

Abstract
Recent advances in our understanding of the molecular pathways that govern the association of inflammation with organ fibrosis and cancer point to the epithelial to mesenchymal transition (EMT) as the common link in the progression of these devastating diseases. The EMT is a crucial process in the development of different tissues in the embryo and its reactivation in the adult may be regarded as a physiological attempt to control inflammatory responses and to ‘heal’ damaged tissue. However, in pathological contexts such as in tumours or during the development of organ fibrosis, this healing response adopts a sinister nature, steering these diseases towards metastasis and organ failure. Importantly, the chronic inflammatory microenvironment common to fibrotic and cancer cells emerges as a decisive factor in the induction of the pathological EMT.
Keywords: inflammation, epithelial–mesenchymal transition, TGF-β, TNF-α, NF-κB
Introduction—EMT in cancer and fibrosis
Although inflammation has been associated with the progression of chronic kidney disease and cancer for decades, the molecular mechanisms involved have remained elusive until recently (Cordon-Cardo & Prives, 1999; Kalluri & Neilson, 2003; Mantovani et al, 2008). The epithelial to mesenchymal transition (EMT) now takes centre stage as the convergence point between inflammation and the progression of degenerative fibrotic diseases and cancer. This cellular process is characterized by the loss in cell polarity and a change in cell shape from cuboidal to fibroblastoid, the downregulation of epithelial markers and the upregulation of mesenchymal markers. The cell acquires the capacity to degrade the basement membrane and migrate through the extracellular matrix to populate different territories either during embryonic development or cancer progression, or to adopt a profibrotic myofibroblast nature in the renal interstitial space (Acloque et al, 2009; Kalluri & Neilson, 2003; Kalluri & Weinberg, 2009). Betty Hay was the first to coin the term ‘epithelial to mesenchymal transformation’ in embryos (Hay, 1968), as well as later describing this cellular behaviour during migration (Hay, 1990) and the importance of the transient nature of this process (Hay, 1991; Fig 1A). Ironically, important elements in the control of the inflammatory response and in the induction of tumour cell death such as TGF-β1 and hypoxia can also act as potent inducers of EMT in an inflammatory microenvironment.
Figure 1. The history of EMT in cancer.
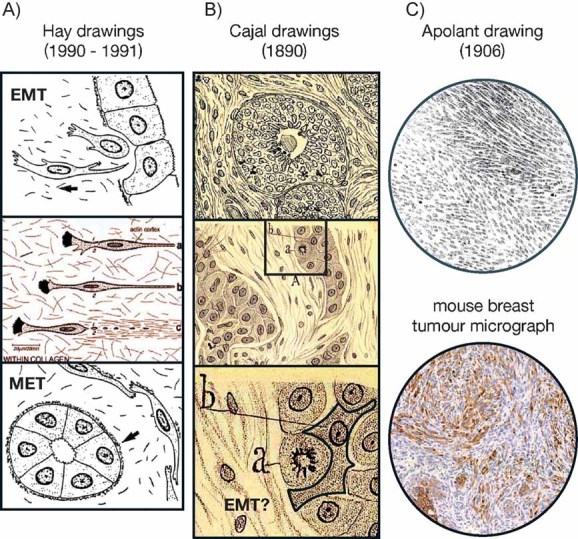
- More than 100 years ago Santiago Ramón y Cajal drew and described the morphological appearance of breast carcinoma so accurately that we can find what we believe to be the first description of EMT (see text). Drawings are adapted from figs. 53 and 48 in Ramon y Cajal (1890). Note the morphology of the cell highlighted as ‘b’.
- At the turn of the 20th century, Hugo Apolant also clearly represented the morphology of EMT-type mouse mammary tumours (Apolant, 1906). The micrograph shows the distribution of cytokeratin 8/18 in both the epithelial and spindle cell populations in a mouse mammary tumour diagnosed as EMT-type. This picture is courtesy of Dr P. Damonte and Dr R. Cardiff, Univ. California at Davis.
