treated for 0, 10, 20, 40 and 60 min with p7/p15 recombinant BID (cBID). The supernatant was analysed on a cytochrome c ELISA and plotted as the percentage of total cytochrome c released. Data represent the average ± SEM of three independent experiments. Statistical analysis was performed using the unpaired Student's t-test, where *p < 0.02; ***p < 0.0005 when compared to control (Graph Pad Prism5 software). ns = non-significant, a.u., arbitrary units; CsH, cyclosporine H; CsA, cyclosporine A; FCCP, carbonylcyanide-p-trifluoromethoxyphenylhydrazone; TMRM, tetramethylrhodamine methyl ester.
We turned to Pink1−/− mouse fibroblasts that allowed additional functional assays. The loss of Δψm was confirmed using the potentiometric dye tetramethylrhodamine methyl ester (TMRM). As defects in Δψm can be compensated by the F1F0 ATP synthase working in the reverse proton pumping mode, we tested whether mitochondrial TMRM fluorescence decreased in response to the ATPase inhibitor oligomycin (Irwin et al, 2003). Pink1−/− cells responded indeed to oligomycin with a dramatic drop in Δψm (Fig 4D), indicating a latent mitochondrial dysfunction (Irwin et al, 2003) and confirming the observations in mutant pink1 fly neurons (Fig 4A). Such a latent mitochondrial dysfunction can be the result of a lower threshold for the opening of the permeability transition pore (PTP), a large conductance, unselective, tightly regulated inner mitochondrial membrane channel that participates in certain forms of cell death (Grimm & Brdiczka, 2007). The PTP inhibitor cyclosporine A (CsA) completely prevented mitochondrial depolarization caused by oligomycin in Pink1−/− cells (Fig 4E), suggesting a role for the PTP in this process. In conclusion, ablation of PINK1 in both a mammalian and a Drosophila model results in a latent dysfunction of the mitochondria, which is sensitive to CsA and causes a disturbance of the electrochemical gradient, the driving force for ATP synthesis.
Such an impairment could be the cause of increased susceptibility to apoptosis and cytochrome c release in Pink1 deficient cells. Pink1−/− cells were indeed more sensitive to two Ca2+-dependent apoptotic stimuli that act through the mitochondrial pathway, the oxidant H2O2 (Fig 4F) and the lipid second messenger arachidonic acid (Fig 4G) (Scorrano et al, 2003). The increased sensitivity to Ca2+-dependent mitochondria-mediated apoptotic stimuli was also observed in purified mitochondria from Pink1−/− liver in vitro. The release of cytochrome c upon stimulation with the proapoptotic BH3-only member of the Bcl-2 family cBID, was slightly, but significantly increased in purified Pink1−/− mitochondria (Fig 4H). Thus, mitochondria lacking Pink1 have a lowered threshold for PTP opening, are more susceptible to Ca2+-dependent apoptotic stimuli and release more cytochrome c in response to cBID (Scorrano et al, 2002).
PINK1 deficit results in reduced Complex I activity
Notably, PTP can be controlled by electron flux through Complex I of the respiratory chain (Fontaine et al, 1998), which is the target of both rotenone and MPTP. We therefore measured rates of oxygen consumption of mitochondria isolated from Pink1+/+ and Pink1−/− mouse liver and energized with substrates feeding electrons at the different respiratory chain complexes. Interestingly, the ratio of ADP-stimulated (State 3) to basal (State 4) oxygen consumption rate was significantly decreased in Pink1−/− mitochondria when they were energized with the Complex I substrates glutamate and malate (Fig 5A), whereas no differences were observed when mitochondria were energized with substrates feeding electrons at Complex II (succinate) or Complex III/IV (TMPD/ascorbate). The deficit in Complex I was confirmed by direct measurements of enzymatic activity of individual respiratory chain complexes (de Paepe et al, 2006) confirming a strong, and specific reduction in Complex I activity in Pink1−/− mitochondrial homogenates (p = 0.00006) (Fig 5B). The other complexes were unaffected (Fig 5C–G, p > 0.05). A reduction of Complex I activity (p = 0.03) was also observed in mitochondria-enriched fractions from mouse brain (Fig 5H), and Drosophila brain and thorax muscle (Fig 5I). A significant reduction in Complex I activity was confirmed in each tissue of two different species, indicating the fundamental relevance of this observation. Thus, ablation of Pink1 results in a primary defect in the catalytic activity of Complex I.
Figure 5. PINK1 deficit results in defects in Complex I of the electron transport chain.
