

Abstract
KATP channels regulate insulin secretion from pancreatic β-cells. Loss- and gain-of-function mutations in the genes encoding the Kir6.2 and SUR1 subunits of this channel cause hyperinsulinism of infancy and neonatal diabetes, respectively. We report two novel mutations in the gating loop of Kir6.2 which cause neonatal diabetes with developmental delay (T293N) and hyperinsulinism (T294M). These mutations increase (T293N) or decrease (T294M) whole-cell KATP currents, accounting for the different clinical phenotypes. The T293N mutation increases the intrinsic channel open probability (Po(0)), thereby indirectly decreasing channel inhibition by ATP and increasing whole-cell currents. T294M channels exhibit a dramatically reduced Po(0) in the homozygous but not in the pseudo-heterozygous state. Unlike wild-type channels, hetT294M channels were activated by MgADP in the absence but not in the presence of MgATP; however, they are activated by MgGDP in both the absence and presence of MgGTP. These mutations demonstrate the importance of the gating loop of Kir channels in regulating Po(0) and further suggest that Mg-nucleotide interaction with SUR1 may reduce ATP inhibition at Kir6.2.
Keywords: channel gating, hyperinsulinism, KATP channel, Kir6.2, neonatal diabetes
INTRODUCTION
Naturally occurring mutations can provide important insights into ion channel function. This is especially true of mutations in the ATP-sensitive potassium (KATP) channel, where mutations can cause either too much or too little insulin secretion, giving rise to hyperinsulinaemic hypoglycaemia of infancy and neonatal diabetes, respectively (for a review, see Ashcroft, 2007).
KATP channels couple the metabolic state of the cell to its electrical activity and are expressed in a wide range of cell types, including neurones, muscle and endocrine tissue (reviewed by Ashcroft, 2007; Nichols, 2006). In pancreatic β-cells, KATP channels have a crucial role in insulin secretion (Fig S1 of Supporting information). Their closure in response to metabolically generated ATP produces membrane depolarization, activation of voltage-gated Ca2+ channels, Ca2+ influx and thereby insulin granule exocytosis (Ashcroft, 2007; Henquin, 2000; Nichols, 2006). Conversely, KATP channel opening leads to membrane hyperpolarization, thus reducing Ca2+ influx and switching off insulin secretion. This metabolic regulation is mediated by changes in the intracellular concentration of adenine nucleotides with ATP inducing KATP channel closure by binding to the Kir6.2 subunit of the channel (Tucker et al, 1997) and Mg-nucleotides (MgATP, MgADP) stimulating channel opening via the SUR1 subunit (Gribble et al, 1997; Nichols et al, 1996; Shyng et al, 1997). The balance between these two opposing effects of nucleotides determines the level of channel activity in the cell.
Mutations in KCNJ11 and ABCC8, the genes encoding the pore-forming (Kir6.2) and regulatory (SUR1) subunits, respectively, of the KATP channel cause impaired insulin secretion. Loss-of-function mutations cause hyperinsulinism of infancy (HI), a condition in which insulin is continuously secreted, independent of the plasma glucose level (De Leon & Stanley, 2007; Dunne et al, 2004). This is a serious condition because the patient can suffer irreversible brain damage as a consequence of the resulting hypoglycaemia. Most of the mutations have been found in SUR1, but 24 have been identified in Kir6.2 (Flanagan et al, 2009).
Gain-of-function mutations in KATP channel genes give rise to neonatal diabetes, a rare disorder characterized by elevated glucose levels within the first six months of life (for reviews, see Ashcroft, 2007; Flanagan et al, 2009; Hattersley & Ashcroft, 2005). All mutations result in neonatal diabetes but this can either be permanent (PNDM) or follow a remitting relapsing course. In some cases, the patient experiences developmental delay, epilepsy and muscle weakness in conjunction with neonatal diabetes, a condition called DEND syndrome (Hattersley & Ashcroft, 2005). In a milder form of this syndrome, known as intermediate DEND syndrome (iDEND), the patients are not affected by epilepsy. Although mutations in either Kir6.2 or SUR1 can cause all these different conditions, both iDEND and DEND syndromes are more commonly associated with Kir6.2 mutations than SUR1 mutations. All Kir6.2 mutations cause a reduced KATP channel sensitivity to inhibition by MgATP, but this is greater for mutations that cause the more severe disease phenotypes (Koster et al, 2008; Masia et al, 2007; Proks et al, 2005b; Shimomura et al, 2006). In β-cells, the increased KATP current impairs insulin release, thus causing diabetes (Girard et al, 2009; Koster et al, 2000). In neurones, it leads to the neurological symptoms of DEND syndrome.
In this paper, we identify two novel KCNJ11 mutations in patients with the opposite phenotypes of iDEND (T293N) and HI (T294M). These mutations lie side-by-side in the gating loop of Kir6.2. Functional analysis revealed that the T293N mutation produces a marked reduction in KATP channel inhibition by MgATP, due to an increase in the intrinsic channel open probability (Po(0)). This leads to a large increase in the resting whole-cell KATP current, which can account for the iDEND phenotype of the patient. In contrast, the T294M mutation reduced Po(0) to unmeasurable levels, thereby reducing the whole-cell current and stimulating insulin secretion.
RESULTS
Patient characteristics and genetics
A novel heterozygous KCNJ11 mutation (c.878C>A; p.Thr293Asn or T293N) was identified in a girl born to second cousins of Turkish descent (Fig 1, case 1). DNA was not available from the parents but neither of them is known to be diabetic. The proband had a birth weight of 2.6 kg and presented with diabetic ketoacidosis at 10 weeks of age. She has developmental delay with severe muscle weakness in the trunk and legs, but no epilepsy, indicating she has iDEND syndrome. She was initially treated with insulin but glibenclamide (1–1.1 mg/kg/day) was added from 15 months of age, which enabled the insulin dose to be reduced from 0.9 to 0.5 U/kg/day. Improvements in her walking, speech and fine motor skills were noted after starting glibenclamide.
Figure 1. Partial pedigrees showing inheritance of KCNJ11 mutations.
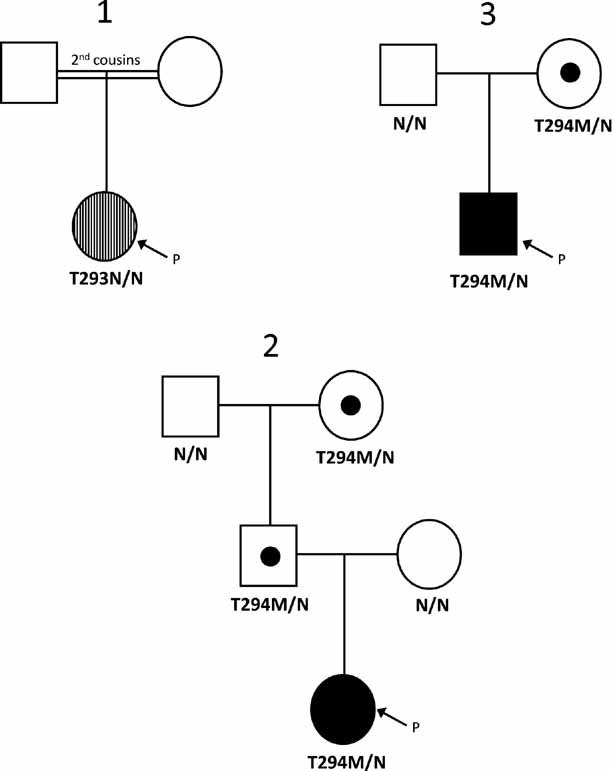
Circles represent females and squares indicate males. An arrow with the letter P points to the proband in each family. Filled symbols denote patients with hyperinsulinism and vertical hatching represents neonatal diabetes. Unaffected heterozygous mutation carriers are denoted by a dot. The genotype is shown below each symbol: N denotes a normal allele and N/N a normal genotype. The residue number and amino acid change are given for mutation carriers.
