

Abstract
Understanding the biological pathways critical for common neurofibromatosis type 1 (NF1) peripheral nerve tumours is essential, as there is a lack of tumour biomarkers, prognostic factors and therapeutics. We used gene expression profiling to define transcriptional changes between primary normal Schwann cells (n = 10), NF1-derived primary benign neurofibroma Schwann cells (NFSCs) (n = 22), malignant peripheral nerve sheath tumour (MPNST) cell lines (n = 13), benign neurofibromas (NF) (n = 26) and MPNST (n = 6). Dermal and plexiform NFs were indistinguishable. A prominent theme in the analysis was aberrant differentiation. NFs repressed gene programs normally active in Schwann cell precursors and immature Schwann cells. MPNST signatures strongly differed; genes up-regulated in sarcomas were significantly enriched for genes activated in neural crest cells. We validated the differential expression of 82 genes including the neural crest transcription factor SOX9 and SOX9 predicted targets. SOX9 immunoreactivity was robust in NF and MPSNT tissue sections and targeting SOX9 – strongly expressed in NF1-related tumours – caused MPNST cell death. SOX9 is a biomarker of NF and MPNST, and possibly a therapeutic target in NF1.
Keywords: MPNST, neurofibroma, NF1, Schwann cell, Sox9
INTRODUCTION
A genetic defect underlies NF1, which is inherited as an autosomal dominant trait affecting 1:3,000 humans (Rasmussen & Friedman, 2000). Analysis of progressive changes downstream of NF1 mutation has been complicated by the spectrum of clinical manifestations in NF1 patients and the diversity of cell types involved. The hallmark of NF1 is the development of peripheral nerve sheath tumours. At least 95% of NF1 patients have multiple dermal NFs, benign tumours that typically appear in adolescence (Rasmussen & Friedman, 2000). Approximately, 30% develop plexiform NFs that are larger, may cause significant morbidity, and can occur congenitally. Questions as fundamental as whether there are molecular differences between dermal and plexiform NF are to date unanswered. Differences between the types of NF are implied as a plexiform NF may transform to an MPNST, a life threatening sarcoma (Evans et al, 2002).
The sequence of biological events driving MPNST formation is unknown. Beyond mutations in both copies of the NF1 tumour suppressor gene (Wimmer et al, 2006), few molecular alterations have been associated with NFs and/or MPNSTs. These alterations include the epidermal growth factor receptor (EGFR), detected in MPNST cell lines and in a subpopulation of NFSCs, as well as amplification of KIT, PDGFRA and PDGFRA mutations, detected in MPNSTs. Loss of tumour suppressor genes, including TP53, RB or INK4A, have been documented in NF1-associated MPNSTs but not NFs (reviewed in Carroll & Ratner, 2008).
Dermal and plexiform NFs are composed of cell types present in normal nerves but in a disorganized form. Axon–Schwann cell contact, which regulates key aspects of normal Schwann cell function, is disrupted in NFs. NFSCs are found distant from axons in a collagen-rich extracellular matrix, admixed with mast cells and fibroblasts (Cichowski & Jacks, 2001). Methods were developed to purify normal human Schwann cells (NHSCs) and NFSCs (Serra et al, 2000). We hypothesized that comparing the gene expression in cultured Schwann cells could identify changes relevant to tumourigenesis because within NFs, only Schwann cells exhibit biallelic NF1 mutations (Serra et al, 2000). NFSCs also show elevated levels of Ras-guanosine triphosphate (GTP) (Sherman et al, 2000), consistent with neurofibromin functioning as a GTPase activating protein (GAP) that inactivates Ras (Le & Parada, 2007), and invade basement membranes and stimulate angiogenesis whereas normal Schwann cells do not (Sheela et al, 1990). Thus, while the data strongly support the view that Schwann cells are the crucial pathogenic cell type in NFs, the molecular changes in NFSCs that drive tumourigenesis are largely unknown.
The critical period(s) in Schwann cell development at which an NF1 mutation results in NF and/or MPNST is also not clear (Carroll & Ratner, 2008; Le et al, 2009; Williams et al, 2008). Schwann cells originate from neural crest stem cells and develop into Schwann cell precursors, then immature Schwann cells and finally mature Schwann cells (Jessen & Mirsky, 2005). The SOX family of transcription factors is important for neural crest stem cell survival (Cheung et al, 2005); SOX10 is required for glial specification in the peripheral nervous system (Britsch et al, 2001). Analysing the expression of these genes might provide insight into the timing of tumourigenesis.
Gene expression in NF1-associated tumours has been analysed by quantitative real time-polymerase chain reaction (qPCR) (Levy et al, 2004), subtractive hybridization (Holtkamp et al, 2004) and cDNA (Miller et al, 2003) and oligonucleotide (Levy et al, 2007; Miller et al, 2006) microarray analyses. Direct comparison of these studies is unfortunately limited due to the multiplicity of platforms and technical variability in sample processing among the different laboratories. To identify a molecular progression model for NF1 peripheral nerve tumourigenesis, we formed the NF1 microarray consortium and analysed primary tumour-derived Schwann cells, MPNST cell lines and NF1 solid tumours.
RESULTS
Creation of a comprehensive gene expression data set consisting of primary tumour-derived Schwann cells, MPNST cell lines and NF1 peripheral nerve tumours
NF tissue samples contain NF1+/− and NF1−/− Schwann cells, fibroblasts, perineurial cells, endothelial cells and mast cells. To avoid this inherent variability and to describe gene expression changes that correspond to a single cell type, we used purified Schwann cells as the basis for our analysis. To ensure data quality, we minimized non-biological variability in sample batch processing by running samples from each experimental group in each processing batch. To minimize the technical variability, a single individual (AH) isolated RNA and we conducted microarray hybridization at a single site. We also hybridized a universal reference RNA in each processing batch along with 11 NF related RNAs as a technical control for batch-to-batch variation. Analysis after each processing batch assessed the power for statistical comparisons (Page et al, 2006). We also used power analysis as a futility analysis in the comparison of dermal and plexiform NFs to determine that a difference between the groups would not be detectable without a far larger sample size (at minimum, 5× more samples).
Schwann cell culture transcription profiles distinguish benign from malignant NF1 tumours but fail to discriminate NF subtypes
To discover gene expression programs that underlie the differences between cultured NHSCs, dermal and plexiform NFSCs (dNFSCs and pNFSCs, respectively) and MPNST cell lines, after referencing (see the Materials and Methods section), analysis of variance (ANOVA) identified 2,827 transcripts (FDR ≤ 0.001) as differentially expressed that we, then, subjected to two-way hierarchical tree clustering (Fig 1 and Table S1 of Supporting Information). Most MPNST cell lines display a unique transcriptional signature and cluster to the far right of the heat map, separately from benign NF-derived Schwann cells. Although we anticipated gene expression signatures that were unique to dNFSCs or pNFSCs, transcripts that passed the ANOVA failed to partition the two NF subtypes. Direct comparison of dNFSCs and pNFSCs also failed to identify a statistically significant signature. Instead, we observed two classes of NFSCs, with genes in Class 1 NFSCs (Fig 1; green bar beneath heat map) less up- or down-regulated than genes in Class 2 NFSCs (Fig 1; blue bar beneath heat map). Class type did not correlate with any known patient parameters or sample handling. Five principal patterns of gene expression were identified and genes were assigned to each using k-means clustering (clusters C1–C5; Fig 1 and Table S2 of Supporting Information; see Table S3 of Supporting Information for biological associations and Table S2 of Supporting Information for detailed cluster and biological association gene lists).
Figure 1. Heat map of transcripts differentially expressed between cultured NHSCs and cultured dNFSCs, pNFSCs and MPNST cell lines.
