

Abstract
Mammalian cells contain three closely related heterochromatin protein 1 (HP1) isoforms, HP1α, β and γ, which, by analogy to their unique counterpart in Schizosaccharomyces pombe, have been implicated in gene silencing, genome stability and chromosome segregation. However, the individual importance of each isoform during normal cell cycle and disease has remained an unresolved issue. Here, we reveal that HP1α shows a proliferation-dependent regulation, which neither HP1β nor γ display. During transient cell cycle exit, the HP1α mRNA and protein levels diminish. Transient depletion of HP1α, but not HP1β or γ, in tumoural and primary human cells leads to defects in chromosome segregation. Notably, analysis of an annotated collection of samples derived from carcinomas reveals an overexpression of HP1α mRNA and protein, which correlates with clinical data and disease outcome. Our results unveil a specific expression pattern for the HP1α isoform, suggesting a unique function related to cell division and tumour growth. The overexpression of HP1α constitutes a new example of a potential epigenetic contribution to tumourigenesis that is of clinical interest for cancer prognosis.
Keywords: cancer, cell cycle, heterochromatin, HP1, proliferation
INTRODUCTION
Cancer has been considered for a long time as a genetic disease induced mainly by hereditary or spontaneous mutations in DNA sequences (Hanahan & Weinberg, 2000; Weinberg, 1996). However, changes affecting chromatin organization have recently been implicated as well in tumourigenesis (Jones & Baylin, 2007), and intense work has been dedicated to understand how these processes relate to each other. Much effort has been put into the characterization of alterations in DNA methylation and different histone modifications (Esteller, 2007). Drugs designed to target these modifications have now been started to be used with some success in cancer treatment (Mulero-Navarro & Esteller, 2008). A present challenge is to find how, beyond DNA and histones, the higher order nuclear organization of chromatin (Misteli, 2007), which is often affected in cancer cells, participates in tumourigenesis. Breast cancer is a particularly interesting model in this respect. Given its clinical and genetic heterogeneity, it cannot be explained as a genetic disease. Thus, to consider if breast cancer cells show particular chromatin alterations that could promote tumourigenesis to proceed is particularly relevant.
In this study, we focus on a key component of constitutive heterochromatin regions, heterochromatin protein 1 (HP1) (Kwon & Workman, 2008; Maison & Almouzni, 2004), highly conserved from Schizosaccharomyces pombe to humans.
The unique HP1 homologue in S. pombe, Swi6, has been implicated in chromatin compaction and gene silencing (Kwon & Workman, 2008), through its interaction with histone H3 methylated on lysine 9 (H3K9me) (Bannister et al, 2001; Ekwall et al, 1996). In addition, Swi6 is involved in mitotic segregation (Ekwall et al, 1995), by promoting sister chromatid cohesion (Bernard et al, 2001) and through the establishment of the centromeric histone H3 variant CENP-ACnp1 (Folco et al, 2008).
Mammalian cells present three HP1 isoforms, HP1α, β and γ, that, by analogy to their fission yeast homologue, have been collectively implicated in gene silencing (Kwon & Workman, 2008). They can all interact with a trimethylated H3K9 (H3K9me3) peptide in vitro (Lachner et al, 2001) and targeting of either HP1α or β to a transgene array results in a local chromatin compaction (Verschure et al, 2005). HP1 isoforms also accumulate at pericentric heterochromatin (Maison & Almouzni, 2004). However, understanding the exact role of mammalian HP1 proteins in chromosome segregation is still at an early stage. Indeed, HP1 proteins interact with components of the centromere and the kinetochore complex (Ainsztein et al, 1998; Obuse et al, 2004; Wheatley et al, 2001) and downregulation (Auth et al, 2006; Obuse et al, 2004) or mislocalization of HP1 isoforms due to either the absence of H3K9me3 (Guenatri et al, 2004) or treatment with the histone deacetylase inhibitor (HDACi) Trichostatin-A (TSA) (Taddei et al, 2001), result in mitotic defects. Intriguingly, a recent report indicates that in contrast to Swi6 in S. pombe, the correct localization of HP1 is not required for the recruitment of cohesins to centromeric regions (Koch et al, 2008). Yet, the HP1α isoform seems to help in protecting cohesins from degradation by recruiting the Shugoshin protein (Yamagishi et al, 2008).
There are several indications that the three mammalian HP1 isoforms, HP1α, β and γ, may not fulfil identical functions. First, they show differences in their nuclear pattern of distribution. The overall nuclear staining of HP1α marks strongly the pericentric heterochromatin, whereas HP1γ shows less specificity for these regions (Gilbert et al, 2003; Minc et al, 1999; Nielsen et al, 2001a). Furthermore, despite their high similarity in structure and function, the three isoforms are not always present together and can interact with different binding partners (Kwon & Workman, 2008; Quivy et al, 2004). Finally, distinct post-translational modifications on individual HP1 isoforms (Lomberk et al, 2006; Minc et al, 1999) may further diversify their functions.
A first possible link between HP1 proteins and tumourigenesis was put forward through the observation that HP1 interacts with the tumour suppressor Retinoblastoma protein (Rb) (Nielsen et al, 2001b; Williams & Grafi, 2000) and participates in the Rb-dependent silencing of cell cycle genes such as Cyclin E (Nielsen et al, 2001b). Similarly, HP1 interacts with the transcriptional co-repressor KAP-1 (Ryan et al, 1999), which is involved in the regulation of the E2F1 (Wang et al, 2007) and p53 (Wang et al, 2005) proteins. Furthermore, HP1α and γ have been found in complex with Chromatin assembly factor 1 (CAF-1) (Murzina et al, 1999; Quivy et al, 2004), of which the intermediate subunit p60 is a validated proliferation marker in breast cancer (Polo et al, 2004). These arguments prompted us to consider how the different HP1 isoforms are regulated in relation to cell proliferation and tumourigenesis. Interestingly, the promoter region of HP1α contains potential target sites for the E2F proteins (Oberley et al, 2003; Weinmann et al, 2002) and myc transcription factors (Kim et al, 2008; Li et al, 2003). Moreover, all the three HP1 isoforms are downregulated in differentiated blood lymphocytes compared to their undifferentiated precursors (Baxter et al, 2004; Gilbert et al, 2003; Istomina et al, 2003; Ritou et al, 2007). Whether this downregulation is a general response to cell cycle exit or a specific feature of blood cell differentiation has not been addressed. Downregulation of HP1α has also been linked to the higher invasive potential of breast cancer cells (Kirschmann et al, 2000; Norwood et al, 2006), but again it is unclear to which aspect of the metastasis process this downregulation relates. Thus, the specific and/or common regulation patterns of the three HP1 isoforms in relation to cell proliferation, quiescence and cancer remain elusive.
To clarify these issues, we decided to carry out a comprehensive study of the behaviour of the distinct HP1 isoforms during cell proliferation, cell cycle exit and tumourigenesis, using both human cell line models and a collection of human tumour-derived tissue samples. We demonstrate that HP1α shows the unique property of displaying a proliferation-dependent expression pattern. Upon transient cell cycle exit, the expression of HP1α, but not β or γ, is reduced. Breast cancer cell lines show overexpression of HP1α, but not β or γ, compared to non-tumoural mammary cells derived from the same patient. Remarkably, HP1α is overexpressed in pancreas, uterus, ovary, prostate and breast carcinomas, as well as in uterine leiomyoma, compared to corresponding non-tumoural tissues. Furthermore, HP1α expression levels in breast carcinomas with a long-term patient follow-up show a significant correlation with disease progression and occurrence of metastasis. Our results demonstrate that HP1α levels are clearly associated with cell proliferation, which is relevant to tumourigenicity and useful for prognosis assessment in breast cancer.
RESULTS
HP1α expression depends on cell proliferation
Recent studies have described a downregulation of HP1 proteins in differentiated blood cells compared to undifferentiated blood cells (Baxter et al, 2004; Gilbert et al, 2003; Istomina et al, 2003; Ritou et al, 2007). This downregulation could be a common response to cell cycle exit, or a specific consequence of (blood) cell differentiation. To address this issue, we examined whether transient cell cycle exit, which is not accompanied by a differentiation process, similarly results in HP1 downregulation. Using two different human primary fibroblast cell lines (WI38 and BJ), in which quiescence is induced by serum starvation, we observed lower protein levels of HP1α, but not β or γ, in quiescent cells compared to proliferating cells (Fig 1A). MCF7 breast carcinoma cells, which are arrested in quiescence by anti-estrogen treatment (Carroll et al, 2000), show a similar downregulation of HP1α, but not β or γ. Thus, using two different means to induce quiescence, we find a specific downregulation of the HP1α isoform, the extent of which correlates with the duration of time the cells have spent in quiescence (not shown). As a control, we verified the downregulation of CAF-1 p60 in all quiescent cells (Polo et al, 2004) (Fig 1A) and assessed the synchronization efficiency by flow cytometry (Fig 1B). Similar to CAF-1 p150 and p60, HP1α downregulation in transient quiescence relates in part to transcriptional regulation, since quiescent BJ cells also show decreased HP1α mRNA levels when compared to asynchronously proliferating cells (determined by quantitative RT-PCR, Fig 1C). Upon exit from the quiescent state, HP1α protein levels gradually increase between 16 and 24 h after release (Fig S1A in Supporting Information), which corresponds to the moment of cell cycle entry (determined by flow cytometry, Fig S1B in Supporting Information). The accumulation of HP1α occurs earlier than the one of CAF-1 p60 (Fig S1A in Supporting Information), mostly observed at the onset of DNA replication.