Two patients with congenital hyperinsulinism (Fig 1, cases 2 and 3) were heterozygous for a second KCNJ11 mutation, T294M (c.881C>T; p.Thr294Met). Case 2 is a female born at 38 weeks gestation with a birth weight of 4.1 kg, who developed severe hypoglycaemia soon after birth. She required high concentrations of intravenous glucose infusions to maintain normoglycaemia and a hypoglycaemia screen at 10 days of age confirmed hyperinsulinism with a plasma insulin concentration of 110 pM at a blood glucose concentration of 1.2 mM. She did not respond to diazoxide treatment and underwent a sub-total pancreatectomy at four weeks of age. Histological analysis showed hyperplasia throughout the pancreas with enlarged nuclei. The T294M mutation was inherited from her unaffected father: her unaffected paternal grandmother also carried the mutation (Fig 1, case 2). Analysis of microsatellite markers across the chromosome 11p15.1–11p15.5 region showed loss of heterozygosity of the maternal allele consistent with a giant focal lesion. The heterozygous germline T294M mutation is therefore homozygous within the pancreas.
Case 3 is a male born at 37 weeks gestation with a birth weight of 4.8 kg. Hyperinsulinaemic hypoglycaemia was diagnosed at one week of age (glucose 2.3 mM, insulin 531 pM) and euglycaemia was achieved with diazoxide therapy (starting dose 10 mg/kg/day, current dose at 19 months of age is 4 mg/kg/day). His mother is also heterozygous for the T294M mutation: she had glycosuria during pregnancy but has no history of hypoglycaemia. A second mutation was not detected within the coding region of the ABCC8 gene.
Effects of T293N and T294M mutations on whole-cell KATP currents
To determine the molecular mechanism of the disease, wild-type and mutant KATP channels were expressed in Xenopus oocytes. Under resting conditions, wild-type KATP channels are closed due to the high intracellular ATP concentration ([ATP]i) (Fig 2A and B). However, substantial currents were activated by the metabolic inhibitor sodium azide, which lowers [ATP]i and thus opens KATP channels. The ability of the KATP channel blockers glibenclamide (Fig 2A and B) and tolbutamide (Fig 2C, Fig S2 of Supporting information) to inhibit these currents confirmed their identity.
Figure 2. Effects of the T293N and T294M mutations on the whole-cell currents.
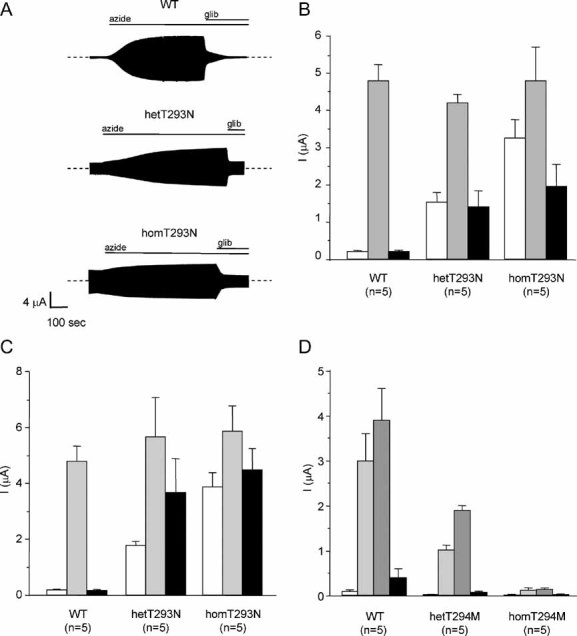
A. Representative wild-type, hetT293M and homT293M whole-cell currents evoked by a voltage step from −10 to −30 mV. The bars indicate the application of 3 mM azide or 100 µM glibenclamide (glib).
B–C. Mean steady-state whole-cell KATP channel current (as indicated) before (white bar) and after the application of 3 mM azide (grey bar) and in the presence of 3 mM azide and either 100 µM glibenclamide (B, black bar) or 0.5 mM tolbutamide (C, black bar). Note B and C were separate sets of experiments using different oocytes. The number of oocytes is indicated below the bar.
D. Mean steady-state whole-cell evoked by a voltage step from −10 mV to −30 mV before (white bar) and after application of 3 mM azide (light grey bar), in the presence of 3 mM azide and 0.34 mM diazoxide (dark grey bar) and in the presence of 3 mM azide and 0.5 mM tolbutamide (black bar).
In contrast to wild-type channels, substantial resting K+ currents were present in oocytes expressing homomeric Kir6.2-T293N/SUR1 (homT293N) channels (Fig 2A–C). These currents were further increased by azide suggesting that homT293N channels are not fully open at resting [ATP]i in the oocyte. Coinjection of wild-type and mutant Kir6.2, to simulate the heterozygous state, produced resting currents (hetT293N) that were much greater than wild-type and further increased by azide (Fig 2A–C). Mutant channels were significantly less sensitive to both glibenclamide and tolbutamide at concentrations that maximally block wild-type channels. Glibenclamide (100 µM) blocked wild-type channels by 96 ± 1% (n = 5) compared with 66 ± 9% (n = 5) for hetT293N channels and 59 ± 4% (n = 5) for homT293N channels (Fig 2B). Inhibition by 0.5 mM tobutamide was 97 ± 1% (n = 5) for wild-type channels, 36 ± 2% (n = 5) for hetT293N channels and 24 ± 3% (n = 5) for homT293N channels (Fig 2C). Thus the T293N mutation appears to induce a greater reduction in block by tolbutamide than glibenclamide.
In contrast to wild-type channels, homT294M channels failed to open in response to either azide or the KATP channel opener diazoxide (Fig 2D). Resting hetT294M currents were also unmeasureable, and although some current was activated by both azide and diazoxide it was significantly less than that observed for wild-type channels (66% and 51% smaller, respectively: Fig 2D, Fig S2 of Supporting information). The currents were fully blocked by tolbutamide.
These differences in the magnitude of the whole-cell currents are consistent with the clinical phenotypes associated with the mutations.
T293N channels exhibit reduced ATP sensitivity
The increase in resting whole-cell KATP currents suggests that the T293N mutation may reduce channel inhibition by ATP, as found for other iDEND mutations. To explore this idea, we measured ATP-concentration response curves in inside-out membrane patches. Because ATP does not interact with SUR1 to stimulate channel activity in the absence of Mg2+ (Gribble et al, 1998), experiments were first carried out in the absence of Mg2+: this enables the effect of the mutation on ATP block at Kir6.2 to be studied in isolation from Mg-nucleotide activation at SUR1.
Figure 3A shows that both homT293N and hetT293N currents were less ATP sensitive: half-maximal block (IC50) was produced by 778 µM, 37 µM and 6 µM ATP for homT293N, hetT293N and wild-type channels respectively (Table 1). The concentration–response curve for homT293N currents was best fit by assuming that a small fraction (4%) of channels are never closed by ATP. These data indicate that the mutation decreases the ATP sensitivity of the channel, at least in part, via Kir6.2.
Figure 3. Effects of the T293N mutation on KATP channel ATP sensitivity.
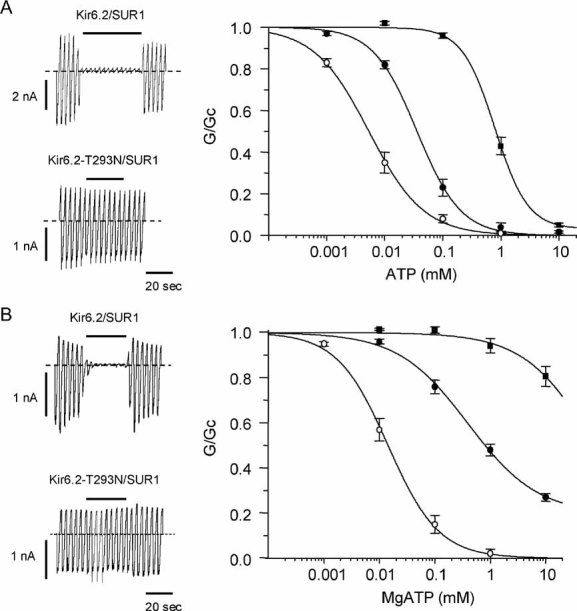
ATP concentration–response relations in the absence (A) and presence (B) of Mg2+.