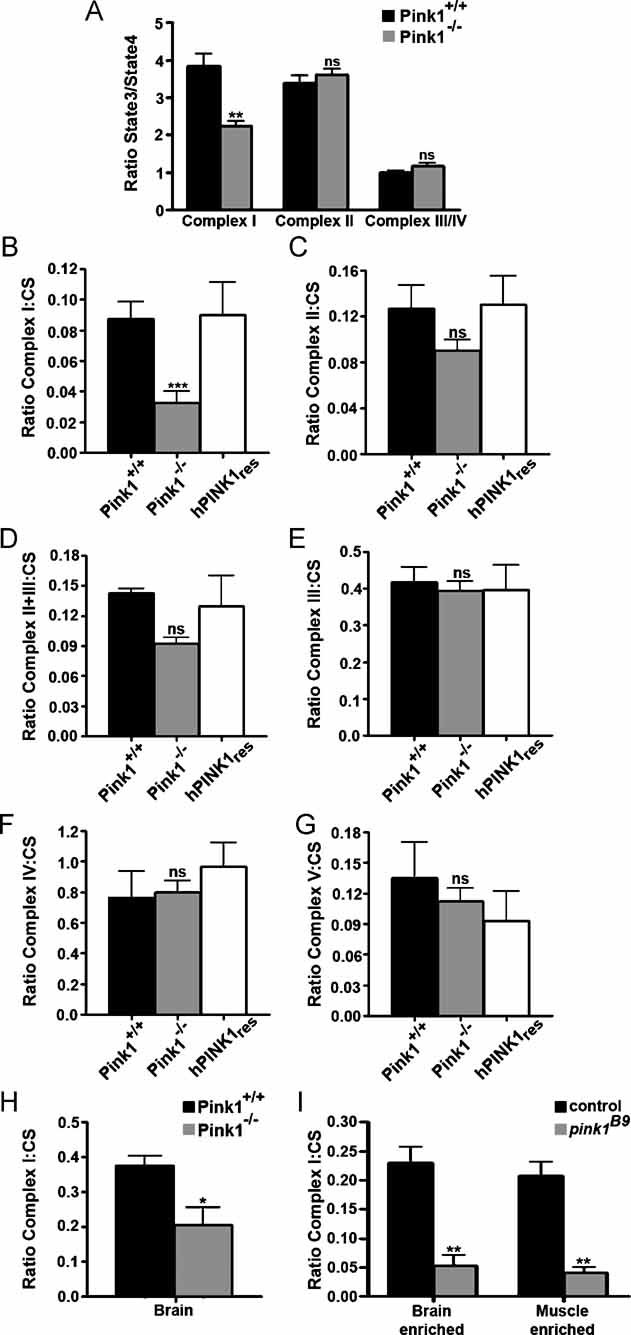
A. To measure the respiration rates between stimulated and basal ADP (ratio State3/State4), mitochondria were isolated from Pink1+/+ and Pink1−/− mice by differential centrifugation and were incubated in the experimental buffer. Mitochondrial oxygen consumption assays were performed in the presence of 5 mM glutamate/2.5 mM malate for the analysis of Complex I-driven respiration; 5 mM succinate in the presence of 2 µM rotenone for Complex II-driven respiration; or 3 mM ascorbate plus 150 µM TMPD in the presence of 0.5 µg/ml antimycin A for Complex IV-driven respiration. Mitochondrial oxygen consumption was measured by using a Clarke-type oxygen electrode (Hansatech Instruments). Data represent the average ± SEM of three independent experiments.
B–G. Respiratory chain measurements performed on mitochondrial homogenates from Pink1+/+, Pink1−/−, and also the rescue cell line with human PINK1 (hPINK1res) fibroblast cells were analysed by spectrophotometric assays for the activity measurement of (B) Complex I (NADH:ubiquinone oxidoreductase, rotenone sensitive), (C) Complex II (succinate:ubiquinone oxidoreductase, malonate sensitive), (D) Complex II+III, (E) Complex III (ubiquinone:cytochrome c oxidoreductase, antimycine sensitive), (F) Complex IV (cytochrome c oxidase). Measurements of (G) Complex V (ATPsynthase, oligomycine sensitive) and citrate synthase enzyme activities were also performed. The protein concentration was 2–4 mg/ml. Values were plotted according to the ratio between the specific complex's activity and citrate synthase activity.
H. Respiratory chain Complex I measurements were performed on mitochondrial homogenates from Pink1+/+ and Pink1−/− mouse brain tissue by spectrophotometric assays (NADH:ubiquinone oxidoreductase, rotenone sensitive). Citrate synthase enzyme activities were also performed. Values were plotted according to the ratio between the specific Complex I activity and citrate synthase activity.
I. Respiratory chain Complex I measurements were performed on fly mitochondrial homogenates from control (w pink1REV) and pink1 mutant (w pink1B9) brain enriched (heads) and muscle-enriched (thorax) tissues by spectrophotometric assays (NADH:ubiquinone oxidoreductase, rotenone sensitive). Citrate synthase enzyme activities were also performed. Values were plotted as a ratio of the specific Complex I activity and citrate synthase activity. Statistical analysis was performed using the unpaired Student's t-test, where *p = 0.03, **p < 0.005 and ***p = 0.00006 when compared to wild-type (Graph Pad Prism5 software). Data represent average ± SEM of at least three independent experiments. ns = non-significant.
We assessed ETC complexes in blue native polyacrylamide gel electrophoresis (BN-PAGE). Protein levels of different Complex I subunits and global assembly of Complex I, as well as of other ETC complexes, were not affected (Supporting Information Fig 1A and B, respectively). Our preliminary experiments also could not reveal significant alterations in expression levels of various individual components of Complex I between Pink1+/+ and Pink1−/− mitochondria (Supporting Information Fig 1C and D). Thus, the deficit in Complex I is functional and not directly reflecting significant alterations in assembly or expression of Complex I.