EMT events have been very well defined during embryonic development for many years (reviewed in Acloque et al, 2009). However, the significance of the EMT in cancer progression has been debated until recently. The main discussion regarding the role of EMT in cancer came from the difficulty in finding morphological evidence from clinical samples, due to the lack of specific markers for many years and the sometimes focal nature of the event. Nevertheless, more than 100 years ago, both Santiago Ramón y Cajal and Hugo Apolant noticed what we would definitely consider an EMT in both human and mouse breast cancer (Fig 1B,C). Indeed, the description of the breast tumours by Cajal in his ‘Manual of Anatomopathology’ is premonitory of the implication of the EMT as the first step in the metastatic cascade. He referred to undifferentiated breast carcinomas as follows: ‘The epithelial islands are not surrounded by a basement membrane… We shall mention the fusiform, pear-like and star-like forms… The cells are not attached to each other… This explains their invasive ability, since free of intercellular cement, they can migrate through the connective tissue’ (Ramon y Cajal, 1890). When we read ‘cement’ in Cajal's words we immediately think of E-cadherin, the molecule that maintains epithelial cells together and that we now know is the main target repressed by the EMT inducers. Figure 1B and C shows fragments of Cajal's and Apolant's drawings illustrating human and mouse breast carcinomas. The reversibility of the EMT process has been well described in the context of embryonic development, where mesenchymal to epithelial processes (MET) are fundamental for the differentiation of tissues and organs once the embryonic migratory cells have reached their destination. Indeed, the MET events certainly take place during metastasis formation as suggested by cell morphology and the re-expression of E-cadherin expression (Bukholm et al, 2000). This EMT–MET double switch for metastasis formation has been discussed by Brabletz et al (2001) while providing a convincing illustration of an EMT process at the invasive front of human colon carcinoma (Fig 2A).
Figure 2. Histological features of EMT in cancer and fibrosis.
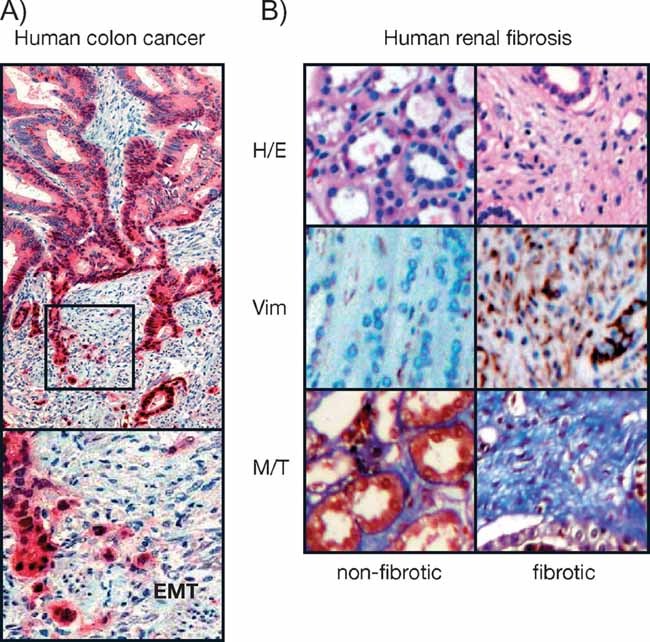
- Immunohistochemical staining of a colorectal carcinoma for β-catenin in red and nuclear counterstaining in blue. The central area of the tumour exhibits polarized epithelial tumour cells lacking nuclear β-catenin, while cells at the invasive front undergo EMT and show nuclear β-catenin (Brabletz et al, 2001). Picture from Dr Thomas Brabletz, Univ. Freiburg (Germany).
- Histological sections showing the halmarks of EMT in the medulla of fibrotic kidney from patients subjected to nephrectomy. Sections are stained with haematoxylin–eosin (H/E) to better appreciate cell morphology and the disappearance of the tubular structures in fibrosis, vimentin expression showing mesenchymal cells (brown, Vim) and fibrotic deposits revealed by the blue Masson–Trichome staining (M/T) (Boutet et al, 2006).