Left: KATP currents recorded in response to voltage ramps from −110 to +110 mV in an inside-out patch excised from oocytes expressing wild-type or homT293N channels. The dashed line indicates the zero current level. The bar indicates application of 100 µM ATP (A) or 1 mM ATP (B).
Right: Mean relationships between [ATP] and KATP conductance (G), expressed relative to that in the absence of nucleotide (Gc), for Kir6.2/SUR1 (○, n = 5), and heterozygous (•, n = 6) or homomeric (▪, n = 5) Kir6.2-T293N/SUR1 channels in the absence of Mg2+ (A); or for wild-type (○, n = 8), hetT293N (•, n = 5) or homT293N (▪, n = 5) currents in the presence of Mg2+ (B). The lines are drawn to equation 1, with the following parameters. A: wild-type (IC50 = 5 µM, h = 0.9), hetT293N (IC50 = 36 µM, h = 1.2), homoT293N (IC50 = 793 µM, h = 1.5, a = 0.03). B: Kir6.2/SUR1 (IC50 = 14 µM, h = 1) and het293N (IC50 = 391 µM, h = 0.7, a = 0.2). The line through the hom293N data is drawn by hand.
Table 1.
Characteristics of mutations
| IC50 (µM; 0 mM Mg2+) | IC50 (µM; 2 mM Mg2+) | %I (3 mM MgATP) | |
|---|---|---|---|
| WT | 6 ± 1 (n = 5) | 17 ± 2 (n = 8) | 0.8 ± 0.3 (n = 8) |
| hetT293N | 37 ± 7 (n = 6) | 324 ± 48 (n = 5) | 34 ± 2 (n = 5) |
| homT293N | 778 ± 92 (n = 5) | n.m. | >90% |
| IC50 (µM; 0 mM Mg2+) | IC50 (µM; 2 mM Mg2+) | %I (3 mM MgATP) | |
|---|---|---|---|
| WT | 6 ± 1 (n = 10) | 16 ± 2 (n = 10) | 0.7 ± 0.3 (n = 10) |
| hetT294M | 6 ± 3 (n = 6) | 7 ± 2 (n = 10) | 1.0 ± 0.4 (n = 10) |
Values in parentheses indicate the number of oocytes. n.m., not meausureable. %I = current recorded in an inside-out patch in the presence of 3 mM MgATP expressed as a percentage of that in the absence of MgATP. The values were calculated from the fit of equation 1 to the data obtained for individual oocytes.
In the intact pancreatic β-cell, the effect of the T293N mutation on ATP inhibition at Kir6.2 will be modified by the stimulatory effect of MgATP at SUR1. Thus, we next compared ATP concentration–response curves in the presence of Mg2+ (Fig 3B). HomT293N currents were significantly less ATP sensitive than in the absence of Mg2+, being blocked less than 20% even at MgATP concentrations as high as 10 mM. The IC50 for hetT293N channels (324 µM) was also less than that of wild-type channels (17 µM; Table 1). As found for other mutations causing neonatal diabetes (Proks et al, 2005a), the increase in the IC50 produced by Mg2+ was greater for hetT293N channels (nine-fold) than for wild-type channels (3-fold) (compare Fig 4A and B). In addition, a large fraction of hetT293N current was unblocked at 3 mM MgATP (34 ± 2%, n = 5).
Figure 4. Effects of Mg2+ on ATP sensitivity.
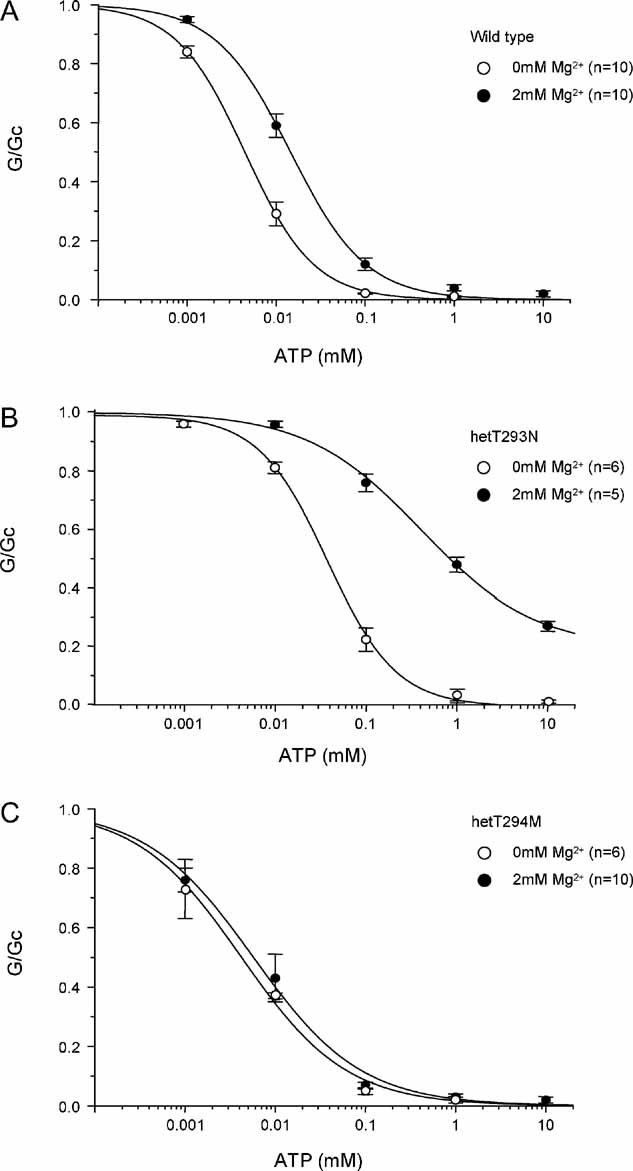
Comparison of ATP concentration–response curves of wild-type (A) het293N (B) and hetT294M (C) channels in the absence (○) and presence (•) of Mg2+. Same data as in Fig 2 and 3.
As Mg2+ is always present inside the cell, and ATP concentrations probably lie above 1 mM in physiological conditions (Gribble et al, 2000), these results suggest that there will be a substantial increase in the resting whole-cell KATP currents for both hetT293N and homT293N channels, as observed experimentally (Fig 2A–C).
T293N channels have an increased intrinsic open probability
Mutations that reduce the ATP sensitivity of the KATP channel can act by impairing ATP binding or transduction, or by influencing the intrinsic (ligand-independent) gating of the channel (Ashcroft 2007; Masia et al, 2007; Proks et al, 2005b; Shimomura et al, 2006). We therefore examined the effect of the T293N mutation on the single-channel kinetics. Experiments were carried out in the absence of ATP, where intrinsic gating can be assessed. We found that the intrinsic Po(0) was markedly increased by the T293N mutation (Fig 5), being 0.87 ± 0.03 (n = 5) for homT293N compared with 0.27 ± 0.04 (n = 6) for wild-type channels. This increase in Po(0) is sufficient to account for the lower ATP sensitivity of the mutant channel (Enkvetchakul et al, 2000; Proks et al, 2005b).
Figure 5. The T293N mutation increases Po(0).