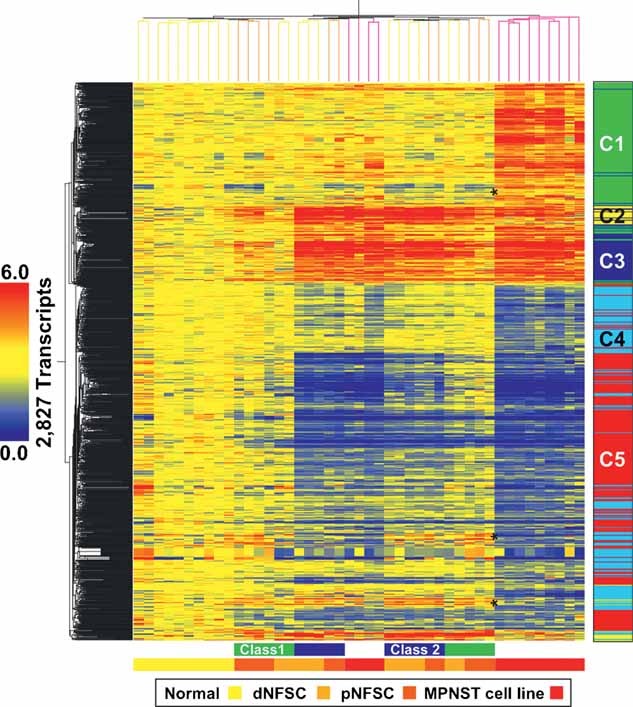
Two-way hierarchical clustering grouped samples as either NHSCs, mixed benign dNFSC and pNFSC or malignant tumour (MPNST) cell lines. Two classes of NFSCs are identifiable, with Class 1 NFSC (green bar beneath the heat map) expression levels attenuated relative to Class 2 NFSC (blue bar beneath the heat map) levels. Genes in clusters C1 and C4, which show decreased or increased expression in benign tumours, respectively, and opposite expression in MPNST cell lines (indicated with asterisks), are abundant in genes associated with cell cycle (AURKA, CDC25B, CDKN2A, CNAP1, INHBA, MCM7, PDGFB) and cell differentiation (ADAM12, ANGPTL4, BMP1, CHL1, IL11, INHBA, PPL, SERPINE2). The bar to the right of the heat map shows five clusters (C1–C5), corresponding to k-means functional clusters listed in Tables S2 and S3 of Supporting Information.
Transcripts differentially expressed in NF1 tumour cell cultures share gene expression signatures with NF1 solid tumours
Each cluster from Fig 1 was re-clustered across NHSCs, primary NFs and primary MPNSTs. We identified sub-cluster(s) that were similarly expressed across cell cultures and their respective solid tumour type. After assigning genes using k-means clustering (Fig 2; Table S5 of Supporting Information), we evaluated clusters C6–C11 to explore the potential biological significance of transcripts deregulated in both NF1 tumour cell cultures and solid tumours (Tables S5 and S6 of Supporting Information). Two-way hierarchical cluster analysis of the transcripts (n = 1,708; 1,108 unique genes) that were similarly expressed in cell cultures and solid tumours again failed to segregate dermal and plexiform NF tumours (Fig 2). The complete list of genes is shown in Table S4 of Supporting Information.
Figure 2. Heat map of transcripts similarly expressed in NF1 cell cultures and tumours.
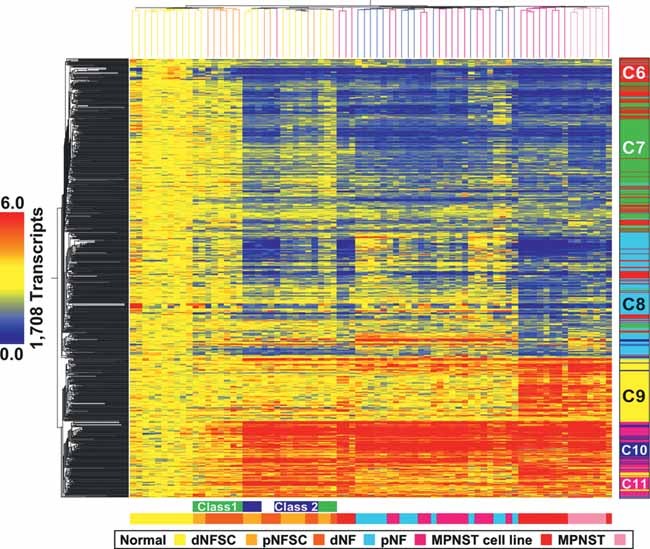
Differentially expressed transcripts in NF1 peripheral nerve cell culture samples were filtered to identify genes with similar patterns of expression in solid tumours. A total of 1,708 transcripts (60%) were identified and clustered across NHSCs, dNFSCs, pNFSCs, MPNST cell lines (annotated as in Fig 1), and dNFs, pNFs and MPNSTs. The bar to the right of the heat map shows five clusters (C6–C11), corresponding to k-means functional clusters listed in Table S5 of Supporting Information.
Analysis of functional enrichment for genes in clusters C6–C11 showed significant associations with nervous system development. We used GATACA1 to identify transcripts from each over-represented ontology or pathway in each cluster associated with nerve development and/or NF or MPNST. Genes in cluster C6 were associated with nervous system development and were down-regulated in most of the sample types (variably down-regulated in class 1 NFSCs and some NFs). Cluster C6 included EMP2, EPB41L3, GFAP, HLA-DQB1, KLK6, L1CAM, LGI1, MBP and NGFR. Genes in cluster C7 were associated with neurogenesis. Cluster C7 consisted of genes down-regulated in MPNST cell lines, MPNSTs and most of the NFs (variable in Class 2 NFSCs and some NFs) and included CDKN2A, CTSD, GJB1, GNAI2, HPCAL1, KNS2, MF12, NES and NFKB1. Genes in cluster C8 were associated with nervous system development. Cluster C8 exhibited decreased expression in most of the MPNST cell lines (variable in MPNSTs and class 2 NFSCs) and contained transcripts that included BCL2, BCL2L2, EDNRB, ERBB3, MPZ, PDGFA, PDGFB and S100β. Transcripts in cluster C9 were associated with morphogenesis and nervous system development. Genes in cluster C9 were up-regulated in MPNST cell lines and MPNSTs and included EN2, HGF, MDK, PAX6, SMAD3 and WT1. Transcripts in clusters C10 and C11 were associated with skeletal development, and C11 with morphogenesis. Genes in cluster C10, variably up-regulated in Class 1 NFSCs and up-regulated in all others, included APOD, CASP1, CD36, EGFR, KIT, LEPR, MME and SOCS3. Cluster C11 was composed of transcripts that were up-regulated in class 2 NFSCs, MPNST cell lines and MPNSTs (variable in class 1 NFSCs and NFs) and contained ADM, CAPN1, FBN2, IGFBP3, PDGFRA, PIAS3, PLAU, PTGES, PTGS2 and TFPI. Cluster C11 also included the neural crest markers TWIST1 and SOX9, and exhibited increased expression in all samples. Additional functional annotation categories for each cluster are shown in Tables S5 and S6 of Supporting Information.
NF1 tumour cell culture and solid tumour expression patterns show broad dysregulation of genes activated in developing Schwann cells
The high representation of genes associated with Schwann cell development in the NF1 tumour signature led us to compare the 1,708 gene signature (1,108 unique genes) to gene orthologues activated in migrating neural crest cells and two stages of Schwann cell development, Schwann cell precursor and immature Schwann cell, based on a published data set (Buchstaller et al, 2004) (Table 1). Strikingly, all down-regulated clusters (C6–C8) showed significant enrichment for gene orthologues activated in immature Schwann cells. Genes up-regulated in MPNST (cluster C9) demonstrated significant over-representation of gene orthologues activated in migrating neural crest cells. The repression of transcripts normally expressed late in Schwann cell development and activation of genes normally expressed early is consistent with significant over-representation of developmental themes identified in our functional analysis (see Tables S4 and S5 of Supporting Information).
Table 1.