Fibrosis is characterized by the presence of an excess of fibrous connective tissue in an organ, and in particular by an excessive deposition of collagen I. It can be the result of a reparative or reactive process that can occur in the lung, the liver, the heart and the kidney among other organs. Under chronic pathological situations, fibrosis progresses to advanced states that lead to defective organ function and final organ failure. In this review we will focus on renal fibrosis as it has been very well studied at the cellular level and the mechanisms leading to the development of fibrosis are thought to be very similar in different organs. Renal fibrosis is the link between the progressive loss of renal function and primary diseases such as glomerulonephritis, diabetes, toxic injury, congenital abnormalities, urinary tract obstruction and chronic rejection of transplanted kidneys (Kalluri & Neilson, 2003; Liu, 2006; Vongwiwatana et al, 2005). Renal fibrosis has been associated with the activation of interstitial fibroblasts to give rise to collagen secreting myofibroblasts. However, different studies have shown that in addition, myofibroblasts can also originate from renal tubular epithelial and endothelial cells that undergo EMT in mouse models of renal fibrosis (Iwano et al, 2002; Zeisberg et al, 2008). Furthermore, the aberrant activation of Snail1, a well-known EMT inducer leads to the appearance of renal fibrosis and renal failure in transgenic mice (Boutet et al, 2006). Interestingly, high Snail1 expression and evidence of EMT has also been found in the kidneys of patients with renal fibrosis (Boutet et al, 2006; Jinde et al, 2001; Rastaldi et al, 2002).
In this review, we will discuss the dual role of TGF-β1 and hypoxia in fibrotic tissues and in the tumour microenvironment. We propose that in the context of a chronic inflammatory condition, TGF-β1 and hypoxia reactivate EMT developmental programmes that converge in the activation of NF-κB also induced by the inflammatory cytokines and oxidative stress. These EMT programmes, in an unsuccessful attempt to repair the injured tissue, turn to a sinister role and lead to the destruction of epithelial homeostasis and accumulation of extracellular matrix in fibrosis, and to the progression of carcinomas towards the metastatic state.
Glossary
- Acute phase proteins
Proteins whose plasma concentrations increase (positive acute phase proteins) or decrease (negative acute phase proteins) in response to inflammation.
- Angiogenesis
Development of blood vessels in the embryo or in an adult tissue.
- Apoptosis
Cell death characterized by shrinkage of the cell, condensation of chromatin and fragmentation of the cell into membrane-covered bodies that are eliminated by phagocytosis.
- Calculi
Abnormal mineral deposit.
- Cytokines
Intercellular protein mediators released by immune cells to regulate the immune response.
- EMT or the epithelial to mesenchymal transition
The conversion of an epithelial to a mesenchymal cell during embryonic development or in several pathologies.
- Fibrosis
Development of excessive fibrous connective tissue in an organ.
- Hypoxia
Exposure of the tissues to oxygen concentrations lower than normal; Deficiency in the amount of oxygen reaching body tissues.
- Inflammation
A poorly specific immune response of body tissues to infection, irritation or other injuries, characterized by pain, swelling, redness and heat.
- Interstitial fibrosis
Accumulation of excessive fibrous connective tissue between the parenchymal cells of an organ.
- Leukocyte infiltration
Accumulation of blood cells in a tissue or organ.
- Lymphocytes
Nearly colourless white blood cells found in the blood, lymph and lymphoid tissues, involved in humoral and cellular immunity.
- Macrophages
Large phagocytes developed from circulating monocytes that migrate to tissues to eliminate cellular debris and particulate antigens. They produce and respond to inflammatory cytokines.
- Metastasis
The spreading of cancer from one part of the body to another.
- Oxidative stress
Pathologic changes seen in living organisms in response to excessive levels of cytotoxic oxidants and oxygen free radicals.
- Phagocytes
Cells with the ability of engulfing and absorbing waste material, cell debris, microorganisms or other foreign bodies in the bloodstream and tissues.
- Renin–angiotensin system
Endocrine system initiated by the release of the enzyme renin, which acting on the circulating protein angiotensinogen produces the decapeptide Ang I, which is transformed to the active hormone, the octapetide Ang II.