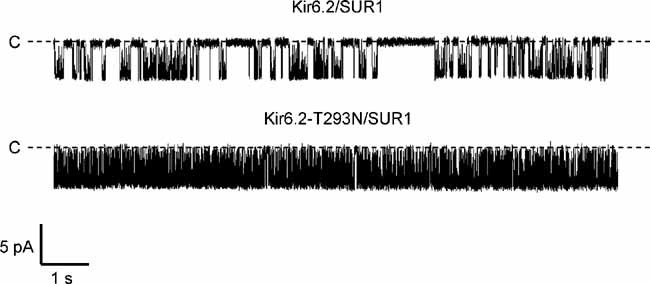
Representative single KATP channel currents recorded at −60 mV from inside-out patches from oocytes expressing SUR1 plus either Kir6.2 or Kir6.2-T293N. The dashed line indicates the zero current level.
The increased Po(0) is produced by an increase in the duration of the bursts of openings and a decrease in the duration and frequency of the long closed states. Mean burst duration was 16 ± 3 ms (n = 5) for wild-type channels and 399 ± 19 ms (n = 5) for homT293N channels. Wild-type channels had two interburst states of 4.2 ± 1.3 and 59 ± 3 ms (n = 5), with relative frequencies of 10 and 5% respectively (the balance being made up of intraburst closed times). In contrast, homT293N channels had a single interburst state of 13 ± 1 ms (n = 5) with a frequency of 0.9%.
T294M channels are expressed at the surface membrane
No currents were detected in inside-out patches excised from oocytes expressing homT294M channels, even in the absence of added nucleotide. This might reflect a reduced density of mutant channels in the plasma membrane or the mutant channels could be present but permanently closed (due to a markedly decreased Po(0)). To determine which of these possibilities was correct, we measured the surface expression of T294M channels in COS-7 cells.
As previously reported (Zerangue et al, 1999), Kir6.2 did not reach the surface membrane in the absence of SUR1, but was easily detected when Kir6.2 and SUR1 were co-expressed (Fig 6A). Co-expression of SUR1 with Kir6.2-T294M resulted in less surface expression, but this was still substantial. This suggests that the lack of homT294M currents observed in excised patches is, in part, due to a lower channel density, caused by impaired trafficking of channels to the plasma membrane. Nevertheless, the fact that the expression is 60% of wild-type indicates many homT294M channels are present, but that they are closed (i.e. they have a Po(0) of zero).
Figure 6. Effects of the T294M mutation on channel density and ATP sensitivity.
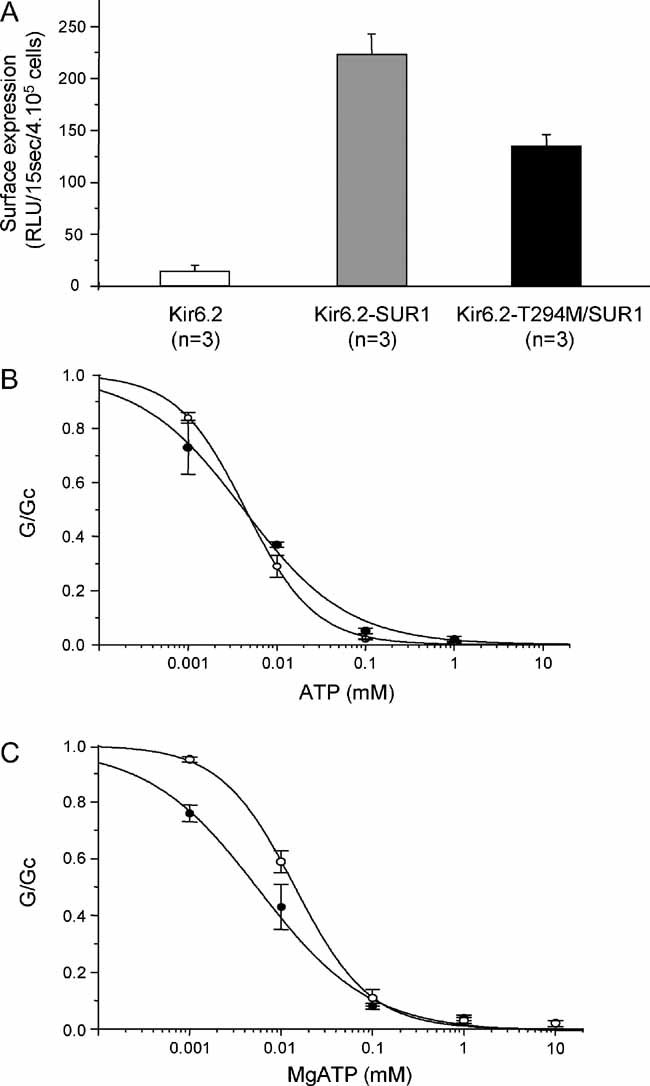
A. Mean surface expression for wild-type and mutant KATP channels in COS-7 cells, measured using a chemiluminnescence assay. Wild-type Kir6.2 was expressed alone or in combination with SUR1.
B,C. Mean relationships between [ATP] and KATP conductance (G) expressed relative to the conductance in the absence of nucleotide (Gc) for Kir6.2/SUR1 (○, n = 10), and hetKir6.2-T294M/SUR1 (•, n = 6) channels in the absence of Mg2+ (B); or for Kir6.2/SUR1 (○, n = 10), and heterozygous (•, n = 10) Kir6.2-T294M/SUR1 currents in the presence of Mg2+ (C). The lines are drawn to equation 1, with the following parameters. B: Kir6.2/SUR1 (IC50 = 5 µM, h = 1.1), hetT294M (IC50 = 4 µM, h = 0.7). C: Kir6.2/SUR1 (IC50 = 14 µM, h = 1.1), hetT294M (IC50 = 6 µM, h = 0.7).
In the heterozygous state, there will be a mixed population of channels that contain a variable number of mutant subunits (from 0 to 4). The Po(0) of hetT294M channels was therefore measured in multi-channel patches (Fig S3 of Supporting information). This represents the average Po(0) of the channel population: it was not different from wild-type, being 0.34 ± 0.03 (n = 15) for hetT294M and 0.27 ± 0.03 (n = 11) for wild-type channels.
ATP-sensitivity of hetT294M channels
Although no homT294M currents were detected in inside-out patches, large hetT294M currents were observed. Figure 6B and C shows that although the ATP sensitivity of hetT294M currents was similar to wild-type in the absence of Mg2+ (IC50 6 µM in both cases), it was greater than wild-type in the presence of Mg2+ (IC50 = 7 and 16 µM, respectively) (Table 1). However, there was no obvious difference in ATP block at ATP concentrations >100 µM. This reflects a decrease in the slope factor (Hill coefficient) of the ATP concentration–inhibition curve of hetT294M channels both in the presence and in the absence of the cation.
Interestingly, unlike both wild-type and hetT293N currents, there was no significant effect of Mg2+ on the ATP sensitivity of hetT294M currents (Fig 4C). This suggests that MgATP and/or MgADP activation by SUR1 is impaired by the T294M mutation. We therefore tested the ability of MgADP (0.1 mM) to activate hetT294M channels both in the absence and presence of 0.1 mM MgATP (Fig 7A). MgADP alone stimulated channel activity to the same extent as for wild-type channels. In striking contrast to wild-type channels, however, MgADP did not activate hetT294M channels preblocked by MgATP.
Figure 7. Mean currents recorded for the wild-type and mutation in the presence of various Mg-nucleotides.