NF-related peripheral nerve cell culture and tumour transcription patterns are enriched for genes regulated during Schwann cell development
| Cluster | Expression pattern | Unique genes | Unique in 9,137 | Migrating neural crest 2,033 of 9,137 unique genes | Schwann cell precursor 778 of 9,137 unique genes | Immature Schwann cell 1,421 of 9,137 unique genes | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expect | Observed | Significance | Expect | Observed | Significance | Expect | Observed | Significance | ||||
| C6 | Down in all, variable in class 1 NFSCs and some NFs | 232 | 88 | 20 | 8 | 1.80E-03 | 7 | 12 | 8.53E-02 | 14 | 26 | 9.08E-04 |
| C7 | Down in all except class 1 NFSCs, variable in class 2 NFSCs and some NFs | 321 | 133 | 30 | 22 | 1.16E-01 | 11 | 19 | 2.65E-02 | 21 | 34 | 2.42E-03 |
| C8 | Down in most MPNST cell lines, variable in MPNSTs and class 2 NFSCs | 245 | 91 | 20 | 6 | 1.08E-04 | 8 | 6 | 7.04E-01 | 14 | 54 | 2.20E-16 |
| C9 | Up in MPNST cell lines and MPNST | 175 | 69 | 15 | 31 | 2.84E-05 | 6 | 2 | 1.25E-01 | 11 | 7 | 2.46E-01 |
| C10 | Up in all, variable in class 1 NFSCs | 90 | 46 | 10 | 5 | 7.41E-02 | 4 | 7 | 1.09E-01 | 7 | 10 | 2.26E-01 |
| C11 | Up class 2 NFSCs, MPNST cell lines and MPNSTs, variable in class 1 NFSCs and NFs | 123 | 71 | 16 | 15 | 8.87E-01 | 6 | 10 | 1.29E-01 | 11 | 12 | 7.42E-01 |
Each of the k-means clusters identified in Fig 2 (C6–C11) was evaluated for over-representation of gene orthologues characteristic of three stages of Schwann cell development. We first generated orthologue gene lists for transcripts up-regulated in migrating neural crest, Schwann cell precursors and immature Schwann cells (Buchstaller et al, 2004). After correcting for redundant probe sets, lack of corresponding gene orthologues and platform-specific gene representation differences, Fisher's exact test was used to calculate the expected overlaps between k-means clusters and each of the orthologue gene lists. All down-regulated clusters (C6–C8) showed significant enrichment for gene orthologues activated late in Schwann cell development (i.e., immature Schwann cells). Up-regulated gene clusters characteristic of MPNST were C9, enriched for gene orthologues activated early in Schwann cell development (i.e., migrating neural crest) and C10, enriched for gene orthologues up-regulated in Schwann cell precursors. Gene orthologues activated in Schwann cell precursors but down-regulated in all samples except Class 1 NFSCs, variable in Class 2 NFSCs and variable in some NFs were also significantly over-represented in cluster C7.
In cluster C11, we identified the neural crest markers TWIST1 and SOX9. TWIST1 inhibits MPNST cell chemotaxis (Miller et al, 2006). Many SOX family members are differentially expressed in NF1 samples relative to normal Schwann cells, including down-regulation of SOX5 (clusters C6 and C7), SOX2, SOX2OT, SOX8, SOX10 and SOX 13 (cluster C8) and up-regulation of SOX11 (cluster C9) and SOX9 (cluster C11).
For technical and biological validation, we measured the expression of 82 genes in at least one sample from each sample type by qPCR. Differential expression of 41/82 genes was confirmed in all samples and 77/82 genes in at least 50% of the samples (Table S7 of Supporting Information). This confirmation rate is comparable to that reported in other studies. The entire data set is publicly available via Gene Expression Omnibus2 (Accession number GSE14038).
SOX9 is over-expressed in NF1-derived peripheral nerve tumours
SOX9 is a neural crest transcription factor required for stem cell survival (Cheung et al, 2005). Our microarray data show over-expression of SOX9 in all NF1 tumour samples, ranging from 1.5- to 137-fold on the microarray. Average SOX9 expression values were at least two-fold higher in MPNST samples relative to NF samples (Fig 3A). Confirmation of SOX9 was conducted by qPCR, either using cDNA synthesized from RNA samples used to generate probes for microarray hybridization (Technical) or independent RNA samples (Biological) (Fig 3B). SOX9 over-expression was then validated at the RNA (Fig 3B) and protein (Fig 3C) levels in independent cultures of human NFSCs and MPNST cell lines.
Figure 3. SOX9 is over-expressed in NF1 tumours relative to Schwann cells.
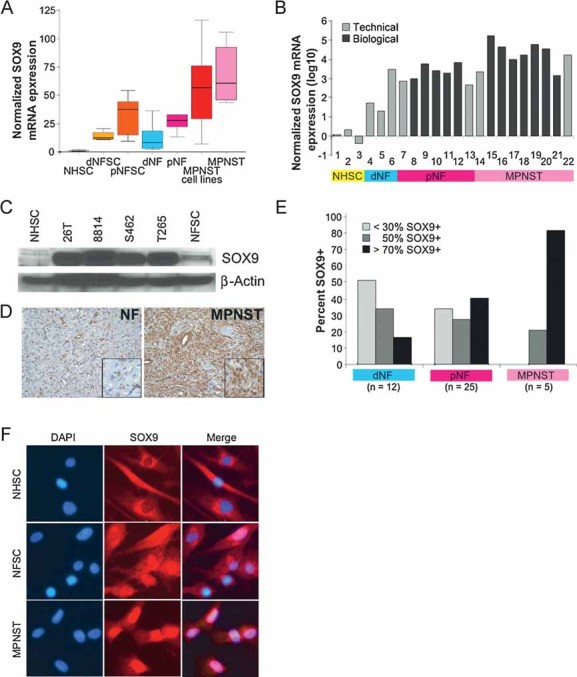
A. Boxplot of SOX9 gene expression microarray data in each of the seven sample types normalized to Schwann cell gene expression (NHSC). Horizontal lines in each bar represent the median SOX9 expression within each sample type and the error bars indicate the range of non-outlier measurements.
B. SOX9 mRNA expression measured by qPCR of individual samples within each sample type and normalized to expression in Schwann cells (NHSC). Fold-change values were transformed to log 10 scale in order to display MPNST samples (ranging from 1,448- to 1,72,950-fold) and NF samples (ranging from 20- to 6,654-fold) on the same graph. Validation of RNA samples that were analysed by microarray (Technical) include: 1–3, NHSCs; 4, dNFSC+/−; 5, dNFSC−/−; 6, dNF; 7, pNFSC; 13, pNF; 14, 22, MPNSTs. Validation of independent RNA samples (Biological) include: 8–12, pNFSCs; 15–21, MPNST cell lines; 15, STS26T; 16, ST8814; 17, S462; 18, T265; 19, 90-8; 20, 88-3; 21, YST1.
C. SOX9 protein expression in Schwann cells (NHSC), MPNST cell lines (26T, 8814, S462, T265) and pNFSC by Western blot (top panel) analysis. β-actin was used as a loading control.
D, E. Immunohistochemical detection of SOX9 in a representative NF and MPNST tissue section with extensive SOX9 expression. Cells stained brown are positive for SOX9. (e) The percentage distribution of SOX9-positive cells in a panel of dNF, pNF and MPNST sections. The majority of samples screened were represented in the microarray analysis with the addition of ten independent samples, including three dNFs, five pNFs and two MPNSTs.
F. Immunofluorescent detection of SOX9 (TRITC = red) and nuclei (DAPI = blue) in cultured NHSCs, NFSC and ST8814 NF1-derived MPNST cells. Similar results were obtained in STS26T sporadic MPNST cells (data not shown). Fields shown are representative of each population. Merging fluorescent images detecting DAPI and TRITC (Merge) highlights nuclear localization of SOX9, mainly in MPNST.
Immunohistochemical analysis of SOX9 protein expression was conducted in a panel of 42 NF1 tumour sections, 10 of which were independent of the gene expression microarray experiment (Fig 3E). Strikingly, SOX9 expression was detected in all the tumours, and the staining intensity was similar in all NF and MPNST samples (strong). In the majority of dermal NFs, <30% of the cells were SOX9-positive. The majority of plexiform NFs contained ≤50% SOX9-positive cells, while >70% of the cells were SOX9 positive in the MPNSTs. Concentration of SOX9 in the nucleus was most prominent in MPNST cells, variable in NFSCs and excluded from the nucleus in normal Schwann cells (Fig 3F).
Differential diagnoses for NFs include schwannomas, benign Schwann cell tumours without NF1 mutations. In a panel of eleven schwannomas stained for SOX9, five were negative or in background only, three had weak positive staining and three had moderate staining in <25% of the cells (data not shown). A differential diagnosis for MPNST is synovial sarcoma. Out of seven synovial sarcomas stained, four were negative, two had weak, focal positivity and only one displayed strong staining in most of the cells. These data support the conclusion that SOX9 is a biomarker for NF and MPSNT.