Relevance of the Complex I deficit for genetic PD
To determine if the defects in Complex I are relevant to the aetiology of PD, we determined whether clinical mutations of PINK1 affect Complex I activity. Pink1−/− cells were rescued with human wild-type or with PINK1-containing clinical mutations (Fig 6A). Human PINK1 (hPink1res) fully complemented Complex I enzymatic activity in Pink1−/− mouse fibroblasts (Fig 5B and 6B). In contrast, two PD PINK1 clinical mutations G309D (Gly309 → Asp) and W437X (Trp437 → Xxx) as well as an artificial mutation K219A (Lys219 → Ala) that inactivates the kinase domain (Silvestri et al, 2005) were not effective (Fig 6A and B). Notably, expression of these PINK1 mutants did not alter the activity of the other ETC complexes (data not shown). Hence, cells expressing only PINK1 carrying PD mutations display a severe Complex I dysfunction similar to that found in mitochondria of Pink1−/− cells. We also measured synaptic function in pink1B9 Drosophila mutants that neuronally express human PINK1 (hpink1) or a clinical mutant G309D (Gly309 → Asp) (hpink1G309D). Expression of human PINK1, but not its clinical mutant G309D rescues mobilization of RP vesicles (Fig 6C and D). Combined together, the data indicate that cellular defects in PINK1 mutants arise from dysfunction of Complex I of the ETC and extend the critical role of Complex I as a factor in the progression of PD from sporadic to genetic forms of the disease.
Figure 6. PINK1 clinical mutations also show reduced Complex I function and concomitant defects in synaptic function.
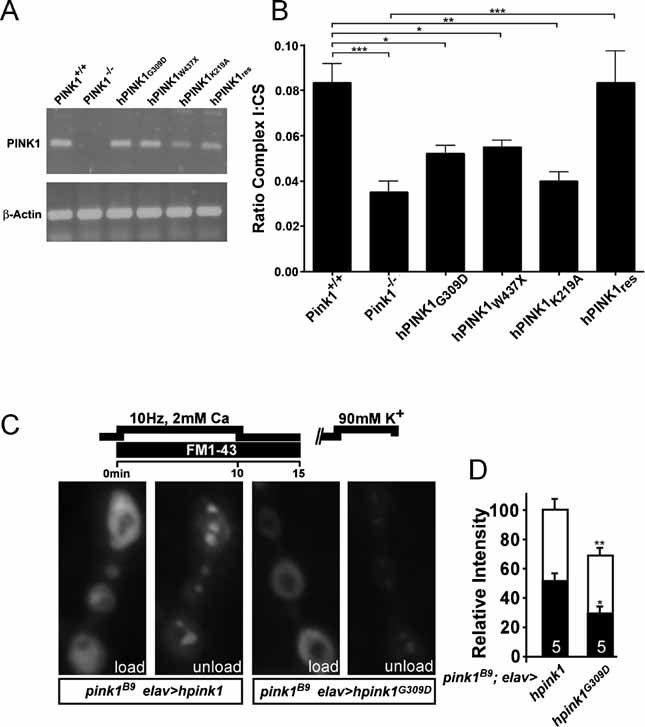
A. PD-related PINK1 mutants were transfected in Pink1−/− fibroblasts and expression levels were analysed by reverse transcriptase-PCR for mRNA detection.
B. Respiratory chain measurements performed on mitochondrial homogenates from Pink1 deficient fibroblast cell lines stably expressing human PINK1 (hPINK1res), PD-related PINK1 mutant hPINK1G309D and hPINK1W437X, and the artificial mutant hPINK1K219A. Spectrophotometric assays for Complex I activity (NADH:ubiquinone oxidoreductase, rotenone sensitive) were performed. Statistical analysis was done using the unpaired Student's t-test, where ***p = 0.00006, where **p < 0.004 and *p < 0.01 (Graph Pad Prism5 software). Data represent the average ± SEM of five independent experiments. ns = non-significant. Values presented for Pink1+/+, Pink1−/− and hPINK1res were previously shown in Fig 4A.
C, D. Reserve pool (RP) labelling in pink1 mutants that neuronally express wild-type human hPINK1 (w pink1B9/Y; elav-GAL4/UAS-hPink1) or the PD-related hPINKG309D mutant (w pink1B9/Y; elav-GAL4/UAS-hPink1G309D). Both the ECP and RP in these animals were labelled by electrically stimulating motor neurons of third instar fillets in HL-3 with 2 mM Ca2+ for 10 min and then leaving the dye with the preparation to rest for 5 min following stimulation (C, ‘load’). ECP vesicles, but not RP vesicles, were subsequently re-mobilized by incubating preparations for 5 min in 90 mM KCl, 2 mM Ca2+ and after washing in Ca2+ free HL-3, the same synapses imaged after loading were imaged again (C, ‘unload’). Fluorescence intensity of loading and unloading, normalized to loading intensity of pink1 mutants that express hPink1 in their neurons, was quantified (D). Note that wild type hPink1 can rescue RP labelling while hPink1G309D cannot. Comparison of RP labelling in w pink1B9 and w pink1B9/Y; elav-GAL4/UAS-hPink1G309D is not statistically significantly different. Statistical analysis was performed using the unpaired Student's t-test, where *p < 0.05; **p < 0.01 when compared to control. ns = non-significant. The number of animals tested is indicated in the bar graphs and error bars indicate SEM. White bars: load; black bars: unload.