Figure 1. The expression of HP1α, but not β or γ, is downregulated in quiescence.
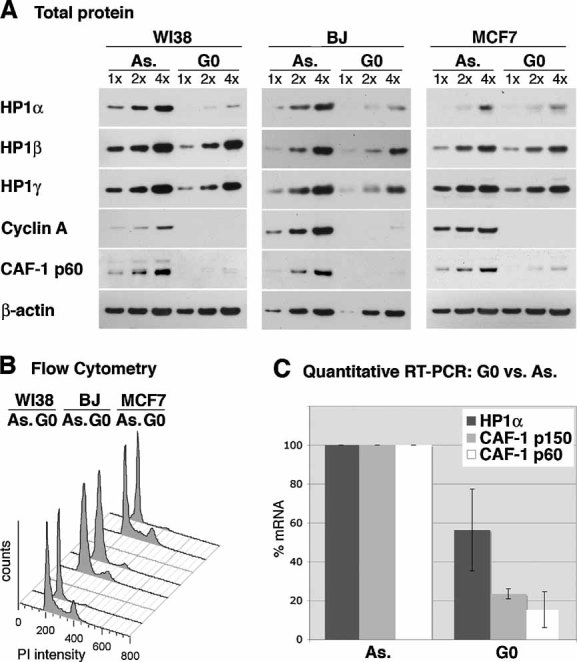
- Total protein levels of HP1α, β and γ are detected by Western blot in asynchronously proliferating (As.) and quiescent (G0) WI38 lung fibroblasts, MCF7 breast cancer cells and BJ primary foreskin fibroblasts. Fibroblasts are arrested in quiescence by serum starvation and MCF7 cells by anti-estrogen treatment. Increasing amounts (x) of total cell extracts are loaded and β-actin serves as a loading control. CAF-1 p60 (Polo et al, 2004) and Cyclin A are used as markers for cell proliferation.
- Flow cytometry analysis of the cell cycle distribution of the cells shown in A.
- HP1α mRNA levels in proliferating (As.) and quiescent (G0) BJ foreskin fibroblasts, as determined by quantitative RT-PCR. Levels are normalized to the reference gene ribosomal protein P0-like protein (RPLPO) (de Cremoux et al, 2004) and levels in proliferating cells are set to 100%. CAF-1 p60 and CAF-1 p150 levels are shown for comparison. The error bar represents data from three independent experiments.
The observed downregulation of HP1α in quiescence could either be specific for the quiescent state or reflect an expression restricted to a specific stage of the cell cycle. We therefore analysed HP1α levels during the cell cycle in synchronized HeLa cells. In contrast to the cell cycle marker Cyclin A, we did not observe significant variation in HP1α levels in the synchronized cell populations (Fig S2A, B in Supporting Information). Similarly, human primary fibroblasts, which display a normal cell cycle regulation, show essentially similar levels of HP1α protein (Fig S2C in Supporting Information) and mRNA in the synchronized cell population (Fig 2D, E). In this respect, HP1α behaves similarly to CAF-1 p60 and p150, which are also ubiquitously expressed during the cell cycle but downregulated in quiescence (Fig 1 and S2 in Supporting Information; (Polo et al, 2004)). In conclusion, HP1α expression levels are high at all stages of the cell cycle in proliferative cells, and HP1α downregulation is specific to the quiescent state.
Figure 2. HP1α is overexpressed in breast cancer cells and associated with chromatin.
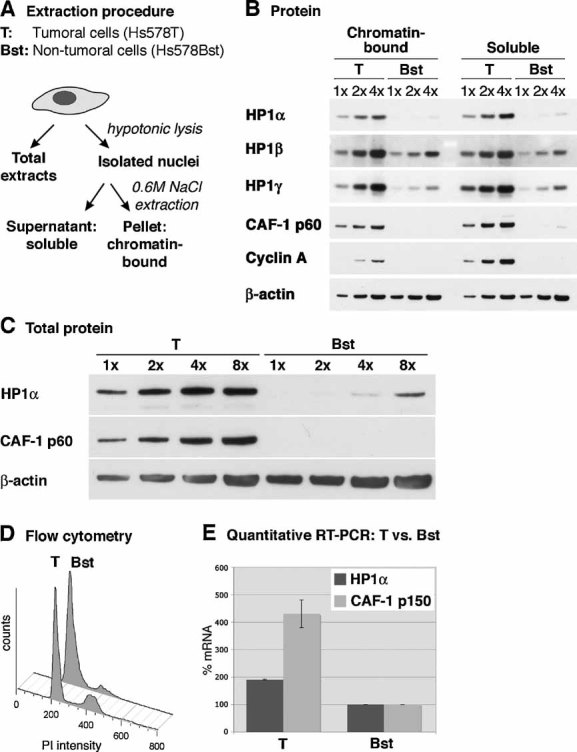
- Scheme of the experimental procedure applied to obtain total, soluble and chromatin-bound cell extracts as used in B.
- HP1α, β and γ protein levels are analysed by Western blotting in soluble and chromatin-bound nuclear extracts from the breast cancer cell line Hs578T (T) and the non-tumoural mammary cell line Hs578Bst (Bst), which are derived from the same patient (Hackett et al, 1977). Cyclin A and CAF-1 p60 are shown for comparison. Increasing amounts (x) of cell extracts are loaded; β-actin serves as a loading control.
- Relative quantification of total HP1α protein levels in tumoural (T) and non-tumoural (Bst) mammary cells, compared to the amounts of CAF-1 p60 (Polo et al, 2004). Increasing amounts (x) of total cell extracts are loaded; β-actin serves as a loading control.
- Flow cytometry analysis of tumoural (T) and non-tumoural (Bst) mammary cells in order to assess polyploidy. Tumoural (T) and non-tumoural (Bst) cells contain 25 and 13% of cells in S-phase, respectively.
- Relative HP1α mRNA levels in tumoural (T) and non-tumoural (Bst) mammary cells, as determined by quantitative RT-PCR. Levels are normalized to the reference gene ribosomal protein P0-like protein (RPLPO) (de Cremoux et al, 2004) and non-tumoural cells are set to 100%. CAF-1 p150 is shown for comparison. Error bars represent data from two independent experiments.
HP1α is overexpressed in breast cancer cells
The proliferation-dependent expression of HP1α suggests a possible differential expression between tumoural and non-tumoural cells, as found for proliferation markers including CAF-1 p60 (Polo et al, 2004). To examine this issue, we used mammary cells derived from the same patient, either tumoural (Hs578T) or non-tumoural (Hs578Bst) (Hackett et al, 1977), to be relevant in our comparison. Besides the total cell extract, we analysed levels of HP1 proteins both in the soluble fraction and in the fraction bound to chromatin (Fig 2A). Indeed, HP1 proteins distribute into different nuclear fractions, distinguished by their capacity to resist extraction with high salt concentrations or with Triton X-100 detergent. The salt- or detergent-resistant pool of HP1 is considered as the active, chromatin-bound pool of HP1 and represents less than 10% of the total HP1 in human and rodent cells, as determined by Western blot quantification (Taddei et al, 2001) or Fluorescence recovery after photobleaching (FRAP) (Cheutin et al, 2003; Dialynas et al, 2007; Festenstein et al, 2003).
Both in the chromatin-bound and in the soluble nuclear fractions, we observed higher HP1α levels in tumoural Hs578T cells than in non-tumoural Hs578Bst cells, whereas HP1β or γ display little differences in expression compared to the loading control β-actin (Fig 2B). Loading of identical amounts of cells also show tumoural overexpression of HP1α, specifically (Fig S3 in Supporting Information). Semi-quantitative Western blot on total cell extracts shows that tumoural Hs578T cells contain approximately eight times more HP1α protein than non-tumoural Hs578Bst cells, compared to the loading control (Fig 2C). We could exclude the possibility that HP1α expression levels reflect the DNA content of the cell, since the tumoural Hs578T cells display only a very moderate aneuploidy in flow cytometry (Fig 2D). The overexpression of HP1α mRNA, detected by quantitative RT-PCR (Fig 2E), further indicates a regulation that involves, at least in part, transcription.
Our analysis of chromatin-bound and soluble fractions suggests that overexpressed HP1α in tumoural cells is partially chromatin-bound and thus possibly important for chromatin organization. This seems to be consistent with its nuclear localization, which is granular and diffuse in the non-tumoural mammary cells but clearly localized into discrete spots in a large fraction of the breast cancer cells (Fig S4A in Supporting Information). These spots largely localize to centromeric regions, detected by the CREST autoimmune serum (Fig S4A in Supporting Information). Yet, the different forms of localization of HP1α were not accompanied by an altered nuclear distribution of H3K9me3 (Fig S4B in Supporting Information), and the different patterns of HP1α staining do not seem to be associated with specific stages of the cell cycle, as detected by staining for cell cycle markers (Fig S4C in Supporting Information). In conclusion, our cell line model shows an overexpression of HP1α, but not HP1β or γ, in tumoural mammary cells compared to non-tumoural mammary cells. A large fraction of HP1α in breast cancer cells is chromatin-bound and localizes to centromeric regions.