Reducing SOX9 expression inhibits MPNST cell survival
To test for a role for SOX9 in tumourigenic cellular behaviour, we used short hairpin RNAs (shRNAs) to reduce SOX9 expression in NF (Fig 4A) and MPNST cells (Fig 4C–E) relative to a non-targeting control. A decrease in SOX9 expression significantly reduced MPNST cell survival (Fig 4F), correlating with a decrease in cell accumulation (Fig 4G) and an increase in cell death (Fig 4H); shRNAs directed towards five other genes (data not shown) did not affect MPNST cell survival. Similar results were obtained in two independent MPNST cell lines. Reducing SOX9 expression with shRNA to a level obtained in MPNST cells (Fig 4A) had a milder effect on NFSC survival (Fig 4B) and little or no effect on normal Schwann cells (data not shown). The reduced effect of SOX9 expression in NFs is not solely a function of the passage number, since NFSCs expressing SOX9 shRNAs for several passages showed only a 10–20% reduction in growth rate. These data suggest that MPNST cells, not NF cells, are critically dependent upon SOX9 expression for survival.
Figure 4. Reducing SOX9 expression in NF1 tumour cells inhibits survival.
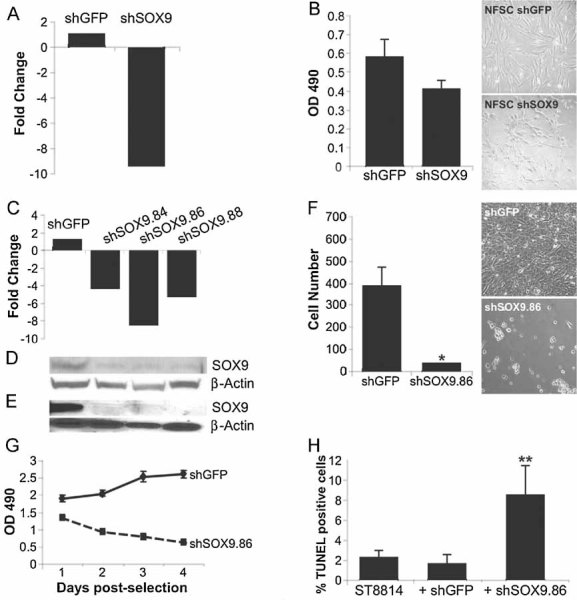
A. Confirmation of reduction in SOX9 RNA expression in NFSCs.
B. NFSCs infected with lentivirus expressing SOX9 shRNA or non-specific shGFP control were plated 7 days post-selection in puromycin to measure cell survival in 4 days using an MTS assay. A trend towards a decrease in cell survival was observed in shSOX9-expressing cells compared to shGFP-expressing cells but was not statistically significant. The corresponding phase contrast images to the right show NFSCs infected with shGFP (NFSC shGFP) or SOX9 shRNA (NFSC shSOX9).
C–E. Confirmation of reduction in SOX9 RNA expression in MPNST cells by qPCR (c) and SOX9 protein expression by western blot (d, e). Expression levels were measured 1 day (d) or 7 days (c, e) post-selection in puromycin and normalized to the non-specific shGFP control. Expression of β-actin was used as a control.
F. Reduction in MPNST cell number (18-fold) in the presence of SOX9 shRNA relative to shGFP control. Cells were plated in triplicate and represent three independent infections (*p = 0.05). The top right panel represents the confluent dish of MPNST cells infected with shGFP lentiviral particles three days post-selection with puromycin; bottom right panel represents dying MPNST cells infected with shSOX9 lentiviral particles three days post-selection with puromycin. Similar results were observed with three different SOX9 shRNAs.
G. MPNST cells were infected with shGFP or shSOX9 lentiviral particles for MTS analysis of cell accumulation during a time course of 1–4 days post-selection in puromycin. Cells were plated in triplicate in the presence or absence of puromycin to account for infection efficiency. All shRNAs infect with similar efficiency at greater than 90%. Values are corrected for infection efficiency and represent three independent infections.
H. MPNST cells treated as described in (c–e) and assayed for apoptosis by TUNEL staining three days post-selection in puromycin. Uninfected and shGFP-infected cells have similar numbers of TUNEL-positive cells with a significant increase in TUNEL-positive cells in the presence of shSOX9 (**p = 0.002).
Transcripts differentially expressed in NF1 tumour cell cultures and solid tumours are enriched for SOX9 target genes
SOX9 transcriptional targets described in the literature for other cell types were not significantly altered in our data set (Panda et al, 2001). However, an analysis of SOX9 binding sites (Mertin et al, 1999) in the promoters of 1,108 unique genes in the NF1 tumour signature revealed >2× potential SOX9 targets relative to the entire genome than expected by chance (39 transcripts as compared to the predicted 18, Fisher's exact test, p = 8.45−06; Table S8 of Supporting Information). To provide preliminary evidence supporting predicted SOX9 transcriptional targets, we analysed the expression of six putative target genes in MPNST cells expressing three independent shSOX9 RNAs as compared to MPNST cells expressing control shRNA. Expression of four out of six (EYA4, DPYSL3, GFRA1 and FOSL2) genes tested were significantly changed in response to reduction in SOX9 expression (Table S9a of Supporting Information). In contrast, neural crest and Schwann cell differentiation markers (TWIST1, SOX10, PMP22 and NGFR) were not significantly changed by SOX9 reduction (data not shown). To test whether increase in SOX9 can affect target gene expression, we infected NHSCs and NFSCs with an adenovirus encoding mouse Sox9 and analysed the expression of the same six putative target genes. The expression of 5/6 genes were significantly altered in NHSCs and 4/6 in NFSCs implying that Sox9 is sufficient to, directly or indirectly, regulate these genes (Table S9b of Supporting Information). Similar to what we observed in MPNST cells expressing shSOX9 RNA, FOSL2, DPYSL3, EYA4 and CHD13 expression were each altered in normal and NFSCs by increased Sox9 expression.
DISCUSSION
We provide a wealth of high-quality, comprehensive data for the NF1 research community. Embedded within the gene signatures of NFs and MPNSTs lie potential biomarkers and molecular targets for future therapeutic development in NF1, including SOX9. Comparing gene expression profiles of NF1 tumour samples to cultured primary Schwann cells resulted in a 1,108 unique gene signature distinguishing NF1 tumour samples from normal Schwann cells. Identifying gene expression differences in cultured MPNST cell lines, NFSCs and normal Schwann cells avoided the complex heterogeneity inherent to solid tumours; using the cell culture gene signature to identify common gene expression patterns, in the solid tumours, which allowed us to identify the Schwann cell component.
The data did not allow us to directly evaluate differences between tumour types that are independent of Schwann cells (e.g. microvasculature, additional cell types). We did observe differences in gene expression between cell cultures and their respective solid tumour type, which could represent the influence of the tumour microenvironment. We anticipate that future studies will use this data set to dissect the molecular contribution of other NF-derived cells, including fibroblasts, mast cells and endothelial cells. This will be important as NF1 +/− mast cells and fibroblasts show NF-relevant properties (reviewed in Cichowski & Jacks, 2001; Le & Parada, 2007) and, in some mouse models of NF1, an Nf1+/− background enhanced tumour development when Nf1 was ablated in Schwann cells (Zhu et al, 2002).
Surprisingly, cluster analysis of global gene expression microarray data did not distinguish dermal and plexiform NFs, although MPNSTs normally arise only from plexiform NFs. Dermal and plexiform NF cells may be inherently the same but exposed to different environments, as the dermal NF is typically cutaneous and the plexiform NF sub-cutaneous. Another possibility is that different molecular alterations initiate the formation of dermal or plexiform NFs but converge on a common molecular pathway. It is also possible that small populations of tumour stem cells with the potential for malignant transformation exist within a plexiform NF tumour cell sub-population but that gene expression within this small subset would not be identified by our analysis.
Two distinct classes of NF samples were identified by transcriptional profiling, designated Class 1 and Class 2. Each class includes Schwann cells from dermal and plexiform NFs. There was no clear patient phenotype associated with these classes, although we recognize that more precise patient information and larger series of cases may reveal a possible correlation. Class 1 transcriptional patterns are more similar to NHSCs than Class 2 transcriptional patterns, and both classes were represented in NFSCs in vitro. A small number of primary tumours also showed a Class 1 pattern, indicating a possible relevance to a specific stage in tumour growth or differentiation that is maintained in vitro; the majority of primary tumours show a Class 2 pattern.
