

Abstract
The role of mitochondria in sporadic Parkinson's disease (PD) has been debated for a little over 20 years since the description of complex I deficiency in the substantia nigra pars compacta (SNpc) of PD patients. However, the identification of recessive pathogenic mutations in the pink1 gene in familial PD cases firmly re-ignited interest in the pathophysiology of mitochondria in PD. PINK1 is a putative mitochondrial serine/threonine kinase, which protects cells against oxidative stress induced apoptosis. The mechanism by which this is achieved and the effect of the pathogenic mutations has been an area of intensive research over the past five years. Significant progress has been made and, in this review, we summarize the physiological roles that have been assigned to PINK1 and the potential mechanisms behind pathogenesis.
Keywords: calcium signalling, mitochondria, oxidative stress, PINK1
Introduction
Since the identification of Pten induced kinase 1 (PINK1) as a Parkinson's disease (PD) associated gene in 2004, a number of studies have sought to identify the biological roles of this serine/threonine kinase in order to aid therapeautic advances for the treatment of PD. PINK1 is a mitochondrially targeted serine/threonine kinase which has been shown to protect cells against oxidative stress induced apoptosis. Mutations associated with PD are located throughout the PINK1 protein but the majority are found within the kinase domain. The location of these mutations, including one that resides within the adenosine triphosphate (ATP) binding pocket of PINK1, suggests that loss of PINK1 kinase activity is responsible for disease initiation. In addition, mutations within the carboxy terminus of the protein have been shown to be important for optimal kinase activity. Table 1 lists all of the mutations identified within the pink1 gene and denotes their locations within the protein with respect to the kinase domain. The PINK1 protein is cleaved upon entry into the mitochondria to produce two protein fragments, one at 54 kDa (ΔN-PINK1) and one at 45 kDa (ΔN2-PINK1). Whether these cleavage products are released or exported from mitochondria after the cleavage event remains an area of current debate and the physiological relevance of cleavage is currently unknown. During the course of this review, we aim to look at the specific cellular roles assigned to PINK1 after 5 years of extensive study, summarize the evidence for each individual role and highlight areas still surrounded by controversy.
Table 1.
Identified mutations in pink1
 |
*mutation results in a stop codon.
⊗, ¥, ≠, Ø, #, ¶, ϕ= both mutations found in a compound heterozygous patient.
ïfound in a compound heterozygote with mutation V317I.
#found in a compound heterozygote with mutation R492X.
¢found in a compound heterozygote with mutation Q456*.
† mutation has been identified in patients and controls.
§mutation was found in a control individual only.
‡amino acid variant found in patients and controls which associates with late onset PD.
PINK1 protects cells against oxidative stress-induced apoptosis
Oxidative stress and impaired electron transport chain function have been linked to the pathogenesis of PD, ever since exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a complex I inhibitor, was found to induce parkinsonism in humans (Langston et al, 1983; Langston & Ballard, 1983). In addition, analysis of post mortem brain tissue from PD patients showed an increase in markers of oxidative damage and complex I deficiency in the SNpc (Dawson & Dawson, 2003; Dexter et al, 1989; Schapira et al, 1989; Wu et al, 2003).
In mitochondria, electrons travel through the electron transport chain (complexes I–IV) to generate energy. Reactive oxygen species (ROS) form as a natural byproduct of oxygen metabolism, where roughly 1–5% of the oxygen consumed is converted into ROS (Westermann, 2008). ROS (such as oxygen ions, free radicals and peroxides) can be useful to the immune system and are known to be involved in cell signalling events but if the levels of ROS overcome the cell's ability to neutralize and eliminate them, they can inflict damage on DNA, lipids and proteins (Zhou et al, 2008a). This situation, known as oxidative stress, can be caused by reduced antioxidant capacity or by the over-production of ROS. For a comprehensive review on ROS production, we recommend reading Balaban et al (2005).
Initial reports that a key function of the PINK1 protein was to protect cells against stress-induced death came from cell culture studies where PINK1-deficient cells were shown to be more susceptible to apoptosis after exposure to mitochondrial toxins (Deng et al, 2005; Valente et al, 2004; Wood-Kaczmar et al, 2008). In addition, over-expression of the wild-type PINK1 protein could protect cells against death mediated by chemical insults such as MPTP and MG-132 but this effect was abrogated when the protein carried either a PD associated mutation or a kinase inactivating mutation (Deng et al, 2005; Haque et al, 2008; Petit et al, 2005; Valente et al, 2004). To confirm the physiological relevance of these studies, patient fibroblasts and immortalized lymphoblasts from individuals carrying a PINK1 mutation have also been reported to display reduced complex I activity and increased levels of oxidative stress (Hoepken et al, 2007). Subsequent examinations of ROS production confirmed that cells lacking PINK1 function have increased basal cytoplasmic and mitochondrial ROS (mROS) production (Gandhi et al, 2009; Piccoli et al, 2008a; Wood-Kaczmar et al, 2008). As the mitochondrial DNA (mtDNA) is located at the same site where ROS production takes place within the mitochondrion, it is vulnerable to ROS-mediated mutations (Westermann, 2008). The increase in ROS may therefore account for the identification of mtDNA mutations within PINK1 PD patients (Bender et al, 2008; Piccoli et al, 2008a).
Utilizing the pink1 knockout mouse (lacking exons 4–7), reductions in complex I were noted in the striatum and, as a novel finding in a PD model, reductions were also noted for complexes II and III (Gautier et al, 2008). In an independent pink1 knockout mouse model (lacking exons 2 and 3 and resulting in a truncation after exon 1), however, Morais et al reported reductions exclusively in complex I in mitochondrially enriched fractions from both the mouse brain and liver (Morais et al, 2009). Despite this discrepancy, both studies noted that the reduced activity of these complexes (I–III in Gautier et al and I in Morais et al) appeared to be caused by a decrease in function rather than reduced levels of the protein complexes within mitochondria (Gautier et al, 2008; Morais et al, 2009). Expression of the wild type PINK1 protein in pink1−/− mouse fibroblasts sufficiently rescued the enzymatic activity of complex I in these cells. In contrast, expression of PINK1 containing the PD associated mutations G309D, W437X and a kinase inactivating mutation K219A failed to rescue the complex I phenotype (Morais et al, 2009). In addition, whilst there were no obvious signs of oxidative stress damage, exposure of littermates to environmental toxins revealed that the neurons of mice lacking the PINK1 protein were more susceptible to oxidative stress (Gautier et al, 2008).
Glossary
- Aggresome
A proteinaceous inclusion body that forms as a consequence of UPS overload or impairment.
- Anterograde mitochondrial transport
The movement of mitochondria from the neuron cell body, along the axons to the dendrites.
- Chaperone proteins
A family of proteins, which assist in the correct folding and/or assembly of proteins or protein complexes within cells.
- Complex I
Also known as NADH dehydrogenase or NADH: quinone reductase is the first enzyme of the respiratory electron transport chain in mitochondria. It catalyses the transfer of electrons from NADH to coenzyme Q.
- Dopaminergic neurons
The main source of DA in the mammalian central nervous system, they are located in the substantia nigra of the brain.