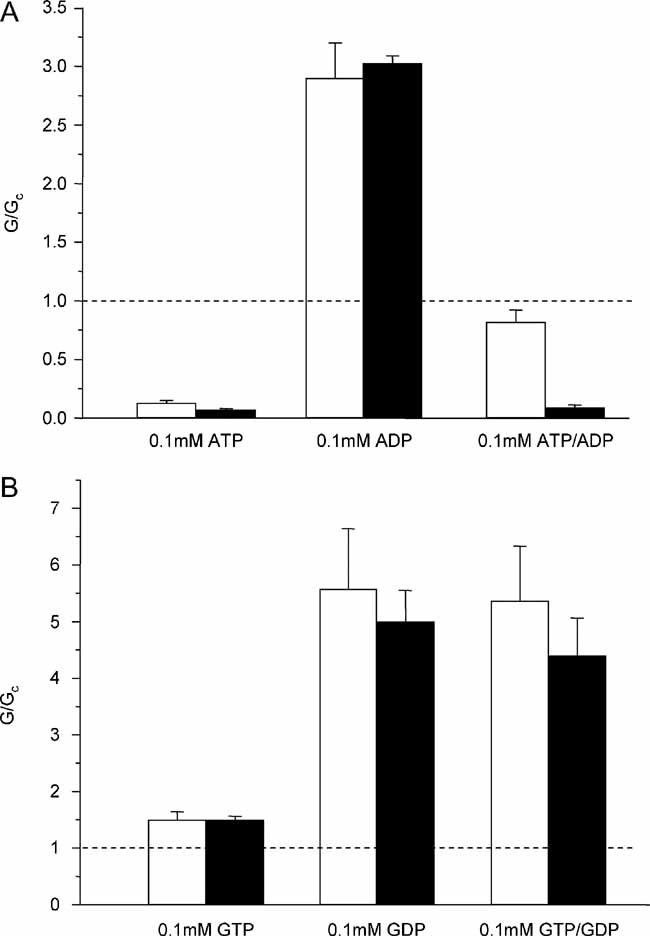
- Mean wild-type (white bar, n = 17) and hetT294M (black bar, n = 12) currents recorded in the presence of 0.1 mM MgATP, 0.1 mM MgADP or 0.1 mM MgATP plus 0.1 mM MgADP.
- Mean wild-type (white bar, n = 6) and hetT294M (black bar, n = 7) currents recorded in the presence of 0.1 mM MgGTP, 0.1 mM MgGDP or 0.1 mM MgGTP plus 0.1 mM MgGDP. Currents (G) are expressed relative to the mean of the currents recorded in control solution (Gc) before and after the application of nucleotide.
As both MgATP and MgADP exert an inhibitory effect at Kir6.2 and a stimulatory effect at SUR1, we next examined the effects of GDP and GTP. At a concentration of 100 µM, these nucleotides have little inhibitory effect at Kir6.2 but stimulate channel activity via SUR1 (Trapp et al, 1997). As Fig 7B shows, MgGDP stimulated channel activity both in the absence and presence of MgGTP. This suggests that nucleotide binding, hydrolysis and transduction by SUR1 are unaffected by the T294M mutation.
DISCUSSION
In this paper, we demonstrate that mutation of two adjacent residues in the cytosolic domain of Kir6.2 either increases (T293N) or decreases (T294M) the KATP current, consistent with the opposite phenotypes of iDEND and hyperinsulinism in patients carrying these mutations.
Our data also show that in the homozygous state, the T293N and T294M mutations have opposite effects on KATP channel open probability. This provides further support for the idea that residues between 292 and 297 serve as a 'gating loop' that forms a cytoplasmic gate to the pore (Nishida et al, 2007; Pegan et al, 2005; Proks et al, 2005b).
Functional implications: the T293N mutation
The large amplitude of hetT293N currents recorded in inside-out patches exposed to physiological levels of MgATP explains the dramatic increase in the resting whole-cell KATP currents. In pancreatic β-cells, a similar increase in the KATP current would be expected to cause membrane hyperpolarization and reduce or abolish the membrane depolarization evoked by glucose. This would prevent electrical activity, Ca2+ influx and insulin secretion, and can thereby account for the diabetic phenotype of the patient (Fig S1D of Supporting information).
The very large currents recorded in the presence of MgATP are entirely consistent with the neurological symptoms of the patient. Previous studies have shown that mutations which cause DEND syndrome give rise to heterozygous KATP currents that are much less sensitive to MgATP inhibition than mutations associated with neonatal diabetes alone (Ashcroft, 2007). This is probably because KATP channels are normally shut in many muscle and nerve tissues and open only under metabolic stress (Nichols, 2006). Thus, a greater reduction in ATP sensitivity is required to increase the resting KATP current sufficiently to influence electrical activity in these cells.
For mutations that cause DEND syndrome, 26–40% of the heterozygous current is not blocked by 3 mM MgATP, compared to 12–20% for mutations causing iDEND, 4–10% (in general) for mutations causing neonatal diabetes alone and <1% for wild-type channels (Ashcroft, 2007). The T293N mutation results in heterozygous currents of a magnitude (34%) similar to those that cause DEND syndrome. It is therefore surprising that the patient does not have epilepsy in addition to severe muscle weakness and developmental delay. However, the L164P mutation, which gives rise to unblocked currents of similar magnitude (34%) at 3 mM MgATP, also produces neonatal diabetes but not epilepsy (Tammaro et al, 2008). The T293N mutation is therefore a second case of a functionally severe mutation that does not cause epilepsy.
Functional implications: the T294M mutation
Case 2 is essentially homozygous for the T294M mutation in her β-cells due to deletion of the maternal allele throughout the whole pancreas. The lack of any azide-activated whole-cell current found for homT294M channels thus explains the congenital hyperinsulinism of this patient. A similar loss of the KATP current in her β-cells would produce a permanent membrane depolarization, irrespective of the blood glucose level. This would stimulate electrical activity and calcium influx, and thus give rise to persistent and unregulated insulin secretion (Fig 1C of Supporting information). The failure of case 2 to respond to diazoxide therapy is consistent with the in vitro results for homT294M channels.
Three heterozygous carriers of the T294M mutation were unaffected: the father and paternal grandmother of case 2, and the mother of case 3. This suggests that the reduction in whole-cell currents seen for het294M channels does not impair insulin secretion enough to produce a phenotype. It also suggests that case 3 must possess a second, as yet unidentified, mutation that contributes to his disease. This is assumed to be a milder mutation that retains sensitivity to diazoxide.
Molecular mechanism of the T293N mutation
The increased KATP current produced by the T293N mutation results from a decreased sensitivity to MgATP block, which is due to two mechanisms. The first is a reduced ability of ATP to close the KATP channel, which is mediated via Kir6.2 as it is present in the absence of Mg2+. This can be accounted for by the large increase in intrinsic Po(0). Second, the enhanced reduction in ATP sensitivity in the presence of Mg2+ argues that the stimulatory actions of MgATP, mediated via SUR1, are enhanced by the mutation. This is also found for some other PNDM mutations (Proks et al, 2005a).
Molecular mechanism of the T294M mutation
Our results indicate that the lack of homT294M currents observed in whole-cell recordings and inside-out patches exposed to nucleotide-free solution reflects both a reduction in channel trafficking to the plasma membrane and a markedly reduced open Po(0). As channel density is 60% of that of wild-type but no currents are seen in either cell-attached or excised membrane patches, we infer that all channels are closed (i.e. Po(0) is close to zero). Thus the T294M mutation appears to affect the intrinsic gating of the channel, but in the opposite direction to the T293N mutation. A KCNJ11 mutation (F55L) associated with HI that markedly decreases, but does not abolish, Po(0) in the homozygous channel has been reported previously: however, this mutation lies within the slide helix (Lin et al, 2006).
There was no change in the Po(0) of hetT294M channels. This suggests that mutant subunits do not exert a measurable influence on Po(0) when combined with wild-type subunits: note that any homT294M channels will be silent and thus not contribute to Po(0) and that abolition of the stimulatory effect of MgADP in the presence of MgATP implies that the heterozygous channel population must contain only a small number of homomeric wild-type channels. The fact that the Po(0) of heteromeric T294M channels is unaffected contrasts with mutations that enhance Po(0), which appear to have an additive effect on the energy of the open state (Proks et al, 2005b).