- Stromal cells
Cells found in the loose connective tissue which make up the support structure of the organs and support the parenchymal cells (functional elements of an organ).
- Unilateral ureteral obstruction (UUO)
Acute obstruction of one of the ureters, mimicking the different stages of ON and leading to tubulointerstitial fibrosis.
Inflammation as an inducer of fibrosis and cancer
Inflammation and cancer
The link between cancer and inflammation has been recognized for decades, as strong associations have been established between chronic inflammatory conditions and tumourigenesis (Cordon-Cardo & Prives, 1999). As such, ulcerative colitis, chronic gastritis, hepatitis and chronic pancreatitis, and their respective relationships with colon, gastric, liver and pancreatic carcinomas exemplify the close connection between inflammation and tumour appearance. Furthermore, non-steroidal anti-inflammatory drugs reduce both the risk of cancer development and its mortality (Mantovani et al, 2008).
Renal fibrosis as an inflammatory disease
Renal fibrosis is a paradigmatic example of organ fibrosis towards degenerative organ disease and final stage kidney damage leading to the destruction of the tissue and death from renal failure. Obstructive nephropathy (ON) in humans, either due to congenital or acquired obstruction of the urinary tract, is the first primary cause of chronic renal failure (CRF) in children and a major cause of kidney failure in adults (Klahr & Morrissey, 2002; Smith et al, 2007). In ON, the evolution of renal disease is similar to that occurring in polycystic kidney disease or renal transplant rejection. Moreover, in its final fibrotic phase, ON is also very similar to the fibrosis secondary to glomerulonephritis, diabetes or hypertension (Chevalier, 2006; Chevalier et al, 2009). Indeed, long-term ON is characterized by tubular atrophy and the appearance of fibrosis (Fig 2B). Studies of ON have benefited from the existence of a recognized experimental model, the unilateral ureteral obstruction (UUO), in mice. UUO does not compromise the life of the animal, as the contralateral kidney maintains or even augments its function offering functional compensation (Klahr & Morrissey, 2002). Since UUO reproduces all the hallmarks of ON and it generates progressive renal fibrosis, it has become the standard experimental model to study the causes and mechanisms underlying tubulointerstitial fibrosis (Chevalier et al, 2009). Even though ON is not an immune disease, it has a major inflammatory component, characterized by the overexpression of inflammatory genes, the release of pro-inflammatory cytokines, the activation of NF-κB and the infiltration of macrophages and lymphocytes (Diamond, 1995; Esteban et al, 2004; Misseri et al, 2005; Silverstein et al, 2003). Similarly, fibrosis can also be considered as the end result of chronic inflammatory reactions induced by a variety of stimuli including persistent infections, autoimmune reactions, allergic responses, chemical insults, radiation, tissue injury and normal ageing (Kalluri & Neilson, 2003). Therefore, ON and UUO can be used as a reference for the development of fibrosis, in part because inflammatory responses have been analysed in depth in this model.
NF-κB activation in fibrosis and cancer
After renal obstruction, an increase in the expression of the prototypical pro-inflammatory cytokines tumour necrosis factor (TNF-α) and interleukin-1 (IL-1) is crucial for the induction of NF-κB, the major inflammatory response pathway, and for recruitment of inflammatory cells to the obstructed kidney (Meldrum et al, 2006; Metcalfe et al, 2008; Misseri et al, 2005; Silverstein et al, 2003; Yamagishi et al, 2001). An increase in leukocyte infiltration, especially macrophages and T lymphocytes, is detected as early as 4–12 h after ureteral obstruction, and it continues to increase over the following days (Diamond, 1995). These cells play a central role in the renal inflammatory response to UUO as the progression of renal injury is closely associated with their accumulation and blocking their recruitment protects against interstitial fibrosis (Lange-Sperandio et al, 2007). The recruitment of circulatory leukocytes is mediated by several mechanisms including the expression of pro-inflammatory, profibrotic and chemoattractant cytokines in the kidney and the corresponding leukocyte chemotactic response to those chemokines (Ferenbach et al, 2007).