- Electron transport chain
Couples the chemical reaction between an electron donor and electron acceptor to the transfer of hydrogen ions across a membrane. It consists of four complexes (complex I: NADH, complex II: succinate dehydrogenase, complex III: cytochrome bc1 and complex IV: cytochrome c oxidase), coenzyme Q, cytochrome c and ATP synthase. The hydrogen ions pumped out of the inner mitochondrial membrane by complexes I, III and IV are used to generate ATP.
- Kinome
Collective name given to all of the protein kinases encoded within an organism's genome.
- Lewy bodies
Abnormal, cytoplasmic, spherical aggregates of protein found within neurons. They are primarily composed of α-synuclein.
- mPTP
A non-selective, high conductance channel that forms at specific sites where the inner and outer mitochondrial membranes meet.
- Na+/Ca2+ exchanger
Membrane protein, which exports calcium from the mitochondria or whole cells whilst simultaneously importing sodium.
- Parkinson's disease
Belongs to a group of conditions known as movement disorders and is characterized by symptoms such as limb tremor, slowness of movement and muscle rigidity. It can be inherited but is most commonly sporadic in nature without obvious genetic faults.
- Phosphatome
The collective name given to all protein phosphatases encoded within an organism's genome.
- SNpc
A brain structure located in the midbrain. It plays an important role in movement, addiction and reward processes.
- Synaptic junctions
The place where a nerve impulse passes, in the form of an electric signal from one neuron to another or to a muscle.
- Type 2 diabetes
Non-insulin-dependent diabetes or adult-onset diabetes is a medical condition characterized by high blood glucose levels and insulin deficiency.
- Ubiquitin
A small, regulatory protein that is covalently attached onto a protein and may induce a number of alterations in protein stability, function and/or localization.
- Ubiquitin-ligase
Enzyme which attaches ubiquitin covalently onto a lysine residue of a target protein.
- UPS
A protein quality control system that uses ubiquitin to tag misfolded cellular proteins for refolding or degradation.
PINK1 has been reported to protect against oxidative stress by phosphorylating the mitochondrial chaperone tumour necrosis factor receptor-associated protein 1 (TRAP1)/heat shock protein 75 (Hsp75) (Pridgeon et al, 2007). PINK1 co-localizes and interacts with TRAP1 in the mitochondrial intermembrane space. Upon phosphorylation, TRAP1 prevents cytochrome c release and H2O2 induced apoptosis by an as yet unknown mechanism and the ability of PINK1 to phosphorylate TRAP1 (in vitro or in cell culture systems) is impaired by kinase inactivating or PD associated mutations. Notably, in the absence of TRAP1, over-expression of wild type PINK1 is unable to protect cells against oxidative stress mediated apoptosis indicating that TRAP1 is essential for the pro-survival effects of PINK1 (Pridgeon et al, 2007). The effect of PINK1 deficiency in relation to oxidative stress and ROS production is summarized in Fig 1.
Figure 1. PINK1 protects against oxidative stress induced apoptosis.

In the absence of PINK1, mitochondrial and cytoplasmic ROS levels increase and cells showed impaired complex I activity within the electron transport chain. Phosphorylation of the TRAP1 protein by PINK1 is required for PINK1's pro-survival effect.
PINK1 and respiration
Animal cells obtain energy in the form of ATP by oxidizing food molecules through the process of respiration. Hydrolysis of ATP supplies the energy needed for a number of cellular processes. Through sub-fractionation of mitochondrial extracts, Liu et al have shown that PINK1 exists in large protein complexes with the respiratory chain complexes I–IV (Liu et al, 2009). Moreover, they showed that mitochondrial respiration—as measured by oxygen consumption—and ATP production can be impaired by loss of PINK1 function or the presence of a PD mutation. As a result of the reduced energy supply, proteasome function may be impaired. Finally, these deficits are associated with increased α-synuclein aggregation (Liu et al, 2009). Piccoli et al have characterized fibroblasts from a patient affected by early-onset parkinsonism carrying a homozygous W437X nonsense mutation in the pink1 gene. When examined, mitochondria from the patient fibroblasts showed a significant decrease in the respiratory activity and ATP production accompanied by ROS accumulation when compared with normal controls. These alterations seem to be due to a decrease in mitochondrial cytochrome c content. However, enzymatic assays and non-denaturing gel electrophoresis showed no significant decrease in the specific activity, content and subunit pattern of the oxidative phosphorylation complexes (Piccoli et al, 2008b). In a number of pathological conditions, the impairment of mitochondrial respiratory chain function is due to mutations in the mitochondrial DNA (mtDNA) (Chinnery & Turnbull, 1999). Although the fibroblasts of patients carrying the W437X mutation did not show variation in either the expression of the oxidative phosphorylation complexes or mtDNA copy number, sequence analysis of the mtDNA revealed two missense mutations in the ND5 and ND6 genes (coding for two subunits of complex I) (Piccoli et al, 2008a). However, Gegg et al have shown that loss of PINK1 expression in human dopaminergic SH-SY5Y cells resulted in a decrease in mtDNA levels and mtDNA synthesis, associated with an impairment of the respiration chain, with complex IV being particularly affected (Gegg et al, 2009).
More recently, live cell imaging techniques have been used to study the precise effects of loss of PINK1 function in both human and mouse neurons (Gandhi et al, 2009). In this study, the authors demonstrate that PINK1 regulates calcium efflux from the mitochondria (discussed in detail in the ‘PINK1 in calcium signalling’ section). As a result of PINK1 deficiency, mitochondria suffer from calcium overload and this stimulates ROS production in the mitochondria and cytosol. Overproduction of free radicals causes inhibition of the glucose transporter, resulting in a lack of substrate delivery, and impaired respiration. Generalized impairment of respiration seems to be a consequence of the lack of substrates for complexes I and II and is associated with reduced oxygen consumption in the PINK1 knockdown and knockout cells (Gandhi et al, 2009). The lowered activity of the oxidative phosphorylation complexes results in a decrease in mitochondrial membrane potential. As a result, the mitochondria switch from the production of ATP, to the consumption of ATP by ATP synthase in order to maintain their membrane potential (Campanella et al, 2008). These data strongly suggest that the respiratory complexes in PINK1 deficiency are intact and that their functional inhibition is in fact secondary to a reduction in substrate supply. The study by Morais et al supports these findings in both PINK1 mouse and Drosophila models (Morais et al). Fig 2 presents a simplified diagram of PINK1 function in cellular respiration.
Figure 2. PINK1 and respiration.

Loss of PINK1 function impairs mitochondrial respiration, results in reduced ATP production and increased ROS levels. The increase in ROS inhibits the glucose transporter and could induce mutations within mitochondrial DNA. Impairment of the glucose transporter reduces substrate delivery to complexes I and II of the respiratory chain. As a direct consequence, ATP synthase is forced to consume, rather than produce, ATP in order to maintain the mitochondrial membrane potential.