DISCUSSION
Available evidence suggests impairment of the mitochondrial respiratory chain in sporadic and toxin-induced PD. However, a more suggestive link to mitochondrial dysfunction comes from studying genetic forms of PD, which taught that pathogenic mutations in the mitochondrial protein PINK1 are sufficient to cause the disease (Valente et al, 2004a,b). Previous studies (Clark et al, 2006; Exner et al, 2007; Park et al, 2006; Poole et al, 2008; Yang et al, 2006) showed altered mitochondrial morphology in loss of function PINK1 cells and animal models, but the exact mode of action of PINK1 remained unclear. We demonstrate here a key defect in the ETC at the level of Complex I caused by Pink1 loss of function, and the first synaptic phenotype caused by Pink1 deficiency. We observed deficits in Complex I driven respiration and specific Complex I enzymatic activity in different tissues of two different species, even before any alteration in mitochondrial morphology could be observed at the light and electron microscopical levels. Moreover, this deficit is completely eliminated when wild type PINK1 but not PINK1 containing clinical mutations is reintroduced, further arguing that this deficit is an upstream event in the pathological cascade induced by PINK1 loss of function.
Recent work has strongly drawn the attention to the mitochondrial fragmentation and other morphological abnormalities associated with PINK1 deficiency (Clark et al, 2006; Deng et al, 2008; Exner et al, 2007; Park et al, 2006; Poole et al, 2008; Yang et al, 2006). We did not observe gross alterations in mitochondrial morphology or synaptic localization, a trait dependent on mitochondrial fission, in both Drosophila and mouse-mutant cell lines (Li et al, 2004; Verstreken et al, 2005). Mitochondrial morphology changes were observed neither at the EM detection level in 2-month old mouse brain and muscle (this study) nor in the striatum of 3-, 4- and 24-month old Pink1−/− mice (Gautier et al, 2008). Furthermore, notwithstanding the many attempts to place Pink1 in the core machinery regulating mitochondrial fusion, a direct link between levels of Pink1 and rate of mitochondrial fusion has not been demonstrated. Since mitochondrial dysfunction, irrespective of the underlying mechanism, is accompanied by fragmentation of the organelle (Dimmer et al, 2008; Rojo et al, 2002), it is likely that the excess mitochondrial fission caused by ablation of Pink1 under certain experimental conditions is an epiphenomenon of the primary mitochondrial dysfunction sustained by reduced Complex I activity (Fig 7). Furthermore, this is entirely consistent with recent data on genetic interactions between the mitochondrial fusion and fission machinery and pink1 (Poole et al, 2008; Yang et al, 2008). Indeed, the data are consistent with an alternative model where Pink1 is not directly involved in mitochondrial dynamics, but they signify a convergence on a similar phenotype.
Figure 7. Schematic representation of upstream and downstream defects caused by PINK1 deficiency.

As discussed in the text and illustrated in this figure, Complex I deficiency can explain almost any alteration previously found to be associated with PINK1 loss of function. The decreased electron transfer at the level of Complex I can lead to reduced Δψm (present study), decreased ATP generation (Clark et al, 2006) and increased ROS production (Gautier et al, 2008). Deficient ATP levels affect dopamine release (Kitada et al, 2007) and RP mobilization (present study) causing reduced RP mobilization. Additionally, reduced Δψm can destabilize cytochrome c release (present study), and dephosphorylation of Drp1 that causes alterations in mitochondrial fusion/fission rates and morphology (Cereghetti et al, 2008). Recruitment of Parkin to such mitochondria (Narendra et al, 2008) will partially act protectively, as it will remove the most severely affected mitochondria, also explaining the genetic interaction between Parkin and Pink (Poole et al, 2008).
Recently, Gautier and co-workers described more general deficiencies of the mitochondrial respiration chain in Pink1 deficient mouse brain (Gautier et al, 2008), appearing as changes in state 3 respiration driven by Complex I and Complex II substrates. However, in our studies, a careful analysis of the ratio between phosphorylating (State 3) and basal rate (State 4) respiration showed no difference between Pink1+/+ and Pink1−/− mitochondria energized with the Complex II substrate succinate. This is further supported by the normal enzymatic activity of Complex II as measured in knockout organelles. Moreover, maximal (uncoupled) respiration was impaired in Pink1−/− mitochondria energized with glutamate, but not with succinate (data not shown), further confirming a specific defect in Complex I rather than a more general problem in ADP:ATP exchange which would be detected irrespective of the substrate used. Gautier et al (2008) proposed that altered mitochondrial dynamics could cause a more general respiration defect, however no evidence was provided. The alternative interpretation (see Fig 7) is equally possible however, as changes in mitochondrial shape might follow a primary mitochondrial dysfunction, for example via calcineurin and fission regulator Drp1 (Cereghetti et al, 2008).