HP1α downregulation results in mitotic defects
The proliferation-dependent expression of HP1α, which is not observed for HP1β or γ, points to a unique function of this isoform and suggests that high amounts of this protein could confer an advantage for cell growth. Interestingly, downregulation of CAF-1 p150 results in a pronounced S-phase arrest in a manner that depends on its interaction with HP1 proteins (Quivy et al, 2008; Quivy et al, 2004). We thus tested whether high expression levels of HP1α, β or γ are required for human cancer cell proliferation. By transfecting HeLa cells with siRNA against HP1α, β or γ, we obtained a specific downregulation of each of the HP1 isoforms (Fig 3A). In contrast to downregulation of its binding partner CAF-1 p150 (Quivy et al, 2008), downregulation of the HP1 isoforms did not result in any obvious effect on cell proliferation, as assessed by flow cytometry (Fig 3B). We obtained similar results for HP1α downregulation in the mammary carcinoma cell line Hs578T (data not shown). However, in agreement with previous reports (Auth et al, 2006; Obuse et al, 2004), we did observe an increased fraction of mitotic profiles displaying lagging chromosomes, misalignments and chromosome bridges (Fig 3C) after downregulation of HP1α. We quantified these as the fraction of aberrant mitoses (Fig 3D). Interestingly, under our experimental conditions, downregulation of HP1β or γ did not give rise to increased mitotic defects, suggesting that only the HP1α isoform is critical for faithful mitosis. Furthermore, defects in mitosis were supported by observation of a three-fold increase in micronuclei formation after downregulation of HP1α, but not HP1β or γ (Fig 3E, F).
Figure 3. Downregulation of HP1α, but not HP1β or γ, leads to mitotic defects in HeLa cells.
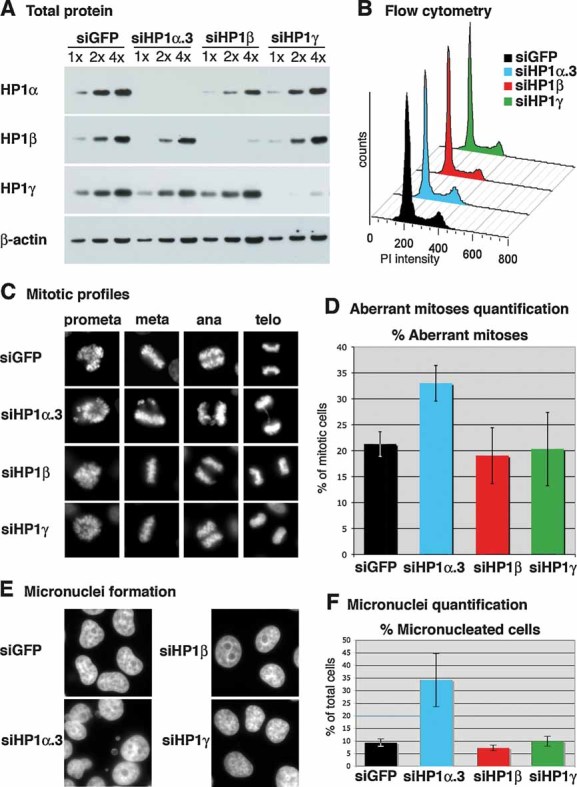
- Western blot analysis of total cell extracts from HeLa cells 72 hours after transfection with siRNA sequences targeting HP1α (named siHP1α.3), HP1β, HP1γ or control siRNA (siGFP). Increasing amounts (x) of total cell extracts are loaded and β-actin is used as a loading control.
- Flow cytometry analysis of the cell cycle distribution of cells shown in A.
- Typical mitotic figures after transfection with siRNA sequences targeting HP1α, HP1β, HP1γ or mock siRNA (siGFP).
- Percentage of aberrant mitotic figures (lagging chromosomes, misalignments, chromosome bridges) in asynchronous HeLa cells 72 h after transfection with siRNA sequences targeting HP1α, HP1β, HP1γ or mock siRNA (siGFP). The error bars represent data from three independent, blind counted, experiments.
- DAPI staining reveals micronuclei formation 72 h after transfection with an siRNA sequence targeting HP1α.
- Quantification of the percentage of micronucleated cells 72 h after transfection with siRNA sequences targeting HP1α, HP1β, HP1γ or control siRNA (siGFP). Error bars represent data from three independent experiments of which two were blind counted.
Since HeLa cells already show a high fraction of deficient mitoses (∼20%) and of micronucleated cells (∼10%), we used primary fibroblasts, proficient for cell cycle control and checkpoint activity for further analysis. Transient transfection with two different siRNAs against HP1α resulted in a specific downregulation of this isoform (Fig S5A in Supporting Information). Again, we did not detect any significant effect on global cell cycle distribution by flow cytometry (Fig S5B in Supporting Information), but we observed a small but reproducible increase in the percentage of prometaphases and a decreased percentage of metaphases by microscopy (Fig S5C in Supporting Information). Interestingly, this effect mimics observations upon mutation of the Drososophila homologue of HP1α (Kellum & Alberts, 1995) or inactivation of the centromeric histone H3 variant CENPA in chicken cells (Regnier et al, 2005). Using live cell imaging, we measured a small but statistically significant (p = 4.9 × 10−7) increase in the duration of mitosis after downregulation of HP1α compared to control (upper graph Fig S5D in Supporting Information). Interestingly, the delay again mostly affected the steps preceding actual chromosome segregation (Fig S5D in Supporting Information, lower graph). In conclusion, our observations in both tumoural and primary cells suggest a role for HP1α in early mitosis, possibly contributing to the correct alignment or the stable attachment of chromosomes at the metaphase plate.
HP1α overexpression in human cancer samples
The proliferation-dependent expression of HP1α and its overexpression in breast cancer cell lines prompted us to study HP1α expression in the physiological context of human cancer. First, we analysed data from published transcriptome studies performed in different tissue types (Andersson et al, 2007; Pyeon et al, 2007; Quade et al, 2004; Ramaswamy et al, 2003; Richardson et al, 2006; Yu et al, 2004), using the Oncomine database (Rhodes et al, 2004). These data showed that HP1α is significantly and consistently overexpressed in several types of malignancies (Fig S6A in Supporting Information), while HP1γ and especially HP1β can be found either up- or downregulated when carrying out similar analyses (not shown). It is remarkable that in leukaemia, only HP1α, but not HP1β or γ, shows an important overexpression that correlates with the time for the disease to relapse (Kirschner-Schwabe et al, 2006) and with the expression of its binding partner CAF-1 p150 and the proliferation marker Ki67 (Fig S6B in Supporting Information).
Our results in cultured cells systematically showed that the difference in HP1α protein levels was more pronounced than the corresponding mRNA levels (Figs 1 and 2), possibly reflecting a post-translational regulation. This encouraged us to analyse the HP1α protein levels in frozen tumoural and non-tumoural human tissue sections by immunohistochemistry. We systematically observed an intense HP1α staining in tumoural cell nuclei in pancreas, uterus, ovary, prostate and breast malignancies (Fig 4A). In the corresponding non-tumoural tissues, HP1α levels were mostly below the limit of detection. This differential expression is specific to HP1α, since HP1β and γ show nuclear staining both in tumoural and non-tumoural tissues (Fig 4B). The nuclei showing intense HP1α immunostaining correspond to carcinoma cells, which also stain positive for the epithelial cytokeratin marker KL-1 (Fig 4C). In addition, a polyclonal antibody against HP1α, which provides a more intense staining than the monoclonal antibody used in Fig 4, also shows a clear overexpression in carcinoma cells with some staining in non-tumoural tissue (Fig S7A in Supporting Information). Staining for H3K9me3, however, did not show a clear difference between non-tumoural and tumoural tissues (Fig S7B in Supporting Information), suggesting that either the HP1α detection is more sensitive or the overexpression occurs independently from H3K9me3.
Figure 4. HP1α protein is overexpressed in multiple types of human cancer.
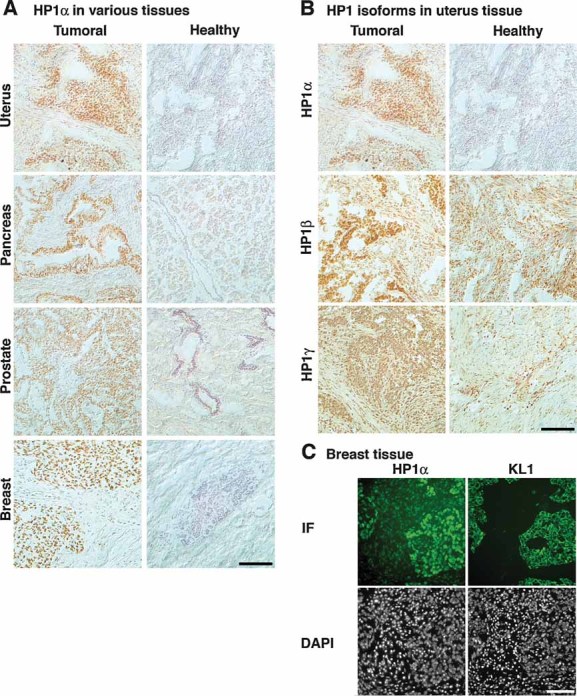
- Frozen human tissue arrays containing sections from tumoural and healthy origin are stained for HP1α by immunohistochemistry and counterstained with hematoxylin.