PINK1 and ubiquitination/protein folding
Most neurodegenerative diseases are associated with the death of specific neuronal populations due to protein folding events leading to misfolding, aggregation and cytotoxicity of certain proteins. In PD, research on protein misfolding and aggregation has taken centre stage following the association of mutations within α-synuclein with familial forms of the disease and the identification of the protein as a major component of Lewy bodies. These aggregates often contain ubiquitin, the signal protein for degradation by the ubiquitin–proteasome system (UPS), and chaperone proteins that are involved in protein folding. Chaperones and the UPS usually work cooperatively to eliminate degraded proteins (Gao & Hu, 2008).
Chaperone proteins, most of which belong to the heat shock protein (Hsp) family, are the first line of defence against protein misfolding and aggregation. In this context, they are likely to be key players in PD pathogenesis. Interestingly, PINK1 has been shown to interact with several chaperone proteins and these are known to affect both PINK1 stability and PINK1's ability to function as a neuroprotective kinase (Fig 3). The interaction between PINK1 and molecular chaperones Hsp90 and Cdc37 is required to stabilize the full-length PINK1 and the 42 kDa PINK1 cleavage product. Mutations that result in reduced PINK1 protein stability have been reported in PD patients; hence it was proposed that inhibition of the PINK1/Hsp90/cdc37 interaction might contribute to the pathogenesis of PD (Moriwaki et al, 2008). Additional studies have shown that this chaperone system influences both the subcellular distribution of PINK1 and the ratio of full-length to cleaved protein (63/55 kDa ratio) (Weihofen et al, 2008). As mentioned in the previous section, PINK1 has also been shown to be necessary for phosphorylation of the mitochondrial chaperone TRAP1/Hsp75.
Figure 3. PINK1 and ubiquitin/protein folding/mitophagy.

PINK1 interacts with the serine protease HtrA2, the E3-ligase Parkin and the molecular chaperone proteins Hsp90 and cdc37. The homologues of HtrA2 are involved in a protein quality control system. Whether HtrA2 is involved in PINK1 degradation has yet to be determined. PINK1's interaction with Parkin stabilizes PINK1 by interfering with PINK1 ubiquitination and subsequent degradation. PINK1 also forms a functional E3 ligase complex with Parkin and DJ-1 to promote the degradation of misfolded Parkin substrates via the UPS. In addition, the interaction between PINK1 and the Hsp90–cdc37 complex results in the stabilization of the full length PINK1 and its 42 kDa cleavage product. The UPS is critically dependent on ATP. Therefore, since loss of PINK1 results in reduced ATP levels, the UPS function may be impaired resulting in the accumulated misfolded and ubiquitinated proteins being removed by autophagy.
In addition to the molecular chaperones, some proteases monitor the quality of proteins in the mitochondria. For example, the ATP-dependent proteases monitor the quality of proteins in the mitochondrial matrix independent of a ubiquitin pathway (Germain, 2008). Another example is HtrA2 (also known as Omi), a mitochondrial serine protease initially identified as a mammalian homologue of the Escherichia coli stress responsive proteases HtrA/DegP and DegS (Faccio et al, 2000). The function of HtrA2 remains unclear but it was proposed to be involved in a protein quality control system within mitochondria, in a manner similar to the homologous stress-adaptive proteins DegP and DegS in bacteria (Spiess et al, 1999; Walsh et al, 2003). Interestingly, PINK1 was shown to interact with HtrA2 and this interaction is likely to play an important role in the regulation of HtrA2 protease activity (Plun-Favreau et al, 2007). Recent evidence suggests that the mitochondrial protein quality control system involving HtrA2 is ubiquitin-dependent (Radke et al, 2008).
The UPS, which is now believed to be the major system of defence against protein misfolding/aggregation, is of major importance in PD aetiology (Olanow & McNaught, 2006). Moreover, a ubiquitin-dependent protein quality control has been proposed to exist in the mitochondria. However, although a number of mitochondrial ubiquitin ligases have been identified (e.g. MITOL, MARCH5 and MULAN), the full spectrum of mitochondrial ubiquitin ligases and their substrates need to be further characterized (Li et al, 2008; Nakamura et al, 2006a; Yonashiro et al, 2006). Parkin is known to be a ubiquitin-ligase, which attaches ubiquitin molecules to substrate proteins. Although Parkin localizes predominantly to the cytosol and cellular vesicles, it has also been shown to associate with the outer mitochondrial membrane. PINK1 has been shown to act upstream of Parkin in a common pathway to maintain mitochondrial function (Yang et al, 2006). Notably, proteasomal inhibition in cells over-expressing wild type PINK1 promotes its sequestration into aggresome-like inclusions that stain positively with antibodies to mitochondrial proteins, amongst which is endogenous Parkin (Muqit et al, 2006). Ultrastructural studies confirmed the presence of intact mitochondria in aggresomes, suggesting that the mechanism by which PINK1 localizes to aggresomes is caused or mediated by mitochondrial recruitment (Muqit et al, 2006). Moreover, a recent report demonstrated the presence of mitochondria within Lewy body-like intraneuronal inclusions in a genetic mouse model of proteasome impairment (Bedford et al, 2008). It is tempting to speculate that PINK1 mutations could impair the function of the UPS. However, PINK1 mutations decrease ATP production (Liu et al, 2009) and could subsequently affect normal UPS function because ubiquitin is bound to its substrates in an ATP dependent manner. This suggests that the effects observed in the UPS may be secondary effects of PINK1 deficiency.
In recent years, it has become clear that ubiquitinated proteins can also be removed via autophagy, especially under conditions of cellular stress. Indeed, increasing evidence suggests that autophagy acts as a compensatory degradation system when the UPS is impaired. This finding is intriguing given the recent data suggesting that loss of PINK1 function promotes autophagy in the mitochondria (mitophagy) and that Parkin function might be required for PINK1 to maintain mitochondrial homeostasis (Dagda et al, 2009). Autophagy is the biological process by which cells remove damaged or redundant components using lysosomes to digest the unwanted elements. Cells deficient in PINK1 were observed to have elevated levels of autophagic vacuoles and lysosomes in comparison to controls; also, stable PINK1 knockdown was able to induce autophagic mitochondrial sequestration, delivery of mitochondria to lysosomes and a decrease in the cellular levels of mitochondria (Dagda et al, 2009). Mitochondrial autophagy could be prevented by the over-expression of PINK1 or ΔN-PINK1 (amino acids 111–581) suggesting that PINK1 kinase activity is required to protect against autophagy. It is of note that some abnormally large mitochondria were observed in cells over-expressing PINK1 but the presence of these abnormal mitochondria did not induce an autophagic response. At present, no hypothesis has been suggested to explain PINK1's function in autophagy. One possibility could be that autophagy is a secondary event induced by the reduction in mitochondrial membrane potential (ΔΨm) and subsequent impaired mitochondrial dynamics. However, additional studies will be required to clarify the situation.
PINK1 in mitochondrial fusion and fission
Mitochondrial fusion and subsequent fission regulate mitochondrial morphology, copy number and overall mitochondrial health. The process of fusion is crucial in maintaining a healthy mitochondrial population. Once the mitochondrial membranes fuse, essentially joining two or more mitochondria, the internal contents such as proteins, metabolites and substrates can be shared between them to compensate for individual deficiencies and maintain a population of functionally optimal mitochondria (Westermann, 2008).