The reduction in hetT294M whole-cell currents presumably reflects, at least in part, a reduced channel density. However, the magnitude of this reduction (∼60%) is greater than can be accounted for by a decrease in channel density, even if this is assumed to be similar to that of homT294 channels (∼40%). This may reflect the fact that unlike wild-type channels, the ATP sensitivity is not reduced by Mg2+. This is due to the inability of MgADP to enhance channel activity in the presence of MgATP.
Several SUR1 mutations causing HI act by preventing MgADP activation of the channel but they do so in both the absence and presence of MgATP (Nichols et al, 1996; Shyng et al, 1998; Yan et al, 2007). This leads to reduced levels of channel activity under conditions of low metabolism, and thus to stimulation of insulin secretion. The T294M mutation is the first mutation to be shown to selectively abolish the ability of MgADP to stimulate channel activity in the presence of MgATP.
Our data suggest that Mg-nucleotide binding, hydrolysis and transduction by SUR1 are unaffected by the T294M mutation, because MgGDP was able to activate the channel in both the absence and presence of MgGTP and because MgADP was able to activate hetT294M currents in the absence of MgATP to the same extent as wild-type channels. We do not know the exact molecular mechanism underlying the inability of MgADP to stimulate hetT294M channels in the presence of MgATP. However, one possibility is suggested by the fact that unlike MgATP, GTP and GDP cause little blockage at Kir6.2 (Trapp et al, 1997).
We hypothesize that (in addition to its well-characterized stimulatory effect on KATP channel activity), SUR1 may impair ATP inhibition of wild-type Kir6.2, but not that of hetT294M, via a Mg-dependent mechanism. This phenomenon would not be evident in the presence of guanine nucleotides, which cause little blockage at Kir6.2 (Trapp et al, 1997). In the presence of MgATP and MgADP, the level of wild-type channel activity will reflect the balance between the stimulatory effects of Mg-nucleotides (mediated by SUR1) and inhibition via Kir6.2: we propose that SUR1 alleviates some of the inhibition of at Kir6.2. HetT294M channels will also be activated by SUR1 and blocked by ATP binding to Kir6.2, but as SUR1 is unable to alleviate inhibition at Kir6.2 the overall current magnitude is less than that of wild-type channels.
The ability of SUR1 to alleviate ATP inhibition at Kir6.2 might also contribute to the reduction in ATP sensitivity seen for wild-type channels in the presence of Mg2+: and suppression of this effect by the T294M mutation could account for the lack of change in ATP sensitivity in Mg2+ solution. It is also intriguing to speculate that the greater reduction in ATP block seen for hetT293N and homT293N channels in the presence of Mg2+ might be due to the opposite effect—a reduction in the ability of SUR1 to enhance ATP block at Kir6.2.
Structural considerations
In a molecular model of Kir6.2, both mutations lie within the gating loop (G-loop) of the cytoplasmic domain of the channel, far away from the ATP-binding site (Antcliff et al, 2005; Fig 8). In the 2.2 Å crystal structure of a chimeric Kir channel, the G-loop forms a gate at the apex of the cytoplasmic pore (Nishida et al, 2007). In its constricted conformation, the pore is too narrow for dehydrated K+ ions to pass through, whereas in the dilated conformation, a mostly hydrated K+ ion should be able to pass. The G-loop encompasses the region over which dilation and contraction occur and in Kir6.2 it includes the residues 289–297. The functional significance of the G-loop in Kir6.2 is demonstrated by the fact that many residues within this loop are mutated in human disease. In addition to the T293 and T294 mutations described here, mutations at E292 (G) and I296 (L) have previously been reported to cause PNDM and DEND syndrome respectively (Proks et al, 2005b; Girard et al, 2006). All mutations in this region affect channel gating with neonatal diabetes mutations increasing Po(0) and, as shown here, HI mutations reducing Po(0).
Figure 8. Location of residues T293 and T294 in a molecular model of Kir6.2.
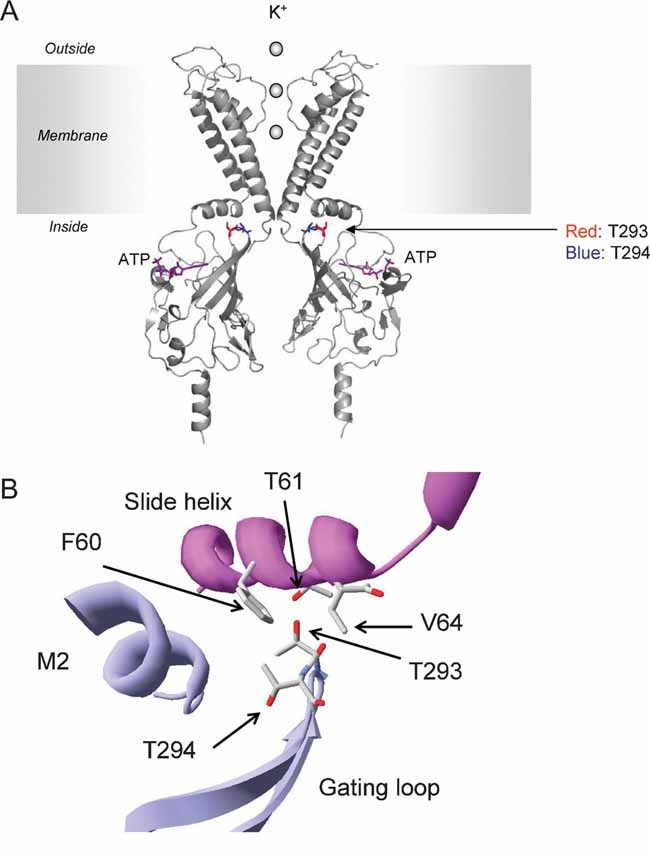
- Location of residues 293 and 294 in a molecular model of Kir6.2 (Antcliff et al, 2005). For clarity, only two transmembrane domains (TMs) and two cytosolic domains are illustrated. T293 and T294 are shown in red and blue, respectively. ATP is shown in purple.
- Close-up of interactions of T293 and T294 with neighbouring residues (indicated in cpk colours). The main chains of the different subunits are indicated in purple and blue.
Our results suggest that in the homomeric channel, the E292G, T293N and I296L mutations stabilize the dilated conformation of the channel, whereas the T294M mutation locks it into the constricted state.
In an atomic resolution model of Kir6.2 (Antcliff et al, 2005), the side chain of residue T293 points up from the gating loop towards the slide helix of the adjacent subunit (Fig 8). The hydroxyl group of the side chain lies within 4 Å of three slide helix residues: F60, T61, and V64. As asparagine has a longer side chain than threonine and is able to form hydrogen bonds, it is likely to make additional contacts with the slide helix.
The side chain of residue T294 points away from the slide helix and the hydroxyl group does not lie within 4 Å of any residue (other than its neighbours). Replacement with methionine extends the length and mass of the side-chain, bringing it within 4 Å of the cytosolic end of the second transmembrane domain of the same subunit.
The Kir6.2 model of Antcliff et al (2005) most closely resembles the dilated (open) conformation of the cytosolic pore of the chimeric Kir channel (Nishida et al, 2007). Thus, we can speculate that interactions between the gating loop and slide helix may promote channel opening, whereas interaction of the gating loop with the lower end of TM2 may promote the closed state.
Clinical implications
Sulphonylureas are an effective therapy for most patients with KCNJ11 mutations that cause neonatal diabetes alone, and many have now successfully transferred from insulin to sulphonylureas (Pearson et al, 2006). In functional studies, KATP channels carrying these mutations remain as sensitive to tolbutamide inhibition as wild-type channels, being 87–96% inhibited by 0.5 mM tolbutamide (Pearson et al, 2006). However, like many mutations that cause DEND syndrome, hetT293N channels were substantially less sensitive to tolbutamide (36% block). Glibenclamide also blocked hetT293N channels less potently (66% block compared to 96% for wild-type channels).