- Frozen uterus tissue sections were stained for HP1α, β and γ by immunohistochemistry and counterstained with hematoxylin. Tissue arrays were processed identically for the three antibodies.
- Two adjacent sections from a frozen medullary breast carcinoma were stained for HP1α and epithelial pan-cytokeratin marker KL-1, by IF. DNA is revealed with DAPI. Areas of HP1α overexpression correspond to regions of epithelial tumour cells. Scale bars: 100 µm.
To determine the importance of HP1α overexpression for tumour growth and disease outcome, we selected cryopreserved breast carcinoma specimens that were collected in 1995. We focused on node negative and metastasis-free invasive carcinoma of size (median 18 mm; range 6–50 mm) that permitted primary conservative tumourectomy. These cases would benefit from new prognostic markers for the indication of adjuvant chemotherapy. Patient's and tumour characteristics are provided in Table SI in Supporting Information. The median follow-up is 146 months (range 30–161). At 10 years, overall survival, the distant recurrence and the disease progression rates were 90 [83–97], 87 [80–95] and 70% [61–81%], respectively. We measured HP1α mRNA expression levels by quantitative RT-PCR in 86 samples and normalized the expression levels to that of the reference gene ribosomal protein P0-like protein (RPLPO) (de Cremoux et al, 2004). We found that high levels of HP1α tend to associate with increased age of the patient (p = 0.0014) and larger, highly mitotic, grade III tumours (non-significant) (Fig 5A). In univariate analysis, HP1α continuous expression was significantly associated with an increased risk of disease progression: a one-unit increase of log 2(HP1α) increased the risk of disease progression by 3% (relative risk (RR) = 1.03 (1.00–1.05), p = 0.041). We determined a cut-off value of 10, which divided patients into two groups (74.4% with HP1α levels ≤10 and 25.6% with HP1α levels >10) and was significantly associated with disease progression (p = 0.0113; RR = 2.47 (1.20–5.09)). At 10 years, 75% [64–87] of the patients with low HP1α levels compared to 57% [39–83] of the patients with high HP1α levels had not shown disease progression. Moreover, this cut-off is also significantly associated with overall survival (p = 0.0134; RR = 3.76 (1.03–13.7)) and the occurrence of distant recurrences (p = 0.011; RR = 3.74 (1.26–11.2)) (Fig 5B).
Figure 5. HP1α expression levels have a prognostic value in breast cancer patients.
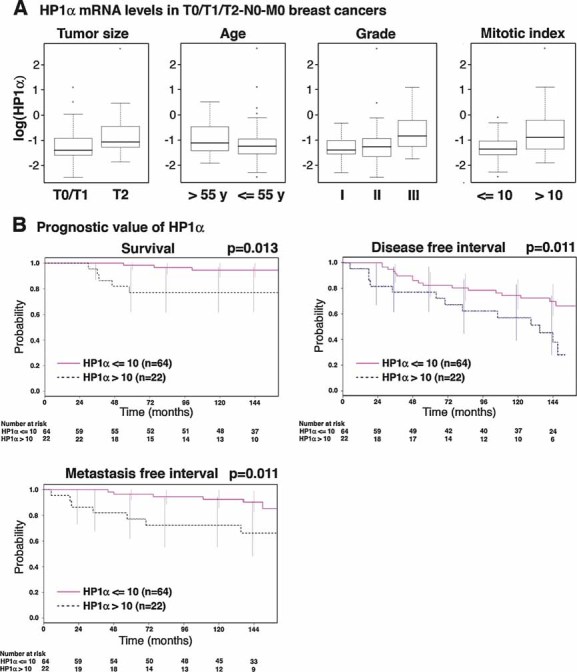
- Box plots representing logarithmic HP1α mRNA expression levels, according to the indicated clinico-pathological factors, in 86 breast cancer samples with a >10 year patient follow-up. Boxes represent the 25–75th percentile; brackets: range; black line: median; black dots: outliers.
- Univariate Kaplan–Meier curves of the survival, the occurrence of metastasis and the disease-free interval (interval before the occurrence of local recurrence, regional lymph node recurrence, controlateral breast cancer or metastasis) in patients expressing high (>10) or low (≤10) levels of HP1α. The number of patients at risk at each time point is indicated below the graphs.
In multivariate analyses, adjusted for known prognostic factors (i.e. patient's age, mitotic index, tumour grade, tumour size, hormone receptor status and Ki67 levels), only HP1α expression is an independent prognostic factor for overall survival (p = 0.0431): high (>10) HP1α levels are associated with an increased risk of death (RR = 3.76 (1.03-13.7)). Similarly, adjusted for the same parameters, only high HP1α expression (p = 0.0077; RR = 3.02 (1.38–6.63)) and high tumour grade (p = 0.0170; RR for grade III = 4.53 (1.51–13.6)) are pejorative for disease progression.
In conclusion, our immunohistochemistry analyses demonstrate that HP1α, but not β or γ, is overexpressed in multiple types of human cancer cells. Furthermore, quantitative RT-PCR analysis carried out for HP1α showed significant correlation with clinico-pathological data and disease outcome. In our series of 86 small breast tumours, high HP1α expression remained the only independent prognostic factor for overall survival after adjustment for classical prognostic markers, such as the mitotic index, tumour grade, tumour size, hormone receptor status or Ki67. We are currently extending our analysis to a second, independent data set to test our cut-off value of 10 and confirm the highly prognostic value of HP1α compared to other markers. Taken together, our data demonstrate that HP1α constitutes a new chromatin-related marker of cell proliferation and tumourigenesis of clinical relevance for prognosis of breast cancer and potentially other types of cancer.
DISCUSSION
A unique regulation of the HP1α isoform
The HP1 family of proteins were identified more than 20 years ago (James & Elgin, 1986), but the common or divergent functions of the three isoforms remain largely unknown. Here, we show that in human cells, the HP1α isoform has unique properties, not shared by HP1β and γ. The protein is expressed in a proliferation-dependent manner, being downregulated during transient cell cycle exit. In line with these findings, cultured cancer cells overexpress HP1α compared to non-tumoural cells. Importantly, this is validated in patient samples. Our data thus demonstrate a unique regulation of HP1α, which is not paralleled by HP1β or γ. The region in which the HP1α gene is located is not often affected by genomic aberrations in breast cancer (Progenetix CGH database (Baudis & Cleary, 2001)). Genomic alterations are thus unlikely to cause HP1α overexpression, which might rather reflect an increased activation by E2F (Oberley et al, 2003; Weinmann et al, 2002) and/or myc transcription factors (Kim et al, 2008; Li et al, 2003).
An interesting question now is whether HP1α overexpression in tumour cells is accompanied by and related to alterations in other heterochromatic marks. We did not observe a difference in nuclear distribution or in staining intensity of H3K9me3 in breast cancer cells and in tumoural tissue samples. However, other factors can promote HP1 recruitment and maintenance in (pericentric) heterochromatin regions, among which is the H3K9-methyltransferase Suv39h1 (Stewart et al, 2005), an RNA component (Maison et al, 2002) and the largest subunit of CAF-1 (p150) (Quivy et al, 2004, 2008). Post-translational modifications of HP1α itself (Lomberk et al, 2006) might also affect its stabilization and recruitment, as has been shown recently for HP1β in the context of DNA damage (Ayoub et al, 2008). Furthermore, it remains unclear how the increased levels of HP1α relate to loss of the heterochromatin mark H4K20me3 in cancer cells (Fraga et al, 2005) or the reported changes in DNA methylation (reviewed in (Esteller, 2007)). Future work should also explore the distinct or common repressive roles of HP1α and the polycomb group proteins. Indeed, HP1α interacts with the Suz12 subunit of the polycomb repressive complex 2 (PRC2) (Cao & Zhang, 2004; Yamamoto et al, 2004), in which all three subunits (Suz12, EZH2 and EED) are downregulated in quiescence (Bracken et al, 2003; Muller et al, 2001) and overexpressed in human cancer (reviewed in (Simon & Lange, 2008)). Considering how these different repressive marks function in cooperation will be necessary to obtain a more complete picture of the aberrant transcriptional repression mechanisms in cancer cells.