The process of fission generates numerous small, morphologically and functionally independent, spherical mitochondria and generally results in the production of two ‘unequal’ daughters (Twig et al, 2008). One daughter, which displays a transient increase in basal ΔΨm, joins the functional population of mitochondria within cells and will subsequently go on to participate in a fusion event. The other daughter, however, displays a transient ΔΨm below that normally observed within healthy organelles and this mitochondrion is subsequently removed from the population by autophagy (Twig et al, 2008).
The shape of mitochondria greatly affects the cell's ability to ensure that the organelles are distributed to areas of high-energy demands. The ability to successfully distribute mitochondria to specific cellular locations, such as synaptic junctions, is of crucial importance in highly polarized cells such as neurons.
A role for PINK1 in the regulation of mitochondrial fission/fusion dynamics has recently been proposed in a series of articles (Deng et al, 2008; Park et al, 2009; Poole et al, 2008; Yang et al, 2008). The initial observation was reported in a series of genetic complementation studies in Drosophila (Park et al, 2009; Poole et al, 2008; Yang et al, 2008). Drosophila PINK1 (dPINK1) RNAi or dPINK1 mutant flies display a number of phenotypes including altered mitochondrial morphology in both flight muscle and dopaminergic neurons. The mitochondrial abnormalities obtained through loss of PINK1 function consist of mitochondrial aggregates, swollen or enlarged mitochondria and the presence of a tubular mitochondrial network within the cells. This phenotype can be modified by genetically complementing the flies with either an extra copy of the fission promoting gene drp-1 or removal of a copy of the fusion promoting gene opa-1 (Poole et al, 2008; Yang et al, 2008). Overall, the combined results of the Drosophila studies would seem to suggest that dPINK1 is involved in promoting mitochondrial fission (Fig 4). In support of this, the study by Morais et al has reported less severe but similar synaptic defects in PINK1 knockout flies to those observed in drp-1 deficient cell lines.
Figure 4. PINK1 in mitochondrial fission/fusion.

The balance between mitochondrial fission and fusion is affected by PINK1. PINK1 functions as a potential pro-fusion protein in mammals and a pro-fission protein in Drosophila. Loss of PINK1 results in increased fission in mammalian mitochondria. This phenotype can be rescued by overexpression of a dominant negative DRP-1 protein. In contrast, loss of PINK1 function drives mitochondrial fusion in Drosophila. This phenotype can be rescued by overexpression of wild type DRP-1.
In contrast, PINK1 is thought to function as a pro-fusion protein in mammalian cells because reduced PINK1 function, via RNAi, in a human dopaminergic stem cell line (197VM) and HeLa cells is associated with fragmented mitochondria and reduced mitochondrial membrane potential (Exner et al, 2007; Wood-Kaczmar et al, 2008) (Fig 4). As confirmation that these mitochondrial abnormalities are physiologically relevant, and not merely an artefact of non-specific RNAi effects, analysis of patient fibroblasts carrying the PINK1 PD associated mutations Q126P or G309D, also revealed an aberrant fragmented mitochondrial morphology (Exner et al, 2007). To date, however, whilst no studies have been able to show a direct, non-complex associated interaction between the PINK1 protein and any of the mitochondrial fusion/fission machinery proteins, over-expression of dominant negative DRP-1 in PINK1 knockdown cells was sufficient to rescue the morphological phenotypes associated with PINK1 deficiency. However, expression of dominant negative DRP-1 failed to reduce ROS production induced by PINK1 deficiency indicating that the morphological changes associated with loss of PINK1 function are downstream of the ROS phenotype (Dagda et al, 2009). Furthermore, we know that loss of PINK1 causes a reduction in the mitochondrial membrane potential. The energy state of the mitochondrial membrane has additional effects on mitochondrial fission and fusion because mitochondria with lower membrane potentials are less likely to undergo fusion events (Twig et al, 2008).
The current evidence given to suggest that PINK1 may play a role in mitochondrial fission/fusion events in humans has essentially been made from direct morphological analyses of cells. While the hypothesis that PINK1 is a pro-fusion protein is exciting, it remains possible that the effects of PINK1 deficiency (or expression of the PINK1 protein carrying a PD mutation) on mitochondrial morphology are merely downstream consequences of a direct action on an entirely distinct process such as ROS production, low ΔΨm and calcium flux (see ‘PINK1 in calcium signalling’ section). In addition, it is worth noting that fission and fusion events are regulated by the cell cycle with an increase in mitochondrial fission being associated with M and S phases (Chan, 2006). To our knowledge, the cell cycle stage has not been determined in any of the current studies prior to examining the role of PINK1 in fission and or fusion events and it may therefore be an additional factor to address in future experiments.
An additional puzzle regarding PINK1 function in fission or fusion is why PINK1 in Drosophila appears to induce the opposite effect than that seen in mammalian cell models or human patient fibroblasts, whereas other fission/fusion homologues display the same phenotypes in both organisms. An exception to PINK1 acting as a pro-fusion protein in mammals has, however, been reported in COS-7 cells, where the authors suggest that the discrepancy in phenotype may be cell-type specific (Yang et al, 2008). If cell-type does indeed play an important role in the morphological assessment of fission/fusion events, this raises the question of why recent studies have not utilized neuronal cell lines in an attempt to recapitulate the polar environment of dopaminergic neurons. In addition, one might expect that if PINK1 is a pro-fusion protein then the knockout mice may display phenotypes similar to those of other pro-fusion protein knockouts. In the case of mfn-1 or mfn-2 knockout mice, cells show severe mitochondrial fragmentation (Chen et al, 2003, 2005). Whilst RNAi studies and morphological assessment of patient fibroblasts suggest a reduction in fusion, no abnormal mitochondrial localization or morphology has been observed in the pink1 knockout mouse models (Kitada et al, 2007; Morais et al, 2009).
At present, Parkin is also a complicating factor in dissecting out a role for PINK1 in fission/fusion. PINK1 and Parkin have been shown to function in the same pathway with PINK1 acting upstream of Parkin (Yang et al, 2006). In addition, it has been reported that PINK1 depletion by RNAi resulted in reduced levels of Parkin protein levels (Yang et al, 2006). However, loss of Parkin function by RNAi, in a wild type PINK1 background, can induce the same mitochondrial phenotypes as those described for PINK1 deficiency (Deng et al, 2008; Mortiboys et al, 2008). Therefore, because loss of Parkin function is sufficient to induce the reported abnormal mitochondrial morphology, and Parkin expression is clearly reduced in the absence of PINK1, it will be hard to dissect out and assign a specific role to PINK1 in mitochondrial dynamics independently of Parkin.
Taking all factors into account, it may therefore be prudent to interpret these mitochondrial morphology effects with caution until further evidence of a direct link between PINK1 function and fission/fusion events is reported. Additional evidence using techniques such as cell–cell fusion, fluorescence recovery after photobleaching (FRAP) or photoactivatable green fluorescent protein (GFP) (previously used to assess fission/fusion in neurons) would help to strengthen the case for PINK1 being a pro-fusion protein in mammals. Notably, photoactivatable GFP has recently been used to assess fusion in Drosophila with regards to Miro (Saotome et al, 2008). These techniques are briefly described in two reviews on mitochondrial fusion and fission dynamics (Berman et al, 2008; Chan, 2006).