A deficient Complex I function may indeed explain the many different phenotypes described in Pink1 deficient mice and flies, as schematically represented in Fig 7. Loss of Complex I would result in the reduction of the mitochondrial electrochemical gradient, observed in our study, and therefore promote the opening of the voltage-dependent PTP, which can in turn cause increased cytochrome c release and cell death (Irwin et al, 2003). Reduced Complex I activity can lead to increased levels of ROS production as found in several studies (Anichtchik et al, 2008; Piccoli et al, 2008). Loss of Complex I in synaptic mitochondria also results in impaired ATP production, which apparently underlies the RP mobilization defects that occur during intense stimulation of the synapse as mentioned here. This RP defect in the pink1 mutants is highly reminiscent of (albeit not as severe as) that observed in drp1 mutants (Verstreken et al, 2005). Drp1 mediates mitochondrial fission and its overexpression in neurons results in excess synaptic mitochondria (Li et al, 2004). Loss of Drp1 leads to depletion of synaptic mitochondria and decreased local mitochondrial ATP production that is used by the myosin ATPase to mediate RP mobilization (Verstreken et al, 2005; Ryan, 1999). Consistent with this hypothesis, ATP levels in pink1B9 mutant flies are reduced compared to controls (Clark et al, 2006), providing an explanation for the neuronal defects we observe in pink1 mutants.
One tantalizing question in PD research is why Pink1 deficiency results in quite robust phenotypes in Drosophila, while in mice the effects are much less pronounced. However, even Drosophila pink1 deficient mitochondria remain quite functional under most conditions and appear to generate sufficient energy to maintain, for instance, basal neuronal transmission. All these strongly indicate that efficient compensation mechanisms allow Pink1 deficient cells to cope with the partial Complex I dysfunction. The analogy with the clinical PINK1 mutations (which as we showed cause similar Complex I defects) is intriguing: while such mutations are present in many cells and tissues of the human body right from the time of conception, it is only late in life, and only in particular brain areas such as the substantia nigra and striatum, that Pink1 deficiency results finally in clinical phenotypes. Thus, one could postulate that compensation mechanisms allow systems to cope for many years with the defect, and additional stress, caused by ageing and/or oxidative dopamine metabolism, probably leads to effective impairment of the system.
In conclusion, our work provides genetic evidence that mutations found in a subset of autosomal recessive PD are impinging on the same central pathogenic event: impairment of Complex I activity. Apparently, the enzymatic activity of Complex I depends on the integrity of the kinase domain of PINK1. Further work is needed to investigate whether Pink1 directly affects the phosphorylation status of Complex I components, or whether it acts indirectly, via intermediate additional proteins, such as phosphorylation substrates of PINK1. Regardless, our work provides additional support to the hypothesis that the dysfunction of Complex I is an important and early event in the pathogenesis of Pink1-associated PD. It is interesting to note that Complex I has also been strongly implicated in the pathogenesis of (some forms of) sporadic PD.
MATERIALS AND METHODS
Generation of conditional Pink1 knockout mice and derived mouse embryonic fibroblast (MEF) cell lines
To generate the targeting vector construct 129/Ola, genomic DNA fragments were amplified by polymerase chain reaction (PCR). Psk62-easyflox was used as a vector. A 5′-fragment consisting of 3 kb of intron 1, which was 1366 basepairs (bp) downstream of the end of exon 1, was inserted. Then a 2 kb floxed homologous arm was inserted. It consisted of a loxP site, a neomycin resistance cassette flanked by frt sites, and exons 2 and 3 and a second loxP site that was 504 base pairs downstream the end of exon 3. A 3.9 kb 3′-homology arm consisting of exons 4, 5, 6, and part of exon 7 was added at the 3′- end of the homologous arm. Deletion of exons 2 and 3 by Cre-mediated recombination leads to a frameshift, thereby creating a truncation of Pink1 protein after exon 1. The linearized vector was electroporated into TBV2 embryonic stem cells. Neomycin resistant clones were screened for homologous recombination by Southern blot. Accurately targeted ES cell clones were expanded and injected into blastocysts originating from C578L/6 female mice. Blastocysts were transferred into pseudopregnant foster mothers to give birth to chimeras. Chimeras were bred to C578L/6 wild type mice and their offspring was screened for germline transmission by Southern blot. Pink1 conditional knockout mice were bred to Cre Deleter animals (Schwenk et al, 1995) with C578L/6 background to create Pink1 complete knockout mice.
The paper explained
PROBLEM:
Parkinson's disease (PD) is a common progressive neurodegenerative disorder. The etiology of PD remains unknown, although there is evidence pointing to the involvement of abnormal protein aggregation, oxidative stress and mitochondrial dysfunction.
The recessive PD gene PINK1 encodes a mitochondrial serine/threonine kinase and it appears to affect the mitochondrial dynamics. A mechanistic link between Pink1 and proteins directly involved in mitochondrial dynamics has however not been demonstrated.