Once activated, NF-κB generates a loop that maintains the inflammatory signals as it controls the expression of genes encoding pro-inflammatory cytokines (e.g. IL-1, IL2, IL-6, TNF-α, etc.), chemokines (e.g. IL-8, MIP-1 α, MCP-1, RANTES, eotaxin, etc.), adhesion molecules (e.g. ICAM, VCAM, E-selectin), inducible enzymes (COX-2 and iNOS), growth factors, some of the acute phase proteins and immune receptors, all of which play critical roles in controlling most inflammatory processes (Blackwell & Christman, 1997; Fig 3). As inflammation plays an important role in fibrosis, inhibition of NF-κB has a big impact on the development of chronic kidney disease. Indeed, NF-κB inactivation prevents inflammatory injury and diminishes the expression of inflammatory genes after UUO (Esteban et al, 2004; Miyajima et al, 2003). Similarly, increasing the levels of the endogenous inhibitor of NF-κB, I-κB, or inhibiting the pathway by the administration of curcumin, reduces renal fibrosis and macrophage influx following UUO or protects against interstitial inflammation in ON, respectively (Kuwabara et al, 2006; Tashiro et al, 2003).
Figure 3. Inflammation, oxidative stress and hypoxia cooperate in the induction of EMT for the progression of organ fibrosis and cancer metastasis.
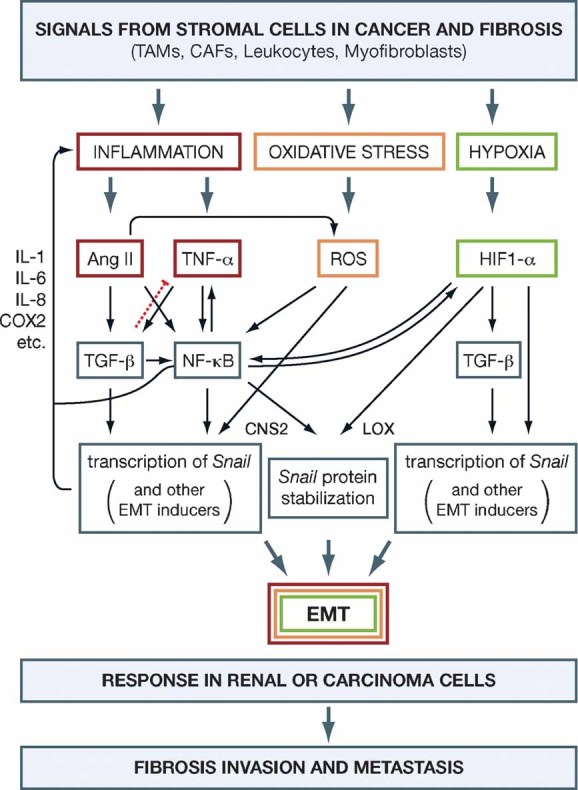
In the fibrotic kidney or in the tumour inflammatory microenvironment TNF-α, Ang II, reactive oxygen species (ROS) and hypoxia converge in the induction of NF-κB, the main inflammatory response pathway. TGF-β is first secreted to control the inflammatory response (dotted red line) but in a chronic inflammatory environment it behaves as a fibrogenic agent and as an inducer of invasiveness. Indeed, the signals released in response to inflammation, oxidative stress ad hypoxia in the stromal or interstitial cells (TAMs, CAFs, leukocytes and myofibroblasts) induce the EMT process in renal and carcinoma cells. TGF-β, NF-κB and hypoxia play a crucial role in the activation of EMT inducers in renal or carcinoma cells. NF-κB and hypoxia also contribute to the EMT process increasing the stability of the inducers. CAFs, cancer associated fibroblasts; CNS2, component of the signalosome; HIF1-α, hypoxia factor 1α; LOX, lysyl-oxidase; TAMs, tumour-associated macrophages.