PINK1 and mitochondrial trafficking
Mitochondria are mobile organelles and cells rearrange their mitochondrial populations according to local ATP needs of each cellular region at a particular time. Regulation of mitochondrial distribution is especially important in neurons, which are particularly dependent on organelle trafficking to balance the changing energy needs of the different regions of the cell (Hollenbeck & Saxton, 2005). Consequently, defects in mitochondrial trafficking/distribution can underlie a neurodegenerative disease (Detmer & Chan, 2007). The majority of the mitochondrial movements have been shown to be microtubule-based. Interestingly, PINK1 has recently been shown to form a multiprotein complex with Miro and Milton, two proteins which (together with kinesin-1) play a crucial function in the anterograde mitochondrial transport along microtubules (from the cell soma to the synapse) (Weihofen et al, 2009) (Fig 4).
Miro and Milton both reside at the outer mitochondrial membrane. PINK1, however, has been shown to reside in the inner membrane of the mitochondria (Gandhi et al, 2006) and contains a predicted transmembrane domain within its amino terminus (TM pred—http://www.ch.embnet.org/software/TMPRED_form.html). Nevertheless, the exact localization of PINK1 is still a matter of debate. Fractionation studies have revealed that the majority of endogenous PINK1, corresponding to the full-length protein, was found in cytoplasmic fractions (Haque et al, 2008). Zhou et al also showed some evidence to suggest that the kinase domain of mitochondrial PINK1 faces the cytosol (Zhou et al, 2008b). In addition, it has been suggested that PINK1 could play a relevant role in PD mechanisms outside the mitochondria (Haque et al, 2008). If PINK1, Miro and Milton are not localized in the same mitochondrial compartment, it is unclear how these proteins would interact. One possibility is that the interaction is not direct and adapter proteins in the mitochondrial inter membrane space may be required for complex association. Alternatively, Miro, Milton and PINK1 could interact en route to the mitochondria from the nucleus. However, if this is the case, it is important to remember that many mitochondrial proteins are not folded correctly until they are imported within the mitochondria (Mills et al, 2008). The significance of the interaction prior to PINK1 mitochondrial import, under these circumstances, would therefore require further investigation. On the other hand, confocal immuno-fluorescence and subcellular fractionation studies have suggested that PINK1 lacking its mitochondria targeting sequence (MTS) (ΔMTS-PINK1) can be found in the mitochondria (Liu et al, 2009; Plun-Favreau et al, 2007; Weihofen et al, 2009; Zhou et al, 2008b). Weihofen et al have therefore suggested that PINK1 could be retained in a MTS independent fashion at the outer membrane of the mitochondria, facing the cytosol, by the Miro–Milton complex. In order to strengthen any of these hypotheses it will be critically important to develop better PINK1 antibodies that allow the detection of endogenous PINK1 at the subcellular level.
Several intracellular signals have been shown to control the movement of mitochondria and thereby influence mitochondrial distribution. The most widely studied of these signals is intracellular calcium which, when elevated, arrests microtubule-based mitochondrial movement in many cell types. The calcium-dependent regulation of mitochondrial motility has been shown to be mediated by the kinesin–Miro–Milton complex (Wang & Schwarz, 2009). A key role of mitochondria is to buffer calcium and prevent calcium overload in the cytosol. Recently, two independent studies have shown that PINK1 regulates calcium efflux from the mitochondria (Gandhi et al, 2009; Marongiu et al, 2009). It could therefore be hypothesized that PINK1, together with Miro and Milton, plays a role in mitochondrial trafficking by regulating mitochondrial calcium efflux (summarized in Fig 5).
Figure 5. PINK1 and mitochondrial trafficking.
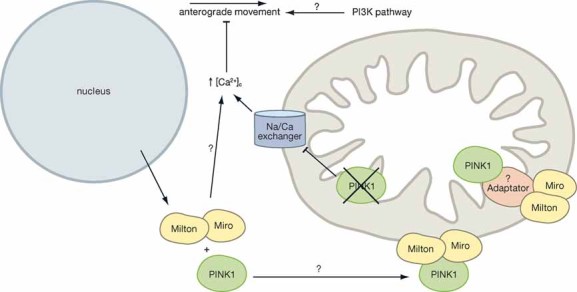
PINK1 interacts with Miro and Milton and may play a role in anterograde mitochondrial transport within cells. It is unclear whether PINK1 and Miro and Milton interact en route to the mitochondria or whether they interact via a possible adapter protein at the mitochondria. Loss of PINK1 function increases calcium levels within the cytosol and this is known to inhibit mitochondrial motility.
Signalling through the PI3-kinase pathway has also been implicated in the regulation of mitochondrial movement (Hollenbeck & Saxton, 2005). Interestingly, PINK1 was originally identified by an analysis of expression profiles from cancer cells after the introduction of exogenous phosphatase and tensin homolog (PTEN), a tumour suppressor that is involved in the regulation of the phosphatidylinositol 3-kinase (PI3K) signalling pathway (Unoki & Nakamura, 2001). Nevertheless, whether the PI3-kinase pathway is important for the regulation of PINK1 and how this might impact on mitochondrial motility still remains to be determined.
PINK1 in calcium signalling
As mentioned previously, two recent studies have highlighted a role for PINK1 in mitochondrial calcium signalling within the mitochondria (Fig 6). Marongiu et al reported that the phenotypes associated with either PINK1 deficiency or expression of the PD associated PINK1 mutant protein W437X (such as the reduced ΔΨm and reduced ATP production) could be restored by specifically blocking mitochondrial calcium uptake (Marongiu et al, 2009). In addition, by directly assessing calcium flux, the authors proposed that expression of PINK1 mutant protein in SH-SY5Y cells may induce mitochondrial calcium overload and that this may explain the mitochondrial phenotypes associated with impaired PINK1 protein function. Gandhi et al have provided a detailed biochemical and physiological analysis on calcium signalling within three independent PINK1 deficient cell populations and have demonstrated that PINK1 directly regulates mitochondrial calcium flux through the Na+/Ca2+ exchanger. The mitochondrial phenotypes associated with PINK1 deficiency, i.e. low ΔΨm, increased ROS production and impaired respiratory complex functions have all been shown to be a consequence of mitochondrial calcium overload. In summary, the study demonstrates that PINK1 deficiency results in the accumulation of calcium in the mitochondria. This accumulation stimulates ROS production, which subsequently inhibits the glucose transporter and results in reduced substrate delivery to the mitochondrial respiratory chain complexes. Furthermore, the reduced calcium buffering capacity of the mitochondria in PINK1 deficient cells, combined with the increased mROS production, lowers the threshold required by the cell to open the mitochondrial permeability transition pore (mPTP) which results in the release of pro-apoptotic factors such as cytochrome c. Notably, dopaminergic neurons utilize calcium channels, rather than sodium channels, to maintain their autonomous pacing and are frequently exposed to large calcium influxes (Chan et al, 2007). The reduced capacity to buffer calcium signals, in the case of PINK1 deficiency, may therefore go some way to explaining the susceptibility of dopaminergic neurons to neuronal death in PD.