RESULTS:
Using Drosophila and mouse models, we show here that an early effect of PINK1 deficiency is the disruption of Complex I function, which results in mitochondrial membrane depolarization, increased sensitivity to apoptotic stress and synaptic transmission deficits in Drosophila neurons. The fundamental importance of Pink1 for energy maintenance under increased demand is further corroborated as this deficit can be rescued by adding ATP to the synapse. The clinical relevance is demonstrated by the fact that human wild type PINK1 but not PINK1-containing clinical mutations can rescue Complex 1 deficiency.
IMPACT:
This study is important as it suggests an alternative hypothesis regarding the role(s) of Pink1 in the mitochondria and in PD. Rather than affecting directly the mitochondrial dynamics, PINK1 mutations affect Complex I activity and this primary effect is upstream of other mitochondrial alterations and PD phenotypes associated with PINK1 mutations.
Heterozygous Pink1 knockout mice (Pink1+/−) were interbred to generate mutant mice and wild type littermate controls. Embryonic day 13 embryos were dissected, heads and red organs were removed and used for genotyping by Southern blot. The rest of the bodies were chopped up in cell culture dishes containing Dulbecco's Modified Eagle's Medium (DMEM) supplied with 50% foetal calf serum (FCS) and 1% penicillin/streptomycin. Cultures were expanded and serum concentration was constantly decreased to 10% FCS after they reached consistent growth. Afterwards the cultures were immortalized by transfection with simian virus 40 (SV40) large T antigen.
Site-directed mutagenesis and generation of stable cell lines
hPINK1 construct was obtained from Origene (USA). Clinical hPINK1 PD mutants G309D (Gly309 → Asp) and W437X (Trp437 → Xxx), and the artificial mutant K219A (Lys219 → Ala) were constructed using the multisite-directed mutagenesis kit according to thesupplier's instructions (Stratagene). To confirm expression of hPINK1 in the transduced fibroblast cell lines, total RNA from these cell lines were extracted using standard methods and prepared for subsequent analysis.
Morphometric analysis
Morphometric analysis was performed as described previously (Cipolat et al, 2004) and examined on an Olympus IX81 inverted microscope equipped with a CellR imaging system (Olympus). For details see the Supporting Information.
Histology
Two-month old mice were perfused through the left ventricle with cacodylate buffer (0.1 M, pH 7.4) followed by Karnovsky fixative (2% paraformaldehyde +2.5% glutaraldehyde) in cacodylate buffer. The brain and heart (muscle) were removed and cut into 2 mm × 3 mm pieces, which were fixed overnight in Karnovsky fixative. The samples were rinsed with cacodylate buffer, post-fixed by immersion in 2% osmium tetroxide for 2 h, dehydrated in a graded ethanol series (pre-stained for 30 min with 1% aqueous uranylacetate in 70% ethanol step), immersed in propylene oxide and embedded in Agar 100 resin (Epon-equivalent, polymerized at 60°C). Ultrathin sections (70 nm) were cut with a Leica Ultracut UCT ultramicrotome and placed on 400-mesh copper Formvar carbon-coated grids. Grids were negatively stained with 4% saturated aqueous uranyl acetate (5 min) and Reynold's lead citrate stain (3 min). Samples were examined through a JEOL JEM-2100 microscope at the K.U. Leuven Electron Microscopy Core Facility of the Department of Human Genetics, Leuven, Belgium.
Flow cytometry
Evaluation of apoptosis was performed as described previously (Frezza et al, 2006), cells were treated with increasing concentrations of H2O2 and arachidonic acid. Cells were analysed by flow cytometry. Viability was measured as the percentage of Annexin-V, propidium iodide (PI) negative cells.
Real-time imaging of mitochondrial membrane potential
For evaluation of the membrane potential, fibroblast cells were grown in 3 cm plastic dishes with glass coverslips (Nunc) and the procedure was performed as described previously (Alirol et al, 2006). For details see the Supporting Information.
Mitochondrial isolation
Mitochondria were isolated from fibroblast cells and from 2-month old mouse liver and brain as described previously (Frezza et al, 2007). Fly heads (brain enriched) and thoraxes (muscle enriched) were homogenized and mitochondrial homogenates were purified as described by Walker et al (2006). For additional details, see the Supporting Information.
Cytochrome c release ELISA
Cytochrome c release in response to recombinant p7/p15 bid-mediated immunodepletion (BID) was determined as described previously (Frezza et al, 2006). For additional details see the Supporting Information.
Respiratory assays
Mitochondrial oxygen consumption was determined for liver mitochondria by using a Clarke-type oxygen electrode (Hansatech Instruments) as described previously (Frezza et al, 2006). Oxidative phosphorylation complex measurements performed on mitochondrial homogenates from fibroblast cells, mouse brain, fly brain and muscle-enriched tissue were analysed by spectrophotometric assays as described previously (de Paepe et al, 2006). See additional details in Supporting Information. For statistical analysis, all the measurements were analysed by Student's t-test.