Of interest are genes that are amplified in array CGH but which we found to be decreased in expression, such as the SOX10 transcription factor (Mantripragada et al, 2008). Silencing of these genes may be relevant to tumour progression. SOX10 is expressed throughout the Schwann cell lineage in neural crest cells and mature Schwann cells (Kuhlbrodt et al, 1998). SOX10 normally promotes cell survival in the neural crest, specification in Schwann cell progenitors and myelin production in mature Schwann cells (Schreiner et al, 2007). Sox10-deficient mice lack glial cells in the peripheral nervous system (Britsch et al, 2001). SOX10 activates transcription of myelin genes, including MPZ (Peirano et al, 2000) and MBP (Wei et al, 2004), also down-regulated in NF1 tumours. While SOX8 can compensate for lack of SOX10 function (Kellerer et al, 2006), SOX8 expression is down-regulated in NF1 tumours. Reduced expression of SOX10 in NF1 tumours suggests that the decreased expression may be necessary for tumour formation. Low SOX10 expression in tumour Schwann cells is also consistent with failure of complete differentiation, based on our finding that gene expression profiles of NF1 tumours down-regulate expression of Schwann cell progenitor and immature Schwann cell genes.
Miller et al (Miller et al, 2006), using the strategy applied here (e.g. Schwann cell referencing) but using chips with lower number of probe sets as compared to those studied here, identified 162 probe sets differently expressed in MPNST cell lines and MPNST tumours compared to normal Schwann cells. Seventy-two unique transcripts overlap with our data set. Sixteen are developmental genes, including TWIST1, SOX9, CUGBP2, FEZ1, GAP43, GAS7, GPM6B, GPR56, LICAM, NGFR, NRP2, PMP22, QKI, SEMA3B, SOX10 and ZFHX1B. In studies that use different referencing strategies and technologies, among 28 genes defined as notably altered in expression 10 met our statistical criteria (FDR ≤ 0.001) and were changed relative to a Schwann cell reference (Holtkamp et al, 2004; Karube et al, 2006; Levy et al, 2004). Among the 10, the developmentally regulated genes DHH, ERBB3, MPZ, S100β, L1CAM and SOX10 are all down-regulated with respect to normal Schwann cells.
Network analysis of the genes that distinguish NF-related cells and tumours from Schwann cells did not reveal perturbation of an obviously known molecular pathway. We expected to identify gene expression changes downstream of Ras and EGFR, because loss of NF1 causes hyperactivity of Ras signalling (Le & Parada, 2007) and EGFR is abnormally expressed in NFs and MPNSTs (DeClue et al, 2000; Perry et al, 2002). It remains possible that transcriptional targets directly downstream of Ras and EGFR in Schwann cells are present in our data set but different from those that have been described in other cell types, or that their expression changes were not sufficient to pass our cut-off criteria, but nonetheless represent biologically significant changes.
Comparison of the NF-related tumour gene expression profile to Schwann cell gene expression throughout development (Buchstaller et al, 2004) indicated that NF cells fail to express genes characteristic of immature Schwann cells and MPNST cells express genes characteristic of more primitive neural crest cells. Our study describes similarity of the differentiation state, but not the cell of origin of NFs or MPNST. De-differentiation of a mature Schwann cell to a Schwann cell precursor-like phenotype cannot be excluded from our data. It is worth mentioning that targeting Nf1 loss in a post-neural crest compartment allowed for mouse NF formation (Wu et al, 2008). Losing the NF1 gene in a neural crest cell, inherently proliferative and migratory, may produce a malignant cellular phenotype (Fig 5). Joseph et al (2008) recently showed that neural crest stem cells do not persist in Nf1; p53 mutant mice; MPNST cells derived from these mice exhibit only some neural crest-like features (Joseph et al, 2008). On the basis of their findings, it may be more likely that MPNSTs form through de-differentiation of the NFSC or Schwann cell precursor, which is itself multipotent (Joseph et al, 2008), to a more primitive, neural crest-like stage.
Figure 5. Model of NF1 peripheral nerve tumour formation.
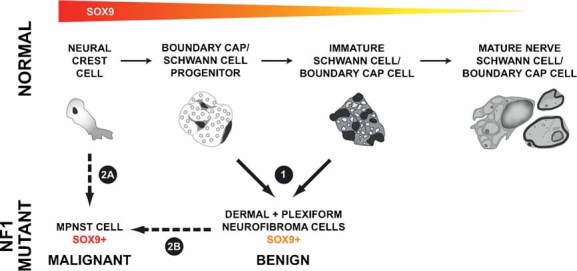
SOX9 expression throughout the Schwann cell lineage is inferred from work in species other than human. Red represents high SOX9 expression, yellow low SOX9 expression. (1) dNF and pNF Schwann cells express intermediate levels of SOX9 and show gene signature characteristic of Schwann cell progenitors/immature Schwann cells. (2A) A neural crest gene signature is characteristic of MPNST cells, and neural crest cells are known to express higher levels of SOX9 than more mature cells. (2A, 2B) Neural crest cells may give rise directly to MPNST, or MPNST may form indirectly via a NF-like cell intermediate.
Consistent with MPNST and NF resembling different stages in Schwann cell development, neural crest transcription factors TWIST1 and SOX9 are expressed at higher levels in MPNST than NFSCs and reducing SOX9 expression kills MPNST cells but has a minimal effect on NF cell survival in vitro. SOX9 in the nucleus of MPNST cells relative to NRSCs is consistent with higher SOX9 transcriptional activity in MPNST cells. SOX9 expression is also up-regulated in neural crest-derived pheochromocytomas that arise in Nf1 mutant mice (Powers et al, 2007). Sox9 may be generally important in stem cells, as it regulates specification of epithelial stem cells (Nowak et al, 2008); in mice lacking Sox9, neural crest cells apoptose (Cheung et al, 2005). High levels of SOX9 expression in MPNSTs appear to result in SOX9 ‘addiction’, consistent with SOX9 being a lineage-survival oncogene in this system (Garraway & Sellers, 2006). Increased levels of SOX9 may not be sufficient to drive proliferation characteristic of MPNSTs, as in preliminary experiments in which we expressed SOX9 in plexiform NFSCs, we observed no change in cell proliferation or cell death (AH and NR, unpublished). Analysis of somatic genetic alterations of SOX9 in tumours and of effects of SOX9 on in vivo tumourigenesis will be necessary to provide definitive evidence that SOX9 is a lineage addicting/survival oncogene. In summary, SOX9 expression provides a biomarker of NFs and MPNSTs. Developing therapeutics aimed at diminishing SOX9 expression or SOX9 transcriptional targets represents a strategy for killing MPNST cells.
MATERIALS AND METHODS
NF acquisition
Diagnosis of NF1 used published criteria (Gutmann et al, 1997). We snap froze tumour tissues at the time of surgery from patients undergoing treatment at Massachusetts General Hospital. Tissues were obtained from the Neuro-Oncology Tissue Repository at Massachusetts General Hospital in accordance with IRB-approved protocols. We obtained blocks of corresponding paraffin-embedded tumours from MGH-pathology files. Histological review of all specimens was performed by a neuropathologist (ASR) and tumours were classified and graded using current WHO classification. For Schwann cell isolation, we froze tumour in 50% Dulbecco's modified Eagle's medium (DMEM), 40% foetal bovine serum (FBS), 10% dimethyl sulphoxide (DMSO) or directly cultured cells. Live purified Schwann cell cultures were shipped to Cincinnati Children's Hospital, incubated in fresh media until 70–80% confluent and cells flash-frozen prior to RNA isolation.
Schwann cell isolation
We generated NHSCs from trauma victims (Casella et al, 1996), and Schwann cells from dermal NFs as described previously (Serra et al, 2000). After mechanical trituration and digestion with enzymes, we resuspended cells in Schwann cell media (SCM/DMEM supplemented with 10% FBS, antibiotics, 0.5 mM IBMX, 10 nM β-heregulin, 0.5 µM forskolin and 2.5 µg/ml insulin) and seeded onto poly-l-lysine and laminin coated plates. For NF1+/− Schwann cells, we kept forskolin constant. For NF1−/− cells, we provided forskolin in pulses of 1 day every 3–4 days in SCM-no forskolin. We assessed the purity of Schwann cell cultures by immunofluorescent staining with an S-100β antibody (Serra et al, 2000). After 3–5 passages, >95% pure Schwann cell cultures were obtained. When a somatic mutation of a tumour was identified, we assessed purity according to genotype (Table S10 of Supporting Information). In NF1−/− Schwann cell cultures, complete loss of heterozygosity (LOH) was observed. We generated plexiform NFSC cultures, 70–95% S100β-positive, as described previously (Wallace et al, 2000). We maintained cells in media lacking forskolin to enrich for NF1−/− cells, and harvested for RNA after 4–6 passages.