The magnitude of the in vitro response to glibenclamide is consistent with the patient's partial response to glibenclamide therapy. Glycaemic control is currently maintained on a combined treatment regime of insulin (0.5 U/kg/day) and glibenclamide (1.1 mg kg/day), but she has shown improvement in neurological functions since the commencement of treatment with sulphonylureas. Similarly, tolbutamide was able to control infantile spasms for 3 months in a child with a Kir6.2-C166F mutation, despite some insulin still being needed to control her diabetes (Bahi-Buisson et al, 2007). These results suggest that even for the most severe mutations it is always worth trying sulphonylurea therapy, as it may improve the neurological symptoms even if it is unable to control the diabetes.
MATERIALS AND METHODS
Mutation detection
Genomic DNA was extracted from peripheral leukocytes using standard procedures. The KCNJ11 minimal promoter region, the single-coding exon of KCNJ11, and the 39 exons of ABCC8 were amplified in six fragments by the polymerase chain reaction (PCR; primer sequences are available on request). Unidirectional sequencing was performed using universal M13 primers and a Big Dye Terminator Cycler Sequencing Kit v3.1 (Applied Biosystems, Warrington, UK) according to the manufacturer's instructions. Reactions were analysed on an ABI 3730 Capillary sequencer (Applied Biosystems, Warrington, UK) and sequences were compared to the published sequence (NM_00525 and to NM_000352.2) using Mutation Surveyor v2.61 (SoftGenetics, PA, USA).
Loss of heterozygosity was investigated by microsatellite analysis of DNA extracted from paraffin-embedded pancreatic tissue and peripheral leukocytes. Six markers (D11S2071, D11S2344, D11S2347, D11S419, D11S1397 and D11S902) were amplified by PCR and allele peak heights were compared using GeneMarker v1.7 (SoftGenetics, PA, USA). Primer sequences are available on request.
Molecular biology and oocyte preparation
Human Kir6.2 (Genbank NM000525; E23 and I337) and rat SUR1 (Genbank L40624) were used. Site-directed mutagenesis of Kir6.2, synthesis of capped mRNA and preparation and injection of Xenopus laevis were performed as previously reported (Proks et al, 2005a,b). Oocytes were coinjected with ∼2 ng of SUR1 mRNA and ∼0.1 ng wild-type or mutant Kir6.2 mRNA. To simulate the heterozygous state, SUR1 was co-expressed with a 1:1 mixture of wild-type and mutant Kir6.2. For each batch of oocytes, all mutations were injected to enable direct comparison of their effects. Oocytes were maintained in Barth's solution and studied 1–7 days after injection.
The paper explained
PROBLEM:
The ATP-sensitive potassium (KATP) channel is a tiny, gated pore in the cell membrane of insulin-secreting β-cells that regulates insulin secretion. When this channel is shut, insulin is secreted and when it is open insulin secretion is prevented. Channel activity is regulated by changes in blood glucose levels: when blood glucose levels rise (as after a meal), ATP is generated by the β-cell, and binds to the channel and causes its closure. This stimulates insulin release and the hormone then restores the blood glucose concentration to its resting level. Mutations in the KATP channel are a common cause of neonatal diabetes (diabetes that develops within the first six months of life) and of hyperinsulinism of infancy (in which insulin secretion is unregulated leading to very low blood glucose levels). The aim of this paper is to identify the molecular mechanism underlying two novel disease-causing mutations in the pore-forming subunit of the KATP channel (Kir6.2 or KCNJ11).
RESULTS:
We describe here a novel Kir6.2 mutation (T293N) that causes neonatal diabetes in conjunction with developmental delay and muscle weakness. Functional analysis revealed that the mutant channel failed to close in response to ATP. This is expected to impair insulin secretion, thus causing diabetes. It might also reduce the activity of brain neurons, thus leading to the neurological problems experienced by the patients. Treatment with glibenclamide (a sulphonylurea drug known to bind to and close the KATP channel) partially blocked the KATP channel in functional studies. It also reduced the insulin requirement of the patient and improved walking, speech and fine motor skills. We also identified a loss-of-function mutation in the same gene at the adjacent residue (T294M) that was associated with hyperinsulinism of infancy. This mutation caused the channel to remain shut even when ATP or metabolism were low. This is expected to cause continuous insulin secretion accounting for the disease phenotype.
IMPACT:
These results provide novel insights into how the opening and closing of the KATP channel is regulated.
Electrophysiology
Whole-cell currents were recorded using a 2-electrode voltage-clamp in response to voltage steps of ±20 mV from a holding potential of −10 mV, in a solution containing (in mM): 90 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES (pH 7.4 with KOH). Metabolic inhibition was induced by 3 mM Na-azide; and 0.5 mM tolbutamide or 100 µM glibenclamide was used to block the KATP channel, as indicated. Diazoxide (340 µM) was used to activate the KATP channels. Macroscopic currents were recorded from giant inside-out patches. The pipette solution contained (mM): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 HEPES (pH 7.4 with KOH). The Mg-free internal (bath) solution contained (mM): 107 KCl, 1 K2SO4, 10 EGTA, 10 HEPES (pH 7.2 with KOH) and K2ATP, as indicated. The Mg-containing solution was made by adding 2 mM MgCl2 to Mg-free solution and using MgATP rather than ATP.
The macroscopic slope conductance was measured between −20 and −100 mV, in response to 3 s voltage ramps from −100 to +110 mV (holding potential −10 mV). To control for possible rundown, Gc was taken as the mean of the conductance in the control solution before and after ATP application. ATP concentration–response curves were fit with a modified Hill equation
| (1) |
where [ATP] is the ATP concentration, IC50 is the [ATP] at which inhibition is half maximal, h is the slope factor (Hill coefficient) and a represents the fraction of unblocked current at saturating [ATP] (a = 0 except where specified).
Single-channel currents were recorded at −60 mV from inside-out patches and analysed by using a combination of Clampfit (Axon Instruments), Origin (OriginLab Corporation) and in-house software (Proks et al, 2005b). For homomeric Kir6.2-T293N/SUR1 channels, the open probability in the absence of ATP (Po(0)) was determined from single-channel patches as the fraction of time spent in the open state for recordings of ∼1 min duration. Open time distributions were fit with a single exponential; up to three exponentials were fitted to dwell-time distributions. Burst durations were determined by using the criterion for the critical time described previously (Magleby & Pallotta, 1983).
For heterozygous Kir6.2-T294M/SUR1 channels, single-channel amplitudes were calculated from an all-points current amplitude histogram. Channel activity (NPo) was measured as the mean current (I) divided by the single-channel amplitude (i) for current records of ∼1 min duration. Po(0) was calculated from NPo/N, where N is the number of channels in the patch estimated from the maximum number of superimposed events.
Cell-surface expression assay
Surface expression was performed on COS-7 cells transiently expressing HA-tagged Kir6.2 and SUR1 (Zerangue et al, 1999). For each experiment, three 35 mm dishes were transfected in parallel. Forty-eight hours post-transfection, cells were fixed in 4% formaldehyde, and incubated with rat monoclonal anti-HA antibody (clone 3F10, Roche) and HRP-conjugated anti-rat secondary antibody (Jackson laboratories). Bound antibodies were detected by femto maximum sensitivity chemiluminescent substrate (Pierce) and quantitated using a Monolight 2010 luminometer. Results are the average of three dishes per experiment, and each data point is the average of three independent experiments.
For more information
OMIM, Online Mendelian Inheritance in Man:
KCNJ11:
http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=600937
Diabetes genes:
Diabetes UK homepage:
American Diabetes Association homepage:
Frances Ashcroft's Laboratory:
http://www.dpag.ox.ac.uk/research/transport/ashcroft/ashcroft_research
Author contributions
Kenju Shimomura performed all patch-clamp studies. Brittany Zadek did the molecular biology for functional studies and Roope Mannikko made Figure 8. Lejla Zubcevic and Christophe Girard performed the surface expression analysis and Mark Lethby the two-electrode voltage-clamp studies. Sarah Flanagan did the genetic analysis. Peter Clayton, Andrew Hattersley, Khalid Hussain, Ritika Kapoor, Oliver Petz, and Mars Skae conducted the clinical investigations.