Figure 6. PINK1 in calcium signalling.

PINK1 directly regulates calcium efflux from mitochondria via the mitochondrial sodium/calcium exchanger. The resulting mitochondrial overload stimulates ROS production. The increase in ROS inhibits the glucose transporter and reduces the substrate supply to the respiratory chain. The combined calcium and ROS increase lowers the mitochondrial membrane potential and results in opening of the mitochondrial PTP pore at a lower stimulus. Opening of the mPTP releases pro-apoptotic factors such as cytochrome c from the mitochondria and induces apoptosis.
An indirect role for PINK1 in calcium signalling has additionally been proposed in zebrafish (Petko et al, 2009). The PINK1 homologue has been shown to interact with the calcium sensing molecule NCS-1. NCS-1 performs multiple roles including the regulation of neurotransmitter release (Pongs et al, 1993), regulation of voltage gated Ca2+ channels (Rousset et al, 2003), down-regulation of D2 dopamine (DA) receptors (Kabbani et al, 2002) and functions as a neuronal pro-survival factor (Nakamura et al, 2006b).
PINK1 and other human diseases
PINK1 and cancer
There is substantial evidence for low cancer rates in patients with PD. This might be related to the involvement of common genes in both diseases (Inzelberg & Jankovic, 2007).
PINK1 has been shown to be down-regulated in the absence of PTEN (Unoki & Nakamura, 2001). The PTEN gene is a tumour suppressor gene encoding a multifunctional phosphatase, which plays an important role in inhibiting the PI3K/Akt pathway and mutations in PTEN have been found in many human cancers. PINK1 therefore appeared to be a novel candidate as a mediator of the PTEN growth-suppressive signalling pathway. Inhibition of the PI3K/Akt pathway and the up-regulation of PINK1 by PTEN suggest the involvement of PTEN in both cancer and PD. It is of note, however, that since the original identification of PINK1 being up-regulated by exogenous PTEN, only one study has subsequently observed this effect (Siddall et al, 2008) and the link between PINK1 and PTEN in PD has yet to be confirmed. Nonetheless, PINK1 has been identified in a sensitized siRNA kinome and phosphatome screen as an essential element for survival, making PINK1 important as a potential cancer drug target (MacKeigan et al, 2005).
Other genes responsible for familial PD, as well as some heat shock proteins, have also been implicated in tumourigenesis. Mutations in the parkin and DJ-1 genes for example, have been reported in different types of cancer (Inzelberg & Jankovic, 2007). Thus, parkin and pink1 might be tumour suppressor genes, whereas DJ-1 is likely to be an oncogene, because of its negative regulatory effects on PTEN. Interestingly, Parkin and DJ-1 have also been shown to be regulated and/or regulators of the PI3K/Akt pathway (Fitzgerald & Plun-Favreau, 2008).
PINK1 and type 2 diabetes
One study has reported a link between transcription of the pink1 gene and type 2 diabetes (Scheele et al, 2007). Specifically, pink1 transcription was suppressed in the skeletal muscle of type 2 diabetic patients. Furthermore, transcription of pink1 was also down-regulated under conditions of inactivity and/or obesity in patients and controls. From this study, the authors proposed a role for PINK1 in glucose metabolism. The recent finding that loss of PINK1 inhibits the glucose transporter is therefore encouraging (Gandhi et al, 2009). Interestingly, pink1 transcription was shown to be up-regulated by FOXO3a—a downstream target of insulin and PI3K/Akt (Mei et al, 2009). These combined observations therefore warrant subsequent studies regarding PINK1's role in this disease.
PINK1 models: fruit fly versus mouse
Different animal models (toxin induced or genetically modified models) have been developed in several species together with specific behaviour tests to assess parkinsonism. In the case of PINK1, knockout models have been developed in the fruit fly Drosophila melanogaster, zebrafish and mice.
Drosophila pink1 (dpink1) knockout flies have thus far been the most useful for assessing PINK1 function—perhaps because of the autosomal recessive nature of PINK1 mutations and compensatory pathways in vertebrates. These fly lines are characteristically viable but sterile or hypofertile, have a motor deficit, a shorter life-span, abnormal flight muscles with impaired function, disorganized mitochondrial morphology, reduced mitochondrial mass, lower concentrations of ATP and a small reduction in the number of dopaminergic neurons (Clark et al, 2006; Park et al, 2006; Yang et al, 2006). Similarly, zebrafish deficient in PINK1 display decreased numbers of dopaminergic neurons, reduced ΔΨm, reduced Parkin protein levels and increased ROS levels (Anichtchik et al, 2008). To date however, only the study by Anichtchik et al has addressed PINK1 function in zebrafish.
By comparison, pink1 inactivation in mice impairs DA release but does not alter (1) DA levels; (2) the number of dopaminergic neurons; (3) DA synthesis; or (4) levels of DA receptors (Gautier et al, 2008; Kitada et al, 2007; Morais et al, 2009). The impairment of DA release in pink1 knockout mice is, however, sufficient to compromise nigrostriatal circuit function. Interestingly, mitochondrial respiration has been shown to be impaired in the striatum of pink1 knockout mice at 3–4 months. Defects in complex I, II and IV were observed in the striatum of the pink1 knockout mice (Gautier et al, 2008). Notably, defects in complexes II and IV are a relatively novel finding in a PD model and most studies to date, including examination of an independent pink1 knockout mouse model, have only observed complex I deficiency (Morais et al, 2009; Plun-Favreau & Hardy, 2008).
However, whilst Drosophila have always proven to be an excellent model for dissecting out signalling pathways and providing mechanistic insights into protein function, they do not necessarily provide the optimum model for therapeautic research. This is highlighted by the fact that treatment of dpink1 knockout flies with antioxidants rescued the dopaminergic cell death phenotypes associated with loss of PINK1 function (Wang et al, 2006). In addition, drug treatments with antioxidant properties were able to rescue the same dopaminergic phenotype in the zebrafish model (Anichtchik et al, 2008). Antioxidants have, however, been used in clinical trials as potential PD therapeautics but the results have been largely inconclusive (Shults & Haas, 2005; Storch et al, 2007).
The most likely reason for a weaker phenotype in mouse models can of course be accredited to the activation of compensation mechanisms present in mammalian species and lacking in lower vertebrates and invertebrates such as zebrafish and Drosophila. Whilst these mouse models may not be the optimum model system to study the mechanisms by which PD proteins perform their functions, they do in fact provide us with information regarding the physiological response to loss of function mutations and the compensatory pathways, which are activated in mammals. The models may therefore recapitulate the early situation in PD patients (perhaps prior to diagnosis) and should therefore provide key information for therapeautics.