NF1 mutation analysis
We carried out mutational analysis of NF1 in Schwann cells from dermal and plexiform NFs and in solid tumours as described previously. Additional details are provided in Table S10 of Supporting Information (Ars et al, 2000; Serra et al, 2001; Upadhyaya et al, 2006).
MPNST cell cultures
We maintained MPNST cell lines ST88-14, STS26T, S520, S462, 88-3, 90-8 and YST1 as described previously (Miller et al, 2006). We isolated RNA from cells under standard growth conditions for each cell line.
RNA isolation and microarray hybridization
We isolated total RNA from frozen tissue and cells using the RNeasy kit (Qiagen) and verified RNA integrity with an Agilent Bioanalyser 2100 (with typical 28S/18S ratios = 2 ± 0.1). We generated biotinylated cDNA probes from a single round of DNA displacement synthesis amplification of 20 ng total RNA (Ovation Biotin RNA Amplification and Labelling System; NuGen) for hybridization to the whole-genome Affymetrix GeneChip HU133 Plus 2.0 using the Affymetrix protocol. We used an Affymetrix Gene Array scanner and GeneChip Operating Software V1.4 to scan and quantify GeneChips using default settings. We processed 86 samples in 9 batches, including: 9 universal tissue reference samples, 10 NHSC samples, 11 dNFSC samples, 11 pNFSC samples, 13 MPNST cell line samples, 13 dNF, 13 pNF and 6 MPNSTs. We used Affymetrix Microarray Suite 5.0 to generate ‘CEL’ files for each sample that were normalized using the Robust Multichip Analysis (RMA) algorithm as implemented in Bioconductor/R (Irizarry et al, 2003).
Microarray data quality control and power analysis
We assessed array images for spatial defect using the GEODEX (Kim et al, 2006) and a deleted residuals approach (Persson et al, 2005). Chips with high spatial artefacts or large deviations from outliers (n = 2) were discarded. One normalization control sample failed quality control and we re-ran the entire batch of samples.
Data analysis strategy
We used a custom GeneChip library file (CDF) based on Refseq target definitions (Hs133P REFSEQ Version 8) to provide accurate interpretation of GeneChip data (Dai et al, 2005). To identify gene expression changes relative to NHSCs, we applied a two-stage referencing strategy: we set the normalized gene expression level value for each transcript in each sample to its ratio relative to the expression of that transcript's measurement in the universal tissue reference; then, we set the normalized gene expression level value for each transcript in each sample to its ratio relative to the median expression of that transcript's measurements across NHSCs. An ANOVA (p ≤ 0.001) was used to compare NHSCs, dNFSCs, pNFSCs and MPNST cell lines. Statistical comparisons and data visualization were performed using GeneSpring GX v7.3.1 (Agilent Technologies). We corrected results from the primary analysis for multiple testing effects by applying the Benjamini and Hochberg false discovery rate correction (Benjamini et al, 2001) (FDR ≤ 0.001).
Transcripts were assigned to empirically defined expression patterns using the k-means clustering algorithm, which generally finds a clustering solution with a smaller within-cluster sum of distances (meaning a more homogeneous cluster) than hierarchical clustering techniques. We used Pearson correlation as a similarity measure and iteratively clustered genes until convergence, then tested an additional five random clusters to ensure that the optimal clustering solution had been identified. To identify genes sharing common gene expression patterns in cultured cells and solid tumours, we re-clustered each cluster across NHSCs, NFs and MPNSTs. We identified three principle patterns of gene expression and genes were assigned to each using k-means clustering. We pooled sub-clusters displaying a similar pattern of gene expression in NFs and MPNSTs as the original cultured cell cluster with all other similarly expressed sub-clusters. A total of 1,708 transcripts were obtained that were subsequently used to generate a heat map including the entire sample set.
We identified statistically over-represented Gene Ontologies and both KEGG and BioCarta pathways (p < 0.05) using the Database for Annotation, Visualization and Integrated Discovery (DAVID) 2007 at the National Institute of Allergy and Infectious Diseases (NIAID) (Huang da et al, 2007). We used the Gene Association to Anatomic and Clinical Abnormalities (GATACA) web server to select genes based upon association with nerve development, NF1 or MPNST.
Schwann cell development orthologues comparison
We used a data set in which Schwann cells were isolated from mouse embryos using PLP promoter-driven GFP at various stages of development (Buchstaller et al, 2004), and re-analysed the files processed by the GEO database-deposited Microarray Suite (Affymetrix). Probe sets were normalized per chip by a median intensity distribution and then referenced to the median of each probeset across the 19 microarrays in the data set. Probe sets were filtered for those identified by Microarray Suite as ‘Present’ in ≥2 of the 19 arrays, resulting in 8,617 probe sets. An ANOVA (p ≤ 0.2) was used to identify the differential expression between E9, E12, E14, E16, E18 and P0. We corrected the results for multiple testing effects by applying the Benjamini and Hochberg false discovery rate correction (FDR ≤ 0.2). The 4,750 identified probe sets were subjected to hierarchical tree clustering using Pearson correlation similarity measure for genes (average linkage) and distance correlation similarity measure for samples (average linkage). Three major clusters were identified with dominant expression at E9 (neural crest), E12–E14 (Schwann cell precursor) and E18–P0 (immature Schwann cell) (Fig S1 of Supporting Information). We eliminated redundant probe sets, lack of corresponding gene orthologues and platform-specific gene representation differences using orthologue gene mapping, and then tabulated the overlap between each developmental signature and each cluster from the NF1 tumour signature. Contingency tables were constructed and we performed Fisher's exact test with R to identify developmental signatures that were statistically significant.
The paper explained
PROBLEM:
Neurofibromatosis type 1 (NF1) is one of the most common inherited human diseases worldwide. Effective therapies are lacking. Peripheral nerve sheath tumours (neurofibromas) are the hallmark of NF1. They consist of benign dermal and plexiform subtypes, but plexiform neurofibroma can transform to malignant peripheral nerve sheath tumour (MPNST), a highly aggressive, life-threatening sarcoma. Neurofibromas are composed of multiple cell types, including Schwann cells, the pathogenic cell type of NF1. The molecular changes that drive tumourigenesis are largely unknown.
RESULTS:
The authors used DNA microarrays to profile the gene expression in normal Schwann cells, Schwann cells cultured from primary benign neurofibromas (dermal and plexiform subtypes), MPNST cell lines and solid tumours. It was found that neurofibromas repress gene programmes normally expressed in late-developing immature Schwann cells, while MPNSTs activate gene programmes normally expressed earlier in the development at the neural crest stage. Strong expression of the transcription factor SOX9 is seen in neurofibroma and MPNST tissue sections, while schwannomas show weak or absent expression. Synovial sarcomas, which may histologically mimic MPNST, are mainly negative. Reduction of SOX9 expression in MPNST cell lines causes cell death.
IMPACT:
SOX9 expression provides a biomarker of neurofibroma and MPNST. Therapeutics aimed at decreasing SOX9 expression or SOX9 transcriptional targets represent a strategy for killing MPNST cells.
SOX9 target gene identification
We used Genome TraFac3 for genome-wide detection of compositionally similar cis-clusters in gene orthologues between mouse and human to determine putative SOX9 target genes, identifying SOX9 cis-elements within orthologue-conserved regions (≥70% sequence similarity based on BlastZ alignment) upstream of each first exon in 2,000 flanking basepairs (Jegga et al, 2007). We identified 394 unique genes containing potential SOX9 binding sites and transcripts dysregulated in NF-related cultures and tumours. We used Fisher's exact test to calculate the statistical significance of over-represented SOX9 target genes in the gene list.
qPCR
We conducted cDNA synthesis (Invitrogen Superscript II) and qPCR (ABI 7500 Sequence Detection System) as described previously (Miller et al, 2006). Primer sequences for target validation are shown in Table S11 of Supporting Information. For technical validation of microarray data, a result of ≥ ± 2-fold in the same direction was considered as confirmation compared to expression of β-actin (Table S7 of Supporting Information).
Western blot analysis
We created cell lysates and conducted Western blotting as described previously (Miller et al, 2006), probed membranes with anti-SOX9 (Santa Cruz Biotechnology, Santa Cruz, CA; 1:700), then re-probed with anti-β-actin (Cell Signaling Technology, Inc. #4967) as a loading control. We detected signals using horseradish peroxidase-conjugated secondary antibodies (BioRad; Hercules, CA) and the ECL Plus developing system (Amersham Biosciences; Piscataway, NJ).