Potential functions of tumoural HP1α overexpression
The unique proliferation-dependent regulation of HP1α suggests the existence of a specific function that would distinguish it from HP1β and HP1γ. Interestingly, the three isoforms distribute differentially within the nucleus: HP1α is most specific for pericentric heterochromatin, whereas HP1β and especially HP1γ show a more diffuse distribution (Gilbert et al, 2003; Minc et al, 1999; Nielsen et al, 2001a). Hence, high HP1α levels in cancer cells might reflect an altered organization of certain heterochromatic regions, which could become more compact and/or more abundant. The increased amount of chromatin-bound HP1α and its different nuclear distributions (Fig 2 and Fig S4 in Supporting Information) in breast cancer cells compared to non-tumoural cells is in agreement with this hypothesis. It is noteworthy that the distinct nuclear distribution of HP1α in breast cancer cells (discrete spots) and non-tumoural cells (granular and diffuse) mimics the localization that has been observed in activated versus resting lymphocytes (Baxter et al, 2004; Grigoryev et al, 2004; Ritou et al, 2007). Yet, this is not solely a consequence of cell cycle status, since we did not observe a different HP1α localization between quiescent and proliferating primary fibroblasts (data not shown).
Besides a structural role in pericentric heterochromatin, the overexpression of HP1α in tumours could also relate to a function in regulating the euchromatic gene expression. Indeed, in Drosophila, HP1 is involved in the regulation of cell cycle genes (De Lucia et al, 2005) and could even actively promote gene expression by stimulating H3K36 demethylation (Lin et al, 2008). Human HP1 plays a role in silencing Cyclin E by the tumour suppressor Rb (Nielsen et al, 2001b), and interacts with the co-repressor protein KAP1, the genomic targets of which include genes involved in crucial cellular pathways (O'Geen et al, 2007). However, there is currently no evidence in mammalian cells that regulation of gene expression would be a unique property of HP1α, not shared by HP1β and γ. Furthermore, if HP1α has a dominant regulatory role in gene expression, a profound impact on cell proliferation and survival would be expected upon its downregulation, which is not in line with our observations so far.
Although we do not formally exclude a more subtle role for HP1α in gene expression, which would deserve a specific study, our results rather favour a mitosis-related advantage of tumoural HP1α overexpression. Indeed, despite the fact that phosphorylation of H3S10 by Aurora B induces the release of HP1 proteins from pericentric chromatin during mitosis (Fischle et al, 2005; Hirota et al, 2005), at least a fraction of HP1α, specifically, remains tightly associated throughout mitosis (Guenatri et al, 2004; Hayakawa et al, 2003; Minc et al, 2001; Schmiedeberg et al, 2004). Hence, HP1α might play a unique role in mitosis. Indeed, we observe a partial colocalization of HP1α with (peri-) centromeric regions in breast cancer cells, suggesting that these heterochromatic regions might constitute the main target sequences of overexpressed HP1α. Furthermore, we show that downregulation of HP1α, but not HP1β or γ, results in mitotic defects, both in HeLa cells and in primary human fibroblasts. Interestingly, in primary fibroblasts, the phenotype includes a prolonged prometaphase, which could reflect a problem in the alignment of chromosomes at the metaphase plate. HP1α was recently shown to be necessary for the recruitment of the shugoshin protein (Yamagishi et al, 2008), which protects centromeric cohesins from degradation and thus prevents premature chromosome segregation. Deficient cohesion can lead to accumulation of cells in prometaphase (Watrin et al, 2006). Hence, a partial depletion of HP1α would affect cohesin protection and result in a prolonged prometaphase in primary cells due to checkpoint activity. In transformed cells, in which the mitotic checkpoint is often less efficient (Weaver & Cleveland, 2005), an increased fraction of aberrant mitoses and micronucleated cells is observed (Fig 3). Thus, elevated levels of HP1α would be more crucial for faithful mitosis in cancer cells when compared to healthy cells, and a positive selection for cancer cells overexpressing HP1α is more likely to occur when the mitotic checkpoint becomes deficient, in order to facilitate the passage through mitosis.
Importance of HP1α in cancer
To evaluate the clinical importance of HP1α expression, we analysed HP1α protein and mRNA levels in human cancer by immunohistochemistry and quantitative RT-PCR. Staining of cryopreserved human tissue samples showed a significant overexpression of HP1α, but not HP1β or γ, in tumoural cells. Thus, HP1α constitutes a potential marker for the diagnosis of multiple types of cancer. In contrast to other proliferation markers, such as Ki67, HP1α stains all tumoural cells and is thus highly suitable for the determination of the exact localization and extent of the tumour.
In order to quantify the HP1α expression and determine its prognostic value, we measured HP1α mRNA levels in 86 small breast tumours with >10 years follow-up. Our data reveal that high HP1α expression correlates with decreased survival and increased occurrence of metastasis over time. Furthermore, multivariate analyses demonstrate that HP1α levels predict disease outcome better than standard prognostic markers. Thus, HP1α constitutes a marker of prognostic value, in breast cancer and potentially in other types of cancer.
Previously, an increased expression of HP1α had been correlated with a decreased invasive potential among several breast cancer cell lines (Kirschmann et al, 2000; Norwood et al, 2006), possibly by silencing of pro-invasive genes. This observation might reflect the inverse correlation that has been suggested between proliferation and invasion (Berglund & Landberg, 2006). Indeed, metastasis requires the acquisition of invasive potential and the adaptation to a new environment, which are often incompatible with high proliferation rates. Thus, a temporal slow-down of tumour proliferation, accompanied by downregulation of HP1α, might permit the expression of pro-invasive genes and the occurrence of metastasis. Yet, the outgrowth of metastases requires cell proliferation and our data suggest that this process is dominant for final patient outcome, since high HP1α expression correlated with earlier diagnosis of metastasis.
Recently, inhibitors of HDAC have been used successfully in cancer treatment (Dokmanovic et al, 2007; Mulero-Navarro & Esteller, 2008). However, their precise mode of action remains elusive. It is interesting that, in proliferating cell lines, a major effect of HDACi is to increase the mobility (Cheutin et al, 2003; Dialynas et al, 2007) and dispersion (Bartova et al, 2005; Dialynas et al, 2007; Taddei et al, 2001) of HP1 proteins. Although the effect of HDACi treatment has not been assessed in vivo on post-therapeautic tissue samples, HP1α might be one of the main targets contributing to the anti-tumour effects of these drugs (Taddei et al, 2005). Without affecting the expression levels of HP1α, HDACi might delocalize the overexpressed HP1α in vivo and abrogate the contribution of HP1α overexpression to cancer cell growth and/or cell division.
In conclusion, HP1α expression levels reflect cell proliferation and are negatively correlated with disease outcome in early breast cancer. Our results favour a role of HP1α in facilitating mitosis, which might be more crucial in cancer cells. We demonstrate a potential clinical application of HP1α as a marker for cancer prognosis. As a consequence, HP1α should now be taken into account in fundamental cancer research to obtain a comprehensive picture of how heterochromatin domains are affected in cancer cells and contribute to tumourigenesis.
MATERIALS AND METHODS
Cell culture and synchronization
Wi38 primary lung fibroblasts (ATCC) and BJ primary foreskin fibroblasts (ATCC) were cultured in MEMα medium (GIBCO), MCF7 breast cancer cells (gift from O. Delattre, Paris) and HeLa cervical carcinoma cells (gift from M. Bornens, Paris) in DMEM (GIBCO), Hs578T breast cancer cells (gift from O. Delattre, Paris) in RPMI medium (GIBCO) containing 10 mM insulin (Sigma) and Hs578Bst healthy mammary cells (ATCC) in DMEM containing 30 ng/ml epidermal growth factor (TEBU). The media were supplemented with 10% foetal calf serum (Eurobio) and 10 mg/ml penicillin and streptomycin (GIBCO).
For synchronization in quiescence, primary cells were grown in serum-free medium for at least 72 h and MCF7 cells for 48 h in a medium containing 10 nM of the anti-estrogen ICI182780 (Fischer Bioblock Scientific) (Carroll et al, 2000). HeLa cells were synchronized in the different stages of the cell cycle as described by Polo et al (2004). BJ cells were synchronized similarly, except that they were blocked for 14 h, released for 10 h, blocked again for 14 h and released for 3, 6 or 12 h for S-phase, G2 and G1, respectively. For mitotic BJ cells, 10 ng/ml nocodozole was added 4 h after the second release from thymidine and cells were harvested 10 h later. Synchronization was checked by flow cytometry as explained by Polo et al (2004), and the data were analysed using FlowJo (Tree Star Inc.).
Transfections and siRNAs
HeLa cells, plated in a medium without antibiotics, were transfected with 30 nM siRNA using Oligofectamine reagent (Invitrogen) and Optimem 1 medium (Gibco). BJ primary cells were transfected at passage 27 (8 passages after reception from ATCC) using nucleofection (Amaxa), according to manufacturer's instructions. Briefly, 4 × 105 cells were transfected with 0.8 nmol siRNA (Dharmacon) or 1 µg of plasmid DNA in 100 µl Nucleofector solution R (Amaxa), using the nucleofection program X-001. Cells were subsequently plated at a density of 7,000 cells/cm2 to permit cell proliferation without reaching confluency within 4 days. siRNA sequences: siGFP: AAGCUGGAGUACAACUACAAC; siHP1α_1: CCUGAGAAAAACUUGGAUUTT (Obuse et al, 2004); siHP1α_3: GGGAGAAGTCAGAAAGTAA; siHP1β: AGGAATATGTGGTGGAAAA; siHP1γ: AGGTCTTGATCCTGAAAGA
Cell extracts
For total cell extracts, after a wash in phosphate buffered saline (PBS), we scraped cells from plates in 1× Laemmli buffer (60 mM Tris-Hcl, pH 6.8; 10% glycerol; 2% sodium dodecyl sulphate (SDS); 1% 2-mercaptoethanol and 0.002% bromophenol blue) and boiled these extracts 10 min before storage at −20°C.