Frances Ashcroft directed the functional experiments, and Andrew Hattersley and Sian Ellard directed the genetic experiments. Frances Ashcroft and Sian Ellard wrote the paper, which was then reviewed by all authors.
Acknowledgments
We thank the Wellcome Trust (076436/Z/05/Z and 081188/A/06/Z), the Royal Society and the European Union (EuroDia, SHM-CT-2006-518513 and EDICT, 201924) for support. FMA is a Royal Society Research Professor. Brittany Zadek was supported by an OXION studentship and Sarah Flanagan by a Sir Graham Wilkins Research Fellowship.
Supplementary information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J. 2005;24:229–239. doi: 10.1038/sj.emboj.7600487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM. ATP-sensitive K+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab. 2007;293:E880–889. doi: 10.1152/ajpendo.00348.2007. [DOI] [PubMed] [Google Scholar]
- Bahi-Buisson N, Eisermann M, Nivot S, Bellanne-Chantelot C, Dulac O, Bach N, Plouin P, Chiron C, de Lonlay P. Infantile spasms as an epileptic feature of DEND syndrome associated with an activating mutation in the potassium adenosine triphosphate (ATP) channel, Kir6.2. J Child Neurol. 2007;22:1147–1150. doi: 10.1177/0883073807306272. [DOI] [PubMed] [Google Scholar]
- De Leon DD, Stanley CA. Mechanisms of Disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;3:57–68. doi: 10.1038/ncpendmet0368. [DOI] [PubMed] [Google Scholar]
- Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley-Green A, Lindley KJ. Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev. 2004;84:239–275. doi: 10.1152/physrev.00022.2003. [DOI] [PubMed] [Google Scholar]
- Enkvetchakul D, Loussouarn G, Makhina E, Shyng SL, Nichols CG. The kinetic and physical basis of KATP channel gating: toward a unified molecular understanding. Biophys J. 2000;78:2334–2348. doi: 10.1016/S0006-3495(00)76779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan SE, Clauin S, Bellanne-Chantelot C, de Lonlay P, Harries LW, Gloyn AL, Ellard S. Update of mutations in the genes encoding the pancreatic beta-cell KATP subunits Kir6.2 (KCNJ11) and sulfonylurea receptor (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat. 2009;30:170–180. doi: 10.1002/humu.20838. [DOI] [PubMed] [Google Scholar]
- Girard C, Shimomura K, Proks P, Absalom N, de Nanclares PG, Ashcroft FM. Functional analysis of six Kir6.2 (KCNJ11) mutations causing neonatal diabetes. Pflugers Arch. 2006;453:323–332. doi: 10.1007/s00424-006-0112-3. [DOI] [PubMed] [Google Scholar]
- Girard CA, Wunderlich FT, Shimomura K, Collins S, Kaizik S, Proks P, Abdulkader F, Clark A, Ball V, Zubcevic L, et al. A mouse model of neonatal diabetes caused by the KATP channel mutation Kir6.2-V59M. J Clin Invest. 2009 doi: 10.1172/JCI35772. Dec 8, DOI: 10.1172/JCI35772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Loussouam G, Tucker SJ, Zhao C, Nichols CG, Ashcroft FM. A novel method for measurement of submembrane ATP concentration. J Biol Chem. 2000;29:30046–30049. doi: 10.1074/jbc.M001010200. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in KATP channel activation by Mg-ADP and diazoxide. EMBO J. 1997;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the β-cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci USA. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: New clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54:2503–2513. doi: 10.2337/diabetes.54.9.2503. [DOI] [PubMed] [Google Scholar]
- Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- Koster JC, Cadario F, Peruzzi C, Colombo C, Nichols CG, Barbetti F. The G53D mutation in Kir6.2 (KCNJ11) is associated with neonatal diabetes and motor dysfunction in adulthood that is improved with sulfonylurea therapy. J Clin Endocrinol Metab. 2008;93:1054–1061. doi: 10.1210/jc.2007-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG. Targeted overactivity of beta cell KATP channels induces profound neonatal diabetes. Cell. 2000;100:645–654. doi: 10.1016/s0092-8674(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. A novel KCNJ11 mutation associated with congenital hyperinsulinism reduces the intrinsic open probability of beta-cell ATP-sensitive potassium channels. J Biol Chem. 2006;28:3006–3012. doi: 10.1074/jbc.M511875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby KL, Pallotta BS. Burst kinetics of single calcium activated potassium channels in cultured muscle. J Physiol. 1983;344:605–623. doi: 10.1113/jphysiol.1983.sp014958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masia R, Koster JC, Tumini S, Chiarelli F, Colombo C, Nichols CG, Barbetti F. An ATP-binding mutation (G334D) in KCNJ11 is associated with a sulfonylurea-insensitive form of developmental delay, epilepsy, and neonatal diabetes. Diabetes. 2007;56:328–336. doi: 10.2337/db06-1275. [DOI] [PubMed] [Google Scholar]
- Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, Gonzalez G, AguilarBryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–4015. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson ER, Flechtner I, Njolstad PR, Neonatal Diabetes International Collaborative Group. Malecki MT, Flanagan S, Larkin B, Ashcroft FM, Klimes I, Codner E, et al. The successful transfer of insulin treated patients with diabetes due to Kir6.2 (KCNJ11) mutations to sulphonylurea tablets. N Eng J Med. 2006;355:467–477. [Google Scholar]
- Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Sleisinger PA, Choe S. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci. 2005;8:279–287. doi: 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- Proks P, Girard C, Ashcroft FM. Enhanced activation by MgATP contributes significantly to the functional effects of KCNJ11 mutations causing neonatal diabetes. Hum Mol Genet. 2005a;14:2717–2726. doi: 10.1093/hmg/ddi305. [DOI] [PubMed] [Google Scholar]
- Proks P, Girard C, Haider S, Gloyn AL, Hattersley AT, Sansom MSP, Ashcroft FM. A novel gating mutation at the internal mouth of the Kir6.2 pore is associated with DEND syndrome. EMBO Rep. 2005b;6:470–475. doi: 10.1038/sj.embor.7400393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura K, Girard CAJ, Proks P, Nazim J, Lippiat JD, Cerutti F, Lorini R, Ellard S, Hattersley A, Barbetti F, et al. Mutations at the same residue (R50) of Kir6.2 (KCNJ11) produce different functional effects. Diabetes. 2006;55:1705–1712. doi: 10.2337/db05-1640. [DOI] [PubMed] [Google Scholar]
- Shyng S, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J Gen Physiol. 1997;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Ferrigni T, Shepard JB, Nestorowicz A, Glaser B, Permutt MA, Nichols CG. Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes. 1998;47:1145–1151. doi: 10.2337/diabetes.47.7.1145. [DOI] [PubMed] [Google Scholar]
- Tammaro P, Flanagan SE, Zadek B, Srinivasan S, Woodhead H, Hameed S, Klimes I, Hattersley AT, Ellard S, Ashcroft FM. A Kir6.2 mutation causing severe functional effects in vitro produces neonatal diabetes without the expected neurological complications. Diabetologia. 2008;51:802–810. doi: 10.1007/s00125-008-0923-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Tucker SJ, Ashcroft FM. Activation and inhibition of K-ATP currents by guanine nucleotides is mediated by different channel subunits. Proc Natl Acad Sci USA. 1997;94:8872–8877. doi: 10.1073/pnas.94.16.8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Yan FF, Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes. 2007;56:2339–2348. doi: 10.2337/db07-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 1999;22:233–254. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.