Immunohistochemistry
We performed SOX9 immunohistochemistry on formalin-fixed, paraffin-embedded sections with polyclonal anti-SOX9 antibody (Abcam, Cambridge, UK). Antigen retrieval was achieved with microwaving in sodium citrate (pH 6), followed by incubation with the primary antibody overnight at 4°C (1:75) and visualization with an avidin–biotin complex (Vectastatin Elite ABC kit; Vector Laboratories, Burlingame, CA) and with 3,3'-diaminobenzidine tetrahydrochloride (Vector Laboratories, Burlingame, CA). We scored SOX9 immunostaining semi-quantitatively for the number of positive cells, as the staining intensity was similar in all samples (strong). The number of positive cells was scored as <25% (+1, some), 25–75% (+2, many) and >75% (+3, maximum). Immunofluorescent detection of SOX9 in cultured cells was conducted using a 1:200 dilution of primary anti-SOX9 antibody followed by a TRITC-conjugated anti-rabbit secondary antibody and 4'-6-diamidino-2-phenylindole (DAPI) visualization of nuclei.
Viral infection
For lentiviral shRNA infection, we infected MPNST cells at 70–90% confluence with lentiviral particles containing shRNAs targeting SOX9 (Open Biosystems; TRC library) or GFP (Addgene). The CCHMC Viral Vector Core4 produced virus using a four plasmid packaging system. We incubated lentiviral particles with the MPNST cells (MOI ∼ 10) in the presence of polybrene (8 µg/ml; Sigma) for 24 h followed by selection in puromycin at a concentration (2 ug/ml) that killed uninfected cells within 3 days. For SOX9 and GFP adenoviral infections (Paul et al, 2003), we incubated viral particles with NHSCs or NFSCs (MOI = 300–500) for 2 h, replaced the growth medium and harvested cells for RNA isolation 48 or 120 h post-infection.
MTS assay
We plated MPNST cells (4 × 103) in triplicate and incubated overnight in a 96-well plate for infection with shGFP or shSOX9 lentiviral particles as described previously. Cells were maintained in the presence or absence of puromycin (2µg/ml) to account for infection efficiency. We conducted MTS assays for viable cell number on days 3–7 post-infection using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega).
Terminal deoxynucleotidyl transferase biotin-dUTP nick end labelling (TUNEL) staining for cell death
We plated MPNST cells (2 × 104) in quadruplicate wells of LabTek chamber slides (Nalge-Nunce International), and then infected them with shSOX9 or shGFP lentiviral particles. After 3 days in the presence or absence of puromycin, we fixed cells in 4% paraformaldehyde for the detection of nuclear DNA fragmentation with the DeadEnd Fluorometric TUNEL System (Promega; Madison, WI). One well without terminal deoxynucleotidyl transferase was used as the negative control. We counted cells in five random fields in triplicate wells. We expressed Fluorescein isothiocyanate (FITC) positive apoptotic cells as a percentage of total cells visualized by DAPI.
Author contributions
Shyra J. Miller helped conceive the project, collected samples, wrote portions of the manuscript and carried out experiments on SOX9 with Atira Hardiman. Ms. Hardiman also purified all the RNAs for the analysis and did all qPCR confirmation. Emily Sites assisted in SOX9 validation experiments. Walter J. Jessen compiled the transcriptional data and carried out the gene expression/bioinformatic analyses, and wrote portions of the manuscript. Sergio Kaiser compiled transcriptional data in the early stages of the project. Bruce J. Aronow oversaw all gene expression/bioinformatic analyses and contributed intellectually to development of the manuscript. Tapan Mehta carried out the futility analysis, under the supervision of Grier Page. David Muir and Margaret R. Wallace oversaw Hua Li who provided the plexiform neurofibroma Schwann cells. Eduard Serra, Eva Lopez and Conxi Lazaro genotyped and provided dermal neurofibroma Schwann cells. Anat Stemmer-Rachamimov provided solid tumor samples and performed SOX9 immunohistochemistry and pathological evaluation of neurofibroma, MPNST, and schwannoma. Marco Giovannini took part in data review. G. Petur Nielsen provided pathological evaluation of synovial sarcoma tissue sections. Meena Upadhyaya carried out NF1 genotyping on solid tumors and plexiform neurofibroma cells. Anil Jegga carried out promoter binding site analysis for SOX9 target genes. Nancy Ratner developed the NF1 Microarray consortium and oversaw all experiments and analyses, and manuscript preparation.
Acknowledgments
We gratefully acknowledge Dr Patrick Wood of the Miami Project to Cure Paralysis for providing human Schwann cells obtained from the cauda equina of organ donors. The cauda equine were provided by the University of Miami Life Alliance Organ Procurement Agency (Dr David Levi, Director). Cincinnati Children's Hospital Affymetrix GeneChip Core (Dr Steve Potter, Sean Smith and Hung-Chi Liang) performed microarray hybridization. We thank Drs Larry Gelbert (Eli Lilly) and Chris Roberts (Rosetta) for extensive input into the development and progress of the NF1 microarray consortium. This study was supported by the DAMD (DOD W81XWH-04-1-0273). MRW was supported by the Hayward Foundation. SM was the recipient of NINDS Translational Neuroscience Award K01-NS049191 and ES was supported by T32 CA 59268.
Supporting Information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Footnotes
For more information
OMIM, Online Mendelian Inheritance in Man:
Neurofibromatosis Type I; NF1:
http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=162200
Children's Tumor Foundation:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, Estivill X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000;9:237–247. doi: 10.1093/hmg/9.2.237. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, Wegner M. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15:66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchstaller J, Sommer L, Bodmer M, Hoffmann R, Suter U, Mantei N. Efficient isolation and gene expression profiling of small numbers of neural crest stem cells and developing Schwann cells. J Neurosci. 2004;24:2357–2365. doi: 10.1523/JNEUROSCI.4083-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590–1605. doi: 10.1002/glia.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella GT, Bunge RP, Wood PM. Improved method for harvesting human Schwann cells from mature peripheral nerve and expansion in vitro. Glia. 1996;17:327–338. doi: 10.1002/(SICI)1098-1136(199608)17:4<327::AID-GLIA7>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Cheung M, Chaboissier MC, Mynett A, Hirst E, Schedl A, Briscoe J. The transcriptional control of trunk neural crest induction, survival, and delamination. Dev Cell. 2005;8:179–192. doi: 10.1016/j.devcel.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeClue JE, Heffelfinger S, Benvenuto G, Ling B, Li S, Rui W, Vass WC, Viskochil D, Ratner N. Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J Clin Invest. 2000;105:1233–1241. doi: 10.1172/JCI7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–314. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, Rubenstein A, Viskochil D. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–57. [PubMed] [Google Scholar]
- Holtkamp N, Reuss DE, Atallah I, Kuban RJ, Hartmann C, Mautner VF, Frahm S, Friedrich RE, Algermissen B, Pham VA, et al. Subclassification of nerve sheath tumors by gene expression profiling. Brain Pathol. 2004;14:258–264. doi: 10.1111/j.1750-3639.2004.tb00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jegga AG, Chen J, Gowrisankar S, Deshmukh MA, Gudivada R, Kong S, Kaimal V, Aronow BJ. GenomeTrafac: a whole genome resource for the detection of transcription factor binding site clusters associated with conventional and microRNA encoding genes conserved between mouse and human gene orthologs. Nucleic Acids Res. 2007;35:D116–D121. doi: 10.1093/nar/gkl1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Joseph NM, Mosher JT, Buchstaller J, Snider P, McKeever PE, Lim M, Conway SJ, Parada LF, Zhu Y, Morrison SJ. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell. 2008;13:129–140. doi: 10.1016/j.ccr.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karube K, Nabeshima K, Ishiguro M, Harada M, Iwasaki H. cDNA microarray analysis of cancer associated gene expression profiles in malignant peripheral nerve sheath tumours. J Clin Pathol. 2006;59:160–165. doi: 10.1136/jcp.2004.023598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellerer S, Schreiner S, Stolt CC, Scholz S, Bosl MR, Wegner M. Replacement of the Sox10 transcription factor by Sox8 reveals incomplete functional equivalence. Development. 2006;133:2875–2886. doi: 10.1242/dev.02477. [DOI] [PubMed] [Google Scholar]