Chromatin-bound cell extracts were made according to the method of Martini et al (1998). We determined protein concentration using Bio-Rad protein assay solution.
Antibodies
The following primary antibodies were used in the present study: Mouse monoclonal anti-HP1α 2HP-1H5 from Euromedex and rabbit polyclonal anti-HP1α H2164 from Sigma (directed against amino acids 67–119 and 177–191, respectively) for immunofluorescence (IF) and immunohistochemistry, mouse monoclonal anti-HP1α 2HP-2G9 for Western blot (Euromedex), mouse monoclonal anti-HP1β 1MOD1A9 (Euromedex), mouse monoclonal anti-HP1γ 2MOD-1G6-AS (Euromedex), anti-CREST human serum (gift from A. Ladurner), rabbit polyclonal anti-CAF-1 p60, mouse monoclonal anti-β-actin AC-15 (Sigma), mouse monoclonal anti-KL1 antibody (Dako), rabbit polyclonal anti-Cyclin A (Santa Cruz), mouse monoclonal anti-H3S10P (Abcam ab14955) and rabbit polyclonal anti-Cdt1 (Santa Cruz 28262).
Secondary antibodies: Fluorescein isothiocyanate (FITC) or Texas Red-coupled antibodies (IF) or horseradish peroxidase-coupled antibodies (Western Blotting) were from Interchim.
Western blotting
After 30 min treatment with 25 U benzonase nuclease (Novagen), cell extracts were loaded on 4–12% gradient gels (Invitrogen), using a 1 × MES migration buffer (Invitrogen), followed by Western blotting procedures as described by Martini et al (1998).
Immunofluorescence (IF)
After fixation in 2% paraformaldehyde, cells are permeabilized in 0.2% Triton X-100 in PBS. IF detection is carried out as in (Martini et al, 1998). Coverslips are mounted in Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector laboratories).
For IF on tissue samples, 8 µm cryosections made from frozen mammary tissues (Curie Institute, Paris, France) were fixed on glass slides in 3% paraformaldehyde and immunostained as above.
Immunohistochemistry
We used 8 µm cryosections from frozen mammary tissues (Curie Institute, Paris, France) or frozen tissue arrays (FMC401, Biomax). Sections, fixed on glass slides in 3% paraformaldehyde and permeabilized in PBS containing 0.5% Triton for 4 min, were incubated for 5 min in 3% H2O2 (Prolabo) for peroxidase inhibition and blocked in PBS containing 1% BSA and 5% non-fat milk. Incubation with primary antibody diluted in blocking solution was followed by revelation with horseradish peroxidase-coupled secondary antibody (DakoCytomation) and diaminobenzidine (DakoCytomation) before counterstaining with hematoxyline (Merck). Slides were dehydrated in increasing ethanol concentrations and toluene before mounting in Entellan mounting medium (Merck).
Live cell imaging
For live cell imaging, BJ cells were transfected twice with siRNA, with a 72 h interval. A plasmid coding for H2B-cherry was introduced in the second transfection, and cells were plated on glass bottom dishes (Mattek). Movies were made using the BioStation system (Nikon). Images were acquired every 20 min, during a period of 24 h, starting 48 h after the second transfection. Movies were analysed using Image J.
RNA extraction, quantitative RT-PCR and primers
We used the mRNeasy mini kit (Qiagen) for total RNA extraction from cell lines or frozen patient samples and produced cDNA using Superscript II reverse transcriptase (Invitrogen) with 1 µg RNA and 3 µg of random primers (Invitrogen) per reaction. We used the Lightcycler 2.0 System (Roche) and the Lightcycler FastStart DNA Master SYBR Green I reaction kit (Roche) for quantitative RT-PCR. For the patient samples, we used the 96-well plate Step One Plus system (Applied Biosystems) and the SYBR Green PCR Master mix (Applied Biosystems). We measured duplicates and carried out three subsequent cDNA dilutions to assess the primer efficiency. We designed primer pairs in order to overlap an intron, so as to distinguish cDNA amplification from putative genomic contamination. Primers: RPLPO forward: GGCGACCTGGAAGTCCAACT; RPLPO reverse: CCATCAGCACCACAGCCTTC; HP1α forward: GATCATTGGGGCAACAGATT; HP1α reverse: TGCAAGAACCAGGTCAGCTT; CAF-1 p150 forward: CAGCAGTACCAGTCCCTTCC; CAF-1 p150 reverse: TCTTTGCAGTCTGAGCTTGTTC; CAF-1 p60 forward: CGGACACTCCACCAAGTTCT; CAF-1 p60 reverse: CCAGGCGTCTCTGACTGAAT. We normalized the quantity of HP1α mRNA according to the human acidic ribosomal phosphoprotein PO (RPLPO) (de Cremoux et al, 2004) by applying x = 100/(E(Cp RPLPO – Cp HP1α)), where E is the mean efficiency of primer pairs and where x reflects the quantity of HP1α mRNA relative to the quantity of RPLPO mRNA in a given sample.
The paper explained
PROBLEM:
Breast cancer is a clinically and genetically diverse disease. How alterations in chromatin organization contribute to its development should therefore be considered. Here, we focus on the three mammalian isoforms of heterochromatin protein 1 (HP1α, β and γ), key components of compact heterochromatin regions, in relation to cell proliferation and breast cancer.
RESULTS:
We reveal that HP1α shows a proliferation-dependent regulation, which neither HP1β nor γ display. During transient cell cycle exit, HP1α mRNA and protein levels diminish and depletion of HP1α leads to defects in chromosome segregation. Importantly, the levels of HP1α mRNA and protein are elevated in breast carcinomas and this upregulation correlates with clinical data and disease outcome. Altogether, we propose that HP1α has a role in mitosis and that this role provides a selective growth advantage to cancer cells.
IMPACT:
Our results suggest a unique function of the HP1α isoform, related to cell division and tumour growth. HP1α overexpression in breast cancer patient samples correlates with disease outcome and should be considered as a new epigenetic marker for prognosis assessment.
Breast cancer patient samples and statistics
This study includes 92 breast cancer samples, selected from the Institut Curie Biological Resources Center for treatment with primary conservative tumourectomy (median tumour size: 18 mm (6–50 mm)). Patients were diagnosed in 1995 and found to be lymph node negative (N0) and metastasis free (M0). Patients were informed of research purposes and did not express opposition. Patient's and tumour characteristics are provided in Table SI in Supporting Information). RNA were extracted from cryopreserved tissue and analysed as described above. RNA of 86 samples were of sufficient quality for further analysis.
Differences between groups were analysed by χ2 or Fisher exact tests for categorical variables and Kruskal–Wallis for continuous variables. Recurrence-free and alive patients were censored at the date of their last known contact. Survival data were defined as the time from diagnosis of breast cancer until the occurrence of disease progression, defined as local recurrence in the treated breast, regional recurrence in lymph node-bearing areas, controlateral breast cancer or distant recurrences. Determination of a cut-off value prognostic for the disease free interval (DFI) was computed using a Cox proportional risks model. A Wald test was used to evaluate the prognostic value of this variable on each event. The overall survival (OS), metastasis free interval and DFI rates were estimated by the Kaplan–Meier method, and groups were compared using a log-rank test. Multivariate analysis was carried out to assess the relative influence of prognostic factors on OS and DFI, using the Cox stepwise forward procedure (Cox 1972). Significance level was 0.05. Analyses were performed using the R software 2.5.0 version.
Author contributions
LDK set up and performed the main experiments of this project and wrote the article, under supervision of GA. GA designed the concept and carried out the follow-up of the project both experimentally and in the writing. AS performed all statistical analyses concerning the breast cancer patient samples, under the supervision of BA. CB performed the Quantitative RT-PCRs on breast cancer patient samples and HR contributed to life cell imaging and siRNA depletion experiments. XS-G selected and provided the breast cancer patient samples along with their specific medical annotations.