Drosophila genetics
The w pink1REV controls and w pink1B9 flies were kindly provided by Jeehye Park and Jongkyeong Chung (Korea Advanced Institute of Science and Technology) (Park et al, 2006) and mutant larvae were selected using green fluorescent protein (GFP) balancers. UAS-hPink1 and UAS-hPink1G309D flies were kindly provided by Zhuohua Zhang (Brenham Institute for Medical Research) (Wang et al, 2006) and were expressed in w pink1B9 mutants using the elav-GAL4 driver (obtained from the Bloomington Stockcenter). UAS-MitoGFP D42-GAL4 flies were kindly provided by William Saxton (Indiana University) and male w pink1B9/Y; UAS mito-GFP D42-GAL4 e/+ larvae were used in our analyses.
JC-1 and MitoGFP labelling
Larval fillets were labelled with JC-1 (Molecular Probes) as described previously (Verstreken et al, 2005). FCCP, an uncoupler, was added at a final concentration of 50 nM. MitoGFP imaging was performed exactly as described by Verstreken et al (2005). Images were captured on a BioRad Radiance microscope or on a Nikon FN-1 microscope with a DS-2MBWc digital camera, 40× water objective, numerical aperture (NA) 0.8 and quantification of labelling intensity was performed using Image-J.
Electrophysiology
Larval electrophysiological recordings were performed as described by Verstreken et al (2003, 2005). Data were recorded with a Multiclamp 700B amplifier (Molecular Devices) and stored using pClamp 10. Concentrations of Ca2+ used in the present study are indicated in the figure legends.
FM1–43
FM1–43 labelling and unloading were performed as described in the figure legends (Verstreken et al, 2005). Images were captured on a Nikon FN-1 microscope with a DS-2MBWc digital camera, 40× water objective, NA 0.8 and quantification of labelling intensity was performed using Image-J. Forward filling of motor neurons with 1 mM ATP was performed as described previously (Verstreken et al, 2005). Briefly, larval fillets with cut motor neurons were incubated in HL-3 with 0.5 mM CaCl2 and 1 mM ATP, and the motor neuron innervating an abdominal hemisegment 2 was sucked into a suction electrode such that the end of the motor neuron is in contact with HL-3 with ATP. The bath solution was then replaced with HL-3 with 1 mM CaCl2 and preparations were incubated for 30 min. Following incubation, 4 µM (final) FM1–43 was added and the filled motor neuron was stimulated to label RP vesicles as described above. KCl unloading was performed in the presence of 1 mM ATP.
Acknowledgments
We thank Sven Vilain for assistance with the Drosophila manipulation. This work was supported by the Fund for Scientific Research, Flanders; KU Leuven; Federal Office for Scientific Affairs (IUAP P6/43/), Belgium; a Methusalem grant of the Flemish Government, VIB; and a Marie Curie Excellence Grant (MEXT-CT-2006-042267). VAM is a Marie Curie postdoctoral researcher. LS is a Senior Telethon Scientist of the Dulbecco-Telethon Institute and research in his laboratory is supported by Telethon Italy and AIRC Italy. CF is supported by a ‘Wintec’ fellowship of AIRC. The work of WW was supported by the Deutsche Forschungsgemeinschaft (SFB596), the Federal Ministry of Education and Research (BMBF) in the framework of the National Genome Research Network (NGFN) Förderkennzeichen 01GS0476, 01GR0430 and 01GN0512.
Supplementary information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
For more information
OMIM, Online Mendelian Inheritance in Man:
Parkinson's disease: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=168600
Pink1: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=608309
The Michael J. Fox Foundation for Parkinson's Research: http://www.michaeljfox.org/
Database of Drosophila Genes and Genomes: http://flybase.org/
Human disease to Drosophila Gene Database: http://superfly.ucsd.edu/homophila/
Bart de Strooper' Laboratoy: http://med.kuleuven.be/cme-mg/lncb/index.html
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Alirol E, James D, Huber D, Marchetto A, Vergani L, Martinou JC, Scorrano L. The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol Biol Cell. 2006;17:4593–4605. doi: 10.1091/mbc.E06-05-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anichtchik O, Diekmann H, Fleming A, Roach A, Goldsmith P, Rubinsztein DC. Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J Neurosci. 2008;28:8199–8207. doi: 10.1523/JNEUROSCI.0979-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson's disease. Ann NY Acad Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci USA. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res. 2008;314:2076–2089. doi: 10.1016/j.yexcr.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- de Paepe B, Smet J, Leroy JG, Seneca S, George E, Matthys D, van Maldergem L, Scalais E, Lissens W, de Meirleir L, et al. Diagnostic value of immunostaining in cultured skin fibroblasts from patients with oxidative phosphorylation defects. Pediatr Res. 2006;59:2–6. doi: 10.1203/01.pdr.0000191294.34122.ab. C. [DOI] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Jankovic J, Guo Y, Xie W, Le W. Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y. Biochem Biophys Res Commun. 2005;337:1133–1138. doi: 10.1016/j.bbrc.2005.09.178. [DOI] [PubMed] [Google Scholar]
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Navoni F, Casarin A, Trevisson E, Endele S, Winterpacht A, Salviati L, Scorrano L. LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum Mol Genet. 2008;17:201–214. doi: 10.1093/hmg/ddm297. [DOI] [PubMed] [Google Scholar]
- Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine E, Eriksson O, Ichas F, Bernardi P. Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation by electron flow through the respiratory chain complex I. J Biol Chem. 1998;273:12662–12668. doi: 10.1074/jbc.273.20.12662. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Gandhi PN, Wang X, Zhu X, Chen SG, Wilson-Delfosse AL. The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. J Neurosci Res. 2008;86:1711–1720. doi: 10.1002/jnr.21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci USA. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm S, Brdiczka D. The permeability transition pore in cell death. Apoptosis. 2007;12:841–855. doi: 10.1007/s10495-007-0747-3. [DOI] [PubMed] [Google Scholar]
- Iaccarino C, Crosio C, Vitale C, Sanna G, Carri MT, Barone P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum Mol Genet. 2007;16:1319–1326. doi: 10.1093/hmg/ddm080. [DOI] [PubMed] [Google Scholar]
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. 2003;35:367–371. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology. 1996;47:S161–170. doi: 10.1212/wnl.47.6_suppl_3.161s. [DOI] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosel S, Hofhaus G, Maassen A, Vieregge P, Graeber MB. Role of mitochondria in Parkinson disease. Biol Chem. 1999;380:865–870. doi: 10.1515/BC.1999.106. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Selective replenishment of two vesicle pools depends on the source of Ca2+ at the Drosophila synapse. Neuron. 2002;35:333–343. doi: 10.1016/s0896-6273(02)00777-8. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lin W, Kang UJ. Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem. 2008;106:464–474. doi: 10.1111/j.1471-4159.2008.05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandemakers W, Morais VA, De Strooper B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci. 2007;120:1707–1716. doi: 10.1242/jcs.03443. [DOI] [PubMed] [Google Scholar]
- Moriwaki Y, Kim YJ, Ido Y, Misawa H, Kawashima K, Endo S, Takahashi R. L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci Res. 2008;61:43–48. doi: 10.1016/j.neures.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, Greenamyre JT. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem. 2005;280:42026–42035. doi: 10.1074/jbc.M508628200. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Parker WD, Jr, Swerdlow RH. Mitochondrial dysfunction in idiopathic Parkinson disease. Am J Hum Genet. 1998;62:758–762. doi: 10.1086/301812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Petit A, Kawarai T, Paitel E, Sanjo N, Maj M, Scheid M, Chen F, Gu Y, Hasegawa H, Salehi-Rad S, et al. Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J Biol Chem. 2005;280:34025–34032. doi: 10.1074/jbc.M505143200. [DOI] [PubMed] [Google Scholar]
- Piccoli C, Sardanelli A, Scrima R, Ripoli M, Quarato G, D'Aprile A, Bellomo F, Scacco S, De Michele G, Filla A, et al. Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem Res. 2008;33:2565–2574. doi: 10.1007/s11064-008-9729-2. [DOI] [PubMed] [Google Scholar]
- Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, et al. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115:1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- Ryan TA. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci. 1999;19:1317–1323. doi: 10.1523/JNEUROSCI.19-04-01317.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+ a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- Shavali S, Brown-Borg HM, Ebadi M, Porter J. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett. 2008;439:125–128. doi: 10.1016/j.neulet.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14:3477–3492. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- Smeitink JA, van den Heuvel LW, Koopman WJ, Nijtmans LG, Ugalde C, Willems PH. Cell biological consequences of mitochondrial NADH: ubiquinone oxidoreductase deficiency. Curr Neurovasc Res. 2004;1:29–40. doi: 10.2174/1567202043480224. [DOI] [PubMed] [Google Scholar]
- Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007:R183–194. doi: 10.1093/hmg/ddm159. 16 (Spec No. 2) [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004a;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol. 2004b;56:336–341. doi: 10.1002/ana.20256. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, Bellen HJ. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron. 2003;40:733–748. doi: 10.1016/s0896-6273(03)00644-5. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Vogel RO, Smeitink JA, Nijtmans LG. Human mitochondrial complex I assembly: a dynamic and versatile process. Biochim Biophys Acta. 2007;1767:1215–1227. doi: 10.1016/j.bbabio.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Walker DW, Hajek P, Muffat J, Knoepfle D, Cornelison S, Attardi G, Benzer S. Hypersensitivity to oxygen and shortened lifespan in a Drosophila mitochondrial complex II mutant. Proc Natl Acad Sci USA. 2006;103:16382–16387. doi: 10.1073/pnas.0607918103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Qian L, Xiong H, Liu J, Neckameyer WS, Oldham S, Xia K, Wang J, Bodmer R, Zhang Z. Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci USA. 2006;103:13520–13525. doi: 10.1073/pnas.0604661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet. 2007;17:602–616. doi: 10.1093/hmg/ddm334. [DOI] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Dawson VL, Dawson TM. Oxidative stress and genetics in the pathogenesis of Parkinson's disease. Neurobiol Dis. 2000;7:240–250. doi: 10.1006/nbdi.2000.0319. [DOI] [PubMed] [Google Scholar]
- Zhang L, Shimoji M, Thomas B, Moore DJ, Yu SW, Marupudi NI, Torp R, Torgner IA, Ottersen OP, Dawson TM, et al. Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum Mol Genet. 2005;14:2063–2073. doi: 10.1093/hmg/ddi211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.