- Kim K, Page GP, Beasley TM, Barnes S, Scheirer KE, Allison DB. A proposed metric for assessing the measurement quality of individual microarrays. BMC Bioinformatics. 2006;7:35. doi: 10.1186/1471-2105-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, Wegner M. Sox10, a novel transcriptional modulator in glial cells. J Neurosci. 1998;18:237–250. doi: 10.1523/JNEUROSCI.18-01-00237.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene. 2007;26:4609–4616. doi: 10.1038/sj.onc.1210261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4:453–463. doi: 10.1016/j.stem.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy P, Bieche I, Leroy K, Parfait B, Wechsler J, Laurendeau I, Wolkenstein P, Vidaud M, Vidaud D. Molecular profiles of neurofibromatosis type 1-associated plexiform neurofibromas: identification of a gene expression signature of poor prognosis. Clin Cancer Res. 2004;10:3763–3771. doi: 10.1158/1078-0432.CCR-03-0712. [DOI] [PubMed] [Google Scholar]
- Levy P, Ripoche H, Laurendeau I, Lazar V, Ortonne N, Parfait B, Leroy K, Wechsler J, Salmon I, Wolkenstein P, et al. Microarray-based identification of tenascin C and tenascin XB, genes possibly involved in tumorigenesis associated with neurofibromatosis type 1. Clin Cancer Res. 2007;13:398–407. doi: 10.1158/1078-0432.CCR-06-0182. [DOI] [PubMed] [Google Scholar]
- Mantripragada KK, Spurlock G, Kluwe L, Chuzhanova N, Ferner RE, Frayling IM, Dumanski JP, Guha A, Mautner V, Upadhyaya M. High-resolution DNA copy number profiling of malignant peripheral nerve sheath tumors using targeted microarray-based comparative genomic hybridization. Clin Cancer Res. 2008;14:1015–1024. doi: 10.1158/1078-0432.CCR-07-1305. [DOI] [PubMed] [Google Scholar]
- Mertin S, McDowall SG, Harley VR. The DNA-binding specificity of SOX9 and other SOX proteins. Nucleic Acids Res. 1999;27:1359–1364. doi: 10.1093/nar/27.5.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SJ, Li H, Rizvi TA, Huang Y, Johansson G, Bowersock J, Sidani A, Vitullo J, Vogel K, Parysek LM, et al. Brain lipid binding protein in axon-Schwann cell interactions and peripheral nerve tumorigenesis. Mol Cell Biol. 2003;23:2213–2224. doi: 10.1128/MCB.23.6.2213-2224.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SJ, Rangwala F, Williams J, Ackerman P, Kong S, Jegga AG, Kaiser S, Aronow BJ, Frahm S, Kluwe L, et al. Large-scale molecular comparison of human schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res. 2006;66:2584–2591. doi: 10.1158/0008-5472.CAN-05-3330. [DOI] [PubMed] [Google Scholar]
- Nowak JA, Polak L, Pasolli HA, Fuchs E. Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell. 2008;3:33–43. doi: 10.1016/j.stem.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page GP, Edwards JW, Gadbury GL, Yelisetti P, Wang J, Trivedi P, Allison DB. The PowerAtlas: a power and sample size atlas for microarray experimental design and research. BMC Bioinformatics. 2006;7:84. doi: 10.1186/1471-2105-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda DK, Miao D, Lefebvre V, Hendy GN, Goltzman D. The transcription factor SOX9 regulates cell cycle and differentiation genes in chondrocytic CFK2 cells. J Biol Chem. 2001;276:41229–41236. doi: 10.1074/jbc.M104231200. [DOI] [PubMed] [Google Scholar]
- Paul R, Haydon RC, Cheng H, Ishikawa A, Nenadovich N, Jiang W, Zhou L, Breyer B, Feng T, Gupta P, et al. Potential use of Sox9 gene therapy for intervertebral degenerative disc disease. Spine. 2003;28:755–763. [PMC free article] [PubMed] [Google Scholar]
- Peirano RI, Goerich DE, Riethmacher D, Wegner M. Protein zero gene expression is regulated by the glial transcription factor Sox10. Mol Cell Biol. 2000;20:3198–3209. doi: 10.1128/mcb.20.9.3198-3209.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry A, Kunz SN, Fuller CE, Banerjee R, Marley EF, Liapis H, Watson MA, Gutmann DH. Differential NF1, p16, and EGFR patterns by interphase cytogenetics (FISH) in malignant peripheral nerve sheath tumor (MPNST) and morphologically similar spindle cell neoplasms. J Neuropathol Exp Neurol. 2002;61:702–709. doi: 10.1093/jnen/61.8.702. [DOI] [PubMed] [Google Scholar]
- Persson S, Wei H, Milne J, Page GP, Somerville CR. Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. Proc Natl Acad Sci USA. 2005;102:8633–8638. doi: 10.1073/pnas.0503392102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers JF, Evinger MJ, Zhi J, Picard KL, Tischler AS. Pheochromocytomas in Nf1 knockout mice express a neural progenitor gene expression profile. Neuroscience. 2007;147:928–937. doi: 10.1016/j.neuroscience.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33–40. doi: 10.1093/oxfordjournals.aje.a010118. [DOI] [PubMed] [Google Scholar]
- Schreiner S, Cossais F, Fischer K, Scholz S, Bosl MR, Holtmann B, Sendtner M, Wegner M. Hypomorphic Sox10 alleles reveal novel protein functions and unravel developmental differences in glial lineages. Development. 2007;134:3271–3281. doi: 10.1242/dev.003350. [DOI] [PubMed] [Google Scholar]
- Serra E, Rosenbaum T, Nadal M, Winner U, Ars E, Estivill X, Lazaro C. Mitotic recombination effects homozygosity for NF1 germline mutations in neurofibromas. Nat Genet. 2001;28:294–296. doi: 10.1038/90148. [DOI] [PubMed] [Google Scholar]
- Serra E, Rosenbaum T, Winner U, Aledo R, Ars E, Estivill X, Lenard HG, Lazaro C. Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell subpopulations. Hum Mol Genet. 2000;9:3055–3064. doi: 10.1093/hmg/9.20.3055. [DOI] [PubMed] [Google Scholar]
- Sheela S, Riccardi VM, Ratner N. Angiogenic and invasive properties of neurofibroma Schwann cells. J Cell Biol. 1990;111:645–653. doi: 10.1083/jcb.111.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman LS, Atit R, Rosenbaum T, Cox AD, Ratner N. Single cell Ras-GTP analysis reveals altered Ras activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. J Biol Chem. 2000;275:30740–30745. doi: 10.1074/jbc.M001702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya M, Spurlock G, Majounie E, Griffiths S, Forrester N, Baser M, Huson SM, Gareth Evans D, Ferner R. The heterogeneous nature of germline mutations in NF1 patients with malignant peripheral serve sheath tumours (MPNSTs) Hum Mutat. 2006;27:716. doi: 10.1002/humu.9429. [DOI] [PubMed] [Google Scholar]
- Wallace MR, Rasmussen SA, Lim IT, Gray BA, Zori RT, Muir D. Culture of cytogenetically abnormal schwann cells from benign and malignant NF1 tumors. Genes Chromosomes Cancer. 2000;27:117–123. [PubMed] [Google Scholar]
- Wei Q, Miskimins WK, Miskimins R. Sox10 acts as a tissue-specific transcription factor enhancing activation of the myelin basic protein gene promoter by p27Kip1 and Sp1. J Neurosci Res. 2004;78:796–802. doi: 10.1002/jnr.20342. [DOI] [PubMed] [Google Scholar]
- Williams JP, Wu J, Johansson G, Rizvi TA, Miller SC, Geiger H, Malik P, Li W, Mukouyama YS, Cancelas JA, et al. Nf1 mutation expands an EGFR-dependent peripheral nerve progenitor that confers neurofibroma tumorigenic potential. Cell Stem Cell. 2008;3:658–669. doi: 10.1016/j.stem.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T, Legius E, Callens T, Beiglbock H, Maertens O, et al. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer. 2006;45:265–276. doi: 10.1002/gcc.20289. [DOI] [PubMed] [Google Scholar]
- Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, Stemmer-Rachamimov AO, Cancelas JA, Ratner N. Plexiform and dermal neurofibromas and pigmentation are caused by nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–116. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.