Acknowledgments
We thank all members of the UMR218 and Sophie Polo for help and discussions, Edith Heard for the KL1 marker; Martial Caly, Anne Vincent Salomon and Carine Ganem for help with patient samples, Patricia Decremoux for helpful discussions concerning Q-RT-PCR and Jennifer Nelson for Q-RT-PCR duplicates. The pCherry-N1 plasmid was kindly provided by Lise Andrieux and Wim Vermeulen (Rotterdam, The Netherlands) and we thank Isabelle Loiodice for cloning of the H2B-cherry. This work was supported by la Ligue Nationale contre le Cancer (Equipe labellisée la Ligue), PIC Programs (‘Retinoblastome’ and ‘Replication, Instabilite chromosomique et cancer’), the European Commission Network of Excellence Epigenome (LSHG-CT-2004-503433), ACI-2007-Cancéropôle IdF ‘Breast cancer and Epigenetics’, ANR ‘CenRNA’ NT05-4_42267 and ANR ‘FaRC’ PCV06_142302. LDK was funded by Cancéropôle Ile-de-France and by Association pour la recherche sur le cancer (ARC).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
For more information
Oncomine transcriptome database: http://www.oncomine.org/main/index.jsp
Progenetix CGH database: http://www.progenetix.net/progenetix/index.html
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Ainsztein AM, Kandels-Lewis SE, Mackay AM, Earnshaw WC. INCENP centromere and spindle targeting: identification of essential conserved motifs and involvement of heterochromatin protein HP1. J Cell Biol. 1998;143:1763–1774. doi: 10.1083/jcb.143.7.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson A, Ritz C, Lindgren D, Eden P, Lassen C, Heldrup J, Olofsson T, Rade J, Fontes M, Porwit-Macdonald A, et al. Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia. 2007;21:1198–1203. doi: 10.1038/sj.leu.2404688. [DOI] [PubMed] [Google Scholar]
- Auth T, Kunkel E, Grummt F. Interaction between HP1alpha and replication proteins in mammalian cells. Exp Cell Res. 2006;28:28. doi: 10.1016/j.yexcr.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–686. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- Bartova E, Pachernik J, Harnicarova A, Kovarik A, Kovarikova M, Hofmanova J, Skalnikova M, Kozubek M, Kozubek S. Nuclear levels and patterns of histone H3 modification and HP1 proteins after inhibition of histone deacetylases. J Cell Sci. 2005;118:5035–5046. doi: 10.1242/jcs.02621. [DOI] [PubMed] [Google Scholar]
- Baudis M, Cleary ML. Progenetix.net: an online repository for molecular cytogenetic aberration data. Bioinformatics. 2001;17:1228–1229. doi: 10.1093/bioinformatics/17.12.1228. [DOI] [PubMed] [Google Scholar]
- Baxter J, Sauer S, Peters A, John R, Williams R, Caparros ML, Arney K, Otte A, Jenuwein T, Merkenschlager M, et al. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J. 2004;23:4462–4472. doi: 10.1038/sj.emboj.7600414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund P, Landberg G. Cyclin e overexpression reduces infiltrative growth in breast cancer: yet another link between proliferation control and tumor invasion. Cell Cycle. 2006;5:606–609. doi: 10.4161/cc.5.6.2569. [DOI] [PubMed] [Google Scholar]
- Bernard P, Maure JF, Partridge JF, Genier S, Javerzat JP, Allshire RC. Requirement of heterochromatin for cohesion at centromeres. Science. 2001;294:2539–2542. doi: 10.1126/science.1064027. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Prall OW, Musgrove EA, Sutherland RL. A pure estrogen antagonist inhibits cyclin E-Cdk2 activity in MCF-7 breast cancer cells and induces accumulation of p130-E2F4 complexes characteristic of quiescence. J Biol Chem. 2000;275:38221–38229. doi: 10.1074/jbc.M004424200. [DOI] [PubMed] [Google Scholar]
- Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, Misteli T. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 2003;299:721–725. doi: 10.1126/science.1078572. [DOI] [PubMed] [Google Scholar]
- de Cremoux P, Bieche I, Tran-Perennou C, Vignaud S, Boudou E, Asselain B, Lidereau R, Magdelenat H, Becette V, Sigal-Zafrani B, et al. Inter-laboratory quality control for hormone-dependent gene expression in human breast tumors using real-time reverse transcription-polymerase chain reaction. Endocr Relat Cancer. 2004;11:489–495. doi: 10.1677/erc.1.00808. [DOI] [PubMed] [Google Scholar]
- De Lucia F, Ni JQ, Vaillant C, Sun FL. HP1 modulates the transcription of cell-cycle regulators in Drosophila melanogaster. Nucleic Acids Res. 2005;33:2852–2858. doi: 10.1093/nar/gki584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dialynas GK, Terjung S, Brown JP, Aucott RL, Baron-Luhr B, Singh PB, Georgatos SD. Plasticity of HP1 proteins in mammalian cells. J Cell Sci. 2007;120:3415–3424. doi: 10.1242/jcs.012914. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- Ekwall K, Javerzat JP, Lorentz A, Schmidt H, Cranston G, Allshire R. The chromodomain protein Swi6: a key component at fission yeast centromeres. Science. 1995;269:1429–1431. doi: 10.1126/science.7660126. [DOI] [PubMed] [Google Scholar]
- Ekwall K, Nimmo ER, Javerzat JP, Borgstrom B, Egel R, Cranston G, Allshire R. Mutations in the fission yeast silencing factors clr4+ and rik1+ disrupt the localisation of the chromo domain protein Swi6p and impair centromere function. J Cell Sci. 1996;109:2637–2648. doi: 10.1242/jcs.109.11.2637. [DOI] [PubMed] [Google Scholar]
- Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16:R50–59. doi: 10.1093/hmg/ddm018. [DOI] [PubMed] [Google Scholar]
- Festenstein R, Pagakis SN, Hiragami K, Lyon D, Verreault A, Sekkali B, Kioussis D. Modulation of heterochromatin protein 1 dynamics in primary Mammalian cells. Science. 2003;299:719–721. doi: 10.1126/science.1078694. [DOI] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- Folco HD, Pidoux AL, Urano T, Allshire RC. Heterochromatin and RNAi are required to establish CENP-A chromatin at centromeres. Science. 2008;319:94–97. doi: 10.1126/science.1150944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- Gilbert N, Boyle S, Sutherland H, de Las Heras J, Allan J, Jenuwein T, Bickmore WA. Formation of facultative heterochromatin in the absence of HP1. EMBO J. 2003;22:5540–5550. doi: 10.1093/emboj/cdg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryev SA, Nikitina T, Pehrson JR, Singh PB, Woodcock CL. Dynamic relocation of epigenetic chromatin markers reveals an active role of constitutive heterochromatin in the transition from proliferation to quiescence. J Cell Sci. 2004;117:6153–6162. doi: 10.1242/jcs.01537. [DOI] [PubMed] [Google Scholar]
- Guenatri M, Bailly D, Maison C, Almouzni G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol. 2004;166:493–505. doi: 10.1083/jcb.200403109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett AJ, Smith HS, Springer EL, Owens RB, Nelson-Rees WA, Riggs JL, Gardner MB. Two syngeneic cell lines from human breast tissue: the aneuploid mammary epithelial (Hs578T) and the diploid myoepithelial (Hs578Bst) cell lines. J Natl Cancer Inst. 1977;58:1795–1806. doi: 10.1093/jnci/58.6.1795. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Haraguchi T, Masumoto H, Hiraoka Y. Cell cycle behavior of human HP1 subtypes: distinct molecular domains of HP1 are required for their centromeric localization during interphase and metaphase. J Cell Sci. 2003;116:3327–3338. doi: 10.1242/jcs.00635. [DOI] [PubMed] [Google Scholar]
- Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- Istomina NE, Shushanov SS, Springhetti EM, Karpov VL, Krasheninnikov IA, Stevens K, Zaret KS, Singh PB, Grigoryev SA. Insulation of the chicken beta-globin chromosomal domain from a chromatin-condensing protein, MENT. Mol Cell Biol. 2003;23:6455–6468. doi: 10.1128/MCB.23.18.6455-6468.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TC, Elgin SC. Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol Cell Biol. 1986;6:3862–3872. doi: 10.1128/mcb.6.11.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellum R, Alberts BM. Heterochromatin protein 1 is required for correct chromosome segregation in Drosophila embryos. J Cell Sci. 1995;108:1419–1431. doi: 10.1242/jcs.108.4.1419. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee JH, Iyer VR. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS ONE. 2008;3:e1798. doi: 10.1371/journal.pone.0001798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschmann DA, Lininger RA, Gardner LM, Seftor EA, Odero VA, Ainsztein AM, Earnshaw WC, Wallrath LL, Hendrix MJ. Down-regulation of HP1Hsalpha expression is associated with the metastatic phenotype in breast cancer. Cancer Res. 2000;60:3359–3363. [PubMed] [Google Scholar]
- Kirschner-Schwabe R, Lottaz C, Todling J, Rhein P, Karawajew L, Eckert C, von Stackelberg A, Ungethum U, Kostka D, Kulozik AE, et al. Expression of late cell cycle genes and an increased proliferative capacity characterize very early relapse of childhood acute lymphoblastic leukemia. Clin Cancer Res. 2006;12:4553–4561. doi: 10.1158/1078-0432.CCR-06-0235. [DOI] [PubMed] [Google Scholar]
- Koch B, Kueng S, Ruckenbauer C, Wendt KS, Peters JM. The Suv39h-HP1 histone methylation pathway is dispensable for enrichment and protection of cohesin at centromeres in mammalian cells. Chromosoma. 2008;117:199–210. doi: 10.1007/s00412-007-0139-z. [DOI] [PubMed] [Google Scholar]