Pending Issues
Identification of the PINK1 cleavage site and the physiological relevance of cleavage
Generation of specific, robust and reliable anti-PINK1 antibodies
Identification of PINK1 substrates
Clarification of PINK1's function in fission/fusion events
Further examination of PINK1 function in calcium signalling
Examination of PINK1's role in mitochondrial bioenergetics
Investigation of additional PD related proteins for calcium signalling abnormalities
Clarification of PINK1's function in autophagy
Final comments
There is extensive evidence to confirm that PINK1 functions to protect neurons against stress-induced cell death. In addition, PINK1 deficiency has been shown to have a profound effect on respiration and ROS production. However, recent evidence has shown that the respiratory complexes are in fact functional in PINK1 deficient cells and many, if not all phenotypes associated with altered respiration can be alleviated simply by increasing the substrate supply to complexes I and II (Gandhi et al, 2009). An exception to this is the reduced calcium efflux from mitochondria. Notably, these experiments have been conducted in human cell models or cultured primary neurons derived from the pink1 knockout mice, therefore it will be interesting to see if the phenotypes can be rescued in vivo using live pink1 knockout mouse and Drosophila model systems.
Finally, one cannot summarize the past 5 years of research into PINK1 function without addressing the continuing problem of PINK1 antibodies to examine the endogenous protein. Several groups, including ours, have reported the identification of endogenous PINK1 protein with PINK1 specific antibodies. However, few have reported the identification of the full-length (63 kDa) and both the cleaved forms of the protein (53 kDa and 45 kDa) (Dagda et al, 2009; Exner et al, 2007; Gandhi et al, 2006; Liu et al, 2009; Muqit et al, 2006; Plun-Favreau et al, 2007; Pridgeon et al, 2007; Siddall et al, 2008; Zhou et al, 2008b). Furthermore, many groups state that the antibodies used do not consistently recognize endogenous PINK1 and some report background bands of the same molecular weight as the physiological PINK1 cleavage products. This essentially renders the antibody useless for examination of anything other than the full-length protein. The lack of a PINK1 specific and consistent antibody capable of detecting all forms of endogenous PINK1 is a prominent obstacle in the PD field and it will have to be an area of intense focus in the next few years if we are to truly understand all aspects of PINK1 function.
The authors declare that they have no conflict of interest.
For more information
PINK1 OMIM link: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=605909
Genetics Home Reference—PINK1: http://ghr.nlm.nih.gov/gene=pink1
Genetics Home Reference—Parkinson's Disease: http://ghr.nlm.nih.gov/condition=parkinsondisease
PINK1 GeneCard: http://www.genecards.org/cgi-bin/carddisp.pl?gene=PINK1
References
- Abou-Sleiman PM, Muqit MM, McDonald NQ, Yang YX, Gandhi S, Healy DG, Harvey K, Harvey RJ, Deas E, Bhatia K, et al. A heterozygous effect for PINK1 mutations in Parkinson's disease? Ann Neurol. 2006;60:414–419. doi: 10.1002/ana.20960. [DOI] [PubMed] [Google Scholar]
- Anichtchik O, Diekmann H, Fleming A, Roach A, Goldsmith P, Rubinsztein DC. Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J Neurosci. 2008;28:8199–8207. doi: 10.1523/JNEUROSCI.0979-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, Gray T, Topham I, Fone K, Rezvani N, et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci. 2008;28:8189–8198. doi: 10.1523/JNEUROSCI.2218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Schwarzkopf RM, McMillan A, Krishnan KJ, Rieder G, Neumann M, Elstner M, Turnbull DM, Klopstock T. Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J Neurol. 2008;255:1231–1235. doi: 10.1007/s00415-008-0892-9. [DOI] [PubMed] [Google Scholar]
- Berman SB, Pineda FJ, Hardwick JM. Mitochondrial fission and fusion dynamics: the long and short of it. Cell Death Differ. 2008;15:1147–1152. doi: 10.1038/cdd.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, Duchen MR. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, Turnbull DM. Mitochondrial DNA and disease. Lancet. 1999;354:I17–I21. doi: 10.1016/s0140-6736(99)90244-1. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of pink1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Jankovic J, Guo Y, Xie W, Le W. Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y. Biochem Biophys Res Commun. 2005;337:1133–1138. doi: 10.1016/j.bbrc.2005.09.178. [DOI] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD. Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccio L, Fusco C, Chen A, Martinotti S, Bonventre JV, Zervos AS. Characterization of a novel human serine protease that has extensive homology to bacterial heat shock endoprotease HtrA and is regulated by kidney ischemia. J Biol Chem. 2000;275:2581–2588. doi: 10.1074/jbc.275.4.2581. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JC, Plun-Favreau H. Emerging pathways in genetic Parkinson's disease: autosomal-recessive genes in Parkinson's disease–a common pathway? FEBS J. 2008;275:5758–5766. doi: 10.1111/j.1742-4658.2008.06708.x. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Muqit MM, Stanyer L, Healy DG, Abou-Sleiman PM, Hargreaves I, Heales S, Ganguly M, Parsons L, Lees AJ, et al. PINK1 protein in normal human brain and Parkinson's disease. Brain. 2006;129:1720–1731. doi: 10.1093/brain/awl114. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Hu H. Quality control of the proteins associated with neurodegenerative diseases. Acta Biochim Biophys Sin (Shanghai) 2008;40:612–618. doi: 10.1111/j.1745-7270.2008.00441.x. [DOI] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci USA. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Schapira AH, Taanman JW. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE. 2009;4:e4756. doi: 10.1371/journal.pone.0004756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain D. Ubiquitin-dependent and -independent mitochondrial protein quality controls: implications in ageing and neurodegenerative diseases. Mol Microbiol. 2008;70:1334–1341. doi: 10.1111/j.1365-2958.2008.06502.x. [DOI] [PubMed] [Google Scholar]
- Haque ME, Thomas KJ, D'Souza C, Callaghan S, Kitada T, Slack RS, Fraser P, Cookson MR, Tandon A, Park DS. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci USA. 2008;105:1716–1721. doi: 10.1073/pnas.0705363105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoepken HH, Gispert S, Morales B, Wingerter O, Del Turco D, Mulsch A, Nussbaum RL, Muller K, Drose S, Brandt U, et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol Dis. 2007;25:401–411. doi: 10.1016/j.nbd.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inzelberg R, Jankovic J. Are Parkinson disease patients protected from some but not all cancers? Neurology. 2007;69:1542–1550. doi: 10.1212/01.wnl.0000277638.63767.b8. [DOI] [PubMed] [Google Scholar]