- Kwon SH, Workman JL. The heterochromatin protein 1 (HP1) family: put away a bias toward HP1. Mol Cells. 2008;26:217–227. [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt's lymphoma cells. Proc Natl Acad Sci USA. 2003;100:8164–8169. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Li B, Swanson S, Zhang Y, Florens L, Washburn MP, Abmayr SM, Workman JL. Heterochromatin protein 1a stimulates histone H3 lysine 36 demethylation by the Drosophila KDM4A demethylase. Mol Cell. 2008;32:696–706. doi: 10.1016/j.molcel.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomberk G, Bensi D, Fernandez-Zapico ME, Urrutia R. Evidence for the existence of an HP1-mediated subcode within the histone code. Nat Cell Biol. 2006;8:407–415. doi: 10.1038/ncb1383. [DOI] [PubMed] [Google Scholar]
- Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol. 2004;5:296–304. doi: 10.1038/nrm1355. [DOI] [PubMed] [Google Scholar]
- Maison C, Bailly D, Peters AH, Quivy JP, Roche D, Taddei A, Lachner M, Jenuwein T, Almouzni G. Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet. 2002;30:329–334. doi: 10.1038/ng843. [DOI] [PubMed] [Google Scholar]
- Martini E, Roche DM, Marheineke K, Verreault A, Almouzni G. Recruitment of phosphorylated chromatin assembly factor 1 to chromatin after UV irradiation of human cells. J Cell Biol. 1998;143:563–575. doi: 10.1083/jcb.143.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minc E, Allory Y, Courvalin JC, Buendia B. Immunolocalization of HP1 proteins in metaphasic mammalian chromosomes. Methods Cell Sci. 2001;23:171–174. doi: 10.1007/978-94-010-0330-8_18. [DOI] [PubMed] [Google Scholar]
- Minc E, Allory Y, Worman HJ, Courvalin JC, Buendia B. Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma. 1999;108:220–234. doi: 10.1007/s004120050372. [DOI] [PubMed] [Google Scholar]
- Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Mulero-Navarro S, Esteller M. Epigenetic biomarkers for human cancer: the time is now. Crit Rev Oncol Hematol. 2008;68:1–11. doi: 10.1016/j.critrevonc.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001;15:267–285. doi: 10.1101/gad.864201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzina N, Verreault A, Laue E, Stillman B. Heterochromatin dynamics in mouse cells: interaction between chromatin assembly factor 1 and HP1 proteins. Mol Cell. 1999;4:529–540. doi: 10.1016/s1097-2765(00)80204-x. [DOI] [PubMed] [Google Scholar]
- Nielsen AL, Oulad-Abdelghani M, Ortiz JA, Remboutsika E, Chambon P, Losson R. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol Cell. 2001a;7:729–739. doi: 10.1016/s1097-2765(01)00218-0. [DOI] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001b;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- Norwood LE, Moss TJ, Margaryan NV, Cook SL, Wright L, Seftor EA, Hendrix MJ, Kirschmann DA, Wallrath LL. A requirement for dimerization of HP1Hsalpha in suppression of breast cancer invasion. J Biol Chem. 2006;28:28. doi: 10.1074/jbc.M512454200. [DOI] [PubMed] [Google Scholar]
- O'Geen H, Squazzo SL, Iyengar S, Blahnik K, Rinn JL, Chang HY, Green R, Farnham PJ. Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS Genet. 2007;3:e89. doi: 10.1371/journal.pgen.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberley MJ, Inman DR, Farnham PJ. E2F6 negatively regulates BRCA1 in human cancer cells without methylation of histone H3 on lysine 9. J Biol Chem. 2003;278:42466–42476. doi: 10.1074/jbc.M307733200. [DOI] [PubMed] [Google Scholar]
- Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biol. 2004;6:1135–1141. doi: 10.1038/ncb1187. [DOI] [PubMed] [Google Scholar]
- Polo SE, Theocharis SE, Klijanienko J, Savignoni A, Asselain B, Vielh P, Almouzni G. Chromatin assembly factor-1, a marker of clinical value to distinguish quiescent from proliferating cells. Cancer Res. 2004;64:2371–2381. doi: 10.1158/0008-5472.can-03-2893. [DOI] [PubMed] [Google Scholar]
- Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007;67:4605–4619. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quade BJ, Wang TY, Sornberger K, Dal Cin P, Mutter GL, Morton CC. Molecular pathogenesis of uterine smooth muscle tumors from transcriptional profiling. Genes Chromosomes Cancer. 2004;40:97–108. doi: 10.1002/gcc.20018. [DOI] [PubMed] [Google Scholar]
- Quivy JP, Gerard A, Cook AJ, Roche D, Almouzni G. The HP1-p150/CAF-1 interaction is required for pericentric heterochromatin replication and S-phase progression in mouse cells. Nat Struct Mol Biol. 2008;15:972–979. doi: 10.1038/nsmb.1470. [DOI] [PubMed] [Google Scholar]
- Quivy JP, Roche D, Kirschner D, Tagami H, Nakatani Y, Almouzni G. A CAF-1 dependent pool of HP1 during heterochromatin duplication. EMBO J. 2004;23:3516–3526. doi: 10.1038/sj.emboj.7600362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Regnier V, Vagnarelli P, Fukagawa T, Zerjal T, Burns E, Trouche D, Earnshaw W, Brown W. CENP-A is required for accurate chromosome segregation and sustained kinetochore association of BubR1. Mol Cell Biol. 2005;25:3967–3981. doi: 10.1128/MCB.25.10.3967-3981.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, Liao X, Iglehart JD, Livingston DM, Ganesan S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–132. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Ritou E, Bai M, Georgatos SD. Variant-specific patterns and humoral regulation of HP1 proteins in human cells and tissues. J Cell Sci. 2007;120:3425–3435. doi: 10.1242/jcs.012955. [DOI] [PubMed] [Google Scholar]
- Ryan RF, Schultz DC, Ayyanathan K, Singh PB, Friedman JR, Fredericks WJ, Rauscher FJ., 3rd KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol Cell Biol. 1999;19:4366–4378. doi: 10.1128/mcb.19.6.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedeberg L, Weisshart K, Diekmann S, Meyer Zu Hoerste G, Hemmerich P. High- and low-mobility populations of HP1 in heterochromatin of mammalian cells. Mol Biol Cell. 2004;15:2819–2833. doi: 10.1091/mbc.E03-11-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Stewart MD, Li J, Wong J. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol Cell Biol. 2005;25:2525–2538. doi: 10.1128/MCB.25.7.2525-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Maison C, Roche D, Almouzni G. Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol. 2001;3:114–120. doi: 10.1038/35055010. [DOI] [PubMed] [Google Scholar]
- Taddei A, Roche D, Bickmore WA, Almouzni G. The effects of histone deacetylase inhibitors on heterochromatin: implications for anticancer therapy? EMBO Rep. 2005;6:520–524. doi: 10.1038/sj.embor.7400441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschure PJ, van der Kraan I, de Leeuw W, van der Vlag J, Carpenter AE, Belmont AS, van Driel R. In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Mol Cell Biol. 2005;25:4552–4564. doi: 10.1128/MCB.25.11.4552-4564.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Ivanov A, Chen L, Fredericks WJ, Seto E, Rauscher F,J, 3rd, Chen J, Cress WD, Ryan RF, Schultz DC, et al. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005;24:3279–3290. doi: 10.1038/sj.emboj.7600791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Rauscher FJ, 3rd, Cress WD, Chen J. Regulation of E2F1 function by the nuclear corepressor KAP1. J Biol Chem. 2007;282:29902–29909. doi: 10.1074/jbc.M704757200. [DOI] [PubMed] [Google Scholar]
- Watrin E, Schleiffer A, Tanaka K, Eisenhaber F, Nasmyth K, Peters JM. Human Scc4 is required for cohesin binding to chromatin, sister-chromatid cohesion, and mitotic progression. Curr Biol. 2006;16:863–874. doi: 10.1016/j.cub.2006.03.049. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7–12. doi: 10.1016/j.ccr.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. How cancer arises. Sci Am. 1996;275:62–70. doi: 10.1038/scientificamerican0996-62. [DOI] [PubMed] [Google Scholar]
- Weinmann AS, Yan PS, Oberley MJ, Huang TH, Farnham PJ. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev. 2002;16:235–244. doi: 10.1101/gad.943102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley SP, Carvalho A, Vagnarelli P, Earnshaw WC. INCENP is required for proper targeting of Survivin to the centromeres and the anaphase spindle during mitosis. Curr Biol. 2001;11:886–890. doi: 10.1016/s0960-9822(01)00238-x. [DOI] [PubMed] [Google Scholar]
- Williams L, Grafi G. The retinoblastoma protein - a bridge to heterochromatin. Trends Plant Sci. 2000;5:239–240. doi: 10.1016/s1360-1385(00)01653-8. [DOI] [PubMed] [Google Scholar]
- Yamagishi Y, Sakuno T, Shimura M, Watanabe Y. Heterochromatin links to centromeric protection by recruiting shugoshin. Nature. 2008;455:251–255. doi: 10.1038/nature07217. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sonoda M, Inokuchi J, Shirasawa S, Sasazuki T. Polycomb group suppressor of zeste 12 links heterochromatin protein 1alpha and enhancer of zeste 2. J Biol Chem. 2004;279:401–406. doi: 10.1074/jbc.M307344200. [DOI] [PubMed] [Google Scholar]
- Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, McDonald C, Thomas R, Dhir R, Finkelstein S, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.