- Kabbani N, Negyessy L, Lin R, Goldman-Rakic P, Levenson R. Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J Neurosci. 2002;22:8476–8486. doi: 10.1523/JNEUROSCI.22-19-08476.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard PA., Jr Parkinson's disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N Engl J Med. 1983;309:310. doi: 10.1056/nejm198308043090511. [DOI] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CA. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS ONE. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Vives-Bauza C, Acin-Perez R, Yamamoto A, Tan Y, Li Y, Magrane J, Stavarache MA, Shaffer S, Chang S, et al. PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson's disease. PLoS ONE. 2009;4:e4597. doi: 10.1371/journal.pone.0004597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, Dallapiccola B, Valente EM, Masliah E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson's disease by disturbing calcium flux. J Neurochem. 2009;103:1561–1574. doi: 10.1111/j.1471-4159.2009.05932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW, You H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc Natl Acad Sci USA. 2009;106:5153–5158. doi: 10.1073/pnas.0901104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RD, Sim CH, Mok SS, Mulhern TD, Culvenor JG, Cheng HC. Biochemical aspects of the neuroprotective mechanism of PTEN-induced kinase-1 (PINK1) J Neurochem. 2008;105:18–33. doi: 10.1111/j.1471-4159.2008.05249.x. [DOI] [PubMed] [Google Scholar]
- Morais VA, Verstreken P, Roething A, Smet J, Snellinx A, Vanbrabant M, Haddad D, Frezza C, Mandemakers W, Vogt-Weisenhorn D, et al. Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med. 2009;1:1–13. doi: 10.1002/emmm.200900006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki Y, Kim YJ, Ido Y, Misawa H, Kawashima K, Endo S, Takahashi R. L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci Res. 2008;61:43–48. doi: 10.1016/j.neures.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, Wood NW, Willems PH, Smeitink JA, Cookson MR, et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol. 2008;64:555–565. doi: 10.1002/ana.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, Eaton S, Payne Smith MD, Venner K, Matilla A, et al. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem. 2006;98:156–169. doi: 10.1111/j.1471-4159.2006.03845.x. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006a;7:1019–1022. doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TY, Jeromin A, Smith G, Kurushima H, Koga H, Nakabeppu Y, Wakabayashi S, Nabekura J. Novel role of neuronal Ca2+ sensor-1 as a survival factor up-regulated in injured neurons. J Cell Biol. 2006b;172:1081–1091. doi: 10.1083/jcb.200508156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, McNaught KS. Ubiquitin-proteasome system and Parkinson's disease. Mov Disord. 2006;21:1806–1823. doi: 10.1002/mds.21013. [DOI] [PubMed] [Google Scholar]
- Park J, Lee G, Chung J. The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun. 2009;378:518–523. doi: 10.1016/j.bbrc.2008.11.086. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Petit A, Kawarai T, Paitel E, Sanjo N, Maj M, Scheid M, Chen F, Gu Y, Hasegawa H, Salehi-Rad S, et al. Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J Biol Chem. 2005;280:34025–34032. doi: 10.1074/jbc.M505143200. [DOI] [PubMed] [Google Scholar]
- Petko JA, Kabbani N, Frey C, Woll M, Hickey K, Craig M, Canfield VA, Levenson R. Proteomic and functional analysis of NCS-1 binding proteins reveals novel signaling pathways required for inner ear development in zebrafish. BMC Neurosci. 2009;10:27. doi: 10.1186/1471-2202-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli C, Ripoli M, Quarato G, Scrima R, D'Aprile A, Boffoli D, Margaglione M, Criscuolo C, De Michele G, Sardanelli A, et al. Coexistence of mutations in PINK1 and mitochondrial DNA in early onset Parkinsonism. J Med Genet. 2008a;33:2565–2574. doi: 10.1136/jmg.2008.058628. [DOI] [PubMed] [Google Scholar]
- Piccoli C, Sardanelli A, Scrima R, Ripoli M, Quarato G, D'Aprile A, Bellomo F, Scacco S, De Michele G, Filla A, et al. Mitochondrial respiratory dysfunction in familiar Parkinsonism associated with PINK1 mutation. Neurochem Res. 2008b;45:596–602. doi: 10.1007/s11064-008-9729-2. [DOI] [PubMed] [Google Scholar]
- Plun-Favreau H, Hardy J. PINK1 in mitochondrial function. Proc Natl Acad Sci USA. 2008;105:11041–11042. doi: 10.1073/pnas.0805908105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, et al. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- Pongs O, Lindemeier J, Zhu XR, Theil T, Engelkamp D, Krah-Jentgens I, Lambrecht HG, Koch KW, Schwemer J, Rivosecchi R, et al. Frequenin–a novel calcium-binding protein that modulates synaptic efficacy in the Drosophila nervous system. Neuron. 1993;11:15–28. doi: 10.1016/0896-6273(93)90267-u. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 Protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radke S, Chander H, Schafer P, Meiss G, Kruger R, Schulz JB, Germain D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem. 2008;283:12681–12685. doi: 10.1074/jbc.C800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset M, Cens T, Gavarini S, Jeromin A, Charnet P. Down-regulation of voltage-gated Ca2+ channels by neuronal calcium sensor-1 is beta subunit-specific. J Biol Chem. 2003;278:7019–7026. doi: 10.1074/jbc.M209537200. [DOI] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- Scheele C, Nielsen AR, Walden TB, Sewell DA, Fischer CP, Brogan RJ, Petrovic N, Larsson O, Tesch PA, Wennmalm K, et al. Altered regulation of the PINK1 locus: a link between type 2 diabetes and neurodegeneration? FASEB J. 2007;21:3653–3665. doi: 10.1096/fj.07-8520com. [DOI] [PubMed] [Google Scholar]
- Shults CW, Haas R. Clinical trials of coenzyme Q10 in neurological disorders. Biofactors. 2005;25:117–126. doi: 10.1002/biof.5520250113. [DOI] [PubMed] [Google Scholar]
- Siddall HK, Warrell CE, Davidson SM, Mocanu MM, Yellon DM. Mitochondrial PINK1-a novel cardioprotective kinase? Cardiovasc Drugs Ther. 2008;22:507–508. doi: 10.1007/s10557-008-6136-5. [DOI] [PubMed] [Google Scholar]
- Spiess C, Beil A, Ehrmann M. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell. 1999;97:339–347. doi: 10.1016/s0092-8674(00)80743-6. [DOI] [PubMed] [Google Scholar]
- Storch A, Jost WH, Vieregge P, Spiegel J, Greulich W, Durner J, Muller T, Kupsch A, Henningsen H, Oertel WH, et al. Randomized, double-blind, placebo-controlled trial on symptomatic effects of coenzyme Q(10) in Parkinson disease. Arch Neurol. 2007;64:938–944. doi: 10.1001/archneur.64.7.nct60005. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene. 2001;20:4457–4465. doi: 10.1038/sj.onc.1204608. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Walsh NP, Alba BM, Bose B, Gross CA, Sauer RT. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell. 2003;113:61–71. doi: 10.1016/s0092-8674(03)00203-4. [DOI] [PubMed] [Google Scholar]
- Wang D, Qian L, Xiong H, Liu J, Neckameyer WS, Oldham S, Xia K, Wang J, Bodmer R, Zhang Z. Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci USA. 2006;103:13520–13525. doi: 10.1073/pnas.0604661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet. 2008;17:602–616. doi: 10.1093/hmg/ddm334. [DOI] [PubMed] [Google Scholar]
- Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking (dagger) Biochemistry. 2009;48:2045–2052. doi: 10.1021/bi8019178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Molecular machinery of mitochondrial fusion and fission. J Biol Chem. 2008;283:13501–13505. doi: 10.1074/jbc.R800011200. [DOI] [PubMed] [Google Scholar]
- Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AS, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE. 2008;3:e2455. doi: 10.1371/journal.pone.0002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci USA. 2003;100:6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura-Hoshino M, Sada K, Hotta H, Yamamura H, et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006;25:3618–3626. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Huang Y, Przedborski S. Oxidative stress in Parkinson's disease: a mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci. 2008a;1147:93–104. doi: 10.1196/annals.1427.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci USA. 2008b;105:12022–12027. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]