Like the injured kidney, inflammation or infection in the context of a neoplasia can trigger the activation of NF-κB, the main transducer of inflammatory responses, which in turn induces the secretion of a plethora of inflammatory cytokines (Fig 3). These cytokines recruit inflammatory cells and, at the tumour–stroma interface, tumour cells together with tumour-associated macrophages (TAMs) and cancer-associated fibroblasts (CAFs) again converge in producing NF-κB and the hypoxia-inducible factor (HIF1-α), generating a microenvironment capable of driving tumour progression (Mantovani et al, 2008). HIF-1 is a potent inducer of EMT in the kidney and also in cancer cells in general (see Box 1 and Fig 3) and the inflammation-mediated increase in NF-κB expression has been associated with several aspects of cancer development, mainly resistance to apoptosis and increased angiogenesis, with some connections to invasiveness (Karin, 2006).
BOX 1: The dual role of hypoxia in fibrosis and cancer.
As TGF-β1, hypoxia has an early beneficial role in fibrosis and cancer, but as if both were Dr Jekyll and Mr Hyde they can take a sinister role that favours the progression of the disease (Bottaro & Liotta, 2003; Massague, 2008). Indeed, TGF-β1 can act as a tumour suppressor or as a promoter of invasion and metastasis, and in the injured kidney, it can behave either as a healing anti-inflammatory cytokine or as a profibrotic agent (see main text). Similarly, while cutting off the oxygen supply seemed a good strategy to kill cancer cells since they massively die at low oxygen concentrations, hypoxia increases the aggressiveness in tumours and promotes renal fibrosis. Interestingly, they share a common mechanism to fulfil their deleterious role, the induction of the EMT.
Hypoxia is an important factor in the development of tissue fibrosis. The obstructed kidney suffers from chronic hypoxia due to a reduced renal blood flow (Hegarty et al, 2001). Hypoxia regulates the expression of a variety of growth factors and cytokines through the activation of the HIF-1α, a potent transcriptional regulator of oxygen-dependent genes, including VEGF and TGF-β1 (Baan et al, 2003) (Fig 3). In accordance with the induction of TGF-β1 by HIF-1α, hypoxia also enhances EMT in vitro, and genetic ablation of epithelial HIF-1α in a mouse model of renal fibrosis was associated with reduced ECM accumulation. Interestingly, in addition to inducing TGF-β1, HIF-1α can also directly activate the expression of EMT inducers Snail, Twist and the members of the Zeb family (Evans et al, 2007; Krishnamachary et al, 2006; Sun et al, 2009; Yang et al, 2008) (Fig 3). This activation can occur in renal cells subjected to hypoxia or that constitutively express HIF-1α, as found in those defective in the von Hippel–Lindau (VHL) tumour suppressor. Furthermore, hypoxia also enhances Snail1 activity by activating the expression of its partners lysyl-oxidases (LOX) both in the kidney after UUO and in cancer cells, thereby promoting fibrogenesis and tumour progression through the reinforcement of the EMT programme (Higgins et al, 2007; Sahlgren et al, 2008) (Fig 3). Importantly, there is a reciprocal activation of HIF-1α and NF-κB (Rius et al, 2008), reinforcing the EMT programme (Fig 3). Thus, rather than dying, many cells in tumours and in the damaged kidney can benefit from a lack of oxygen through the induction of EMT, favouring the progression of cancer and fibrosis.
NF-κB can also directly activate the expression of potent EMT inducers, including Snail1 and Zeb factors (Bachelder et al, 2005; Barberà et al, 2004; Chua et al, 2007; Julien et al, 2007; Kim et al, 2007) (Fig 3). Thus, there seems to be an important connection between cancer, inflammation and EMT. This connection has been reinforced recently by the finding that TNF-α induces Snail1 promoter activity and EMT in cancer cells (Dong et al, 2007) and that it can also stabilize Snail1 protein (Wu et al, 2009). Snail1 is a highly unstable protein, targeted for degradation by phosporylation and ubiquitylation mediated by GSK-3β and SCFβ-Trcp, respectively (Zhou et al, 2004). TNF-α induces Snail1 protein stabilization by activating the expression of the COP9 signalosome 2 (CNS2), which in turn inhibits the binding of Snail1 to GSK-3β and β-Trcp (Wu et al, 2009; Fig 3). Interestingly, both transcriptional and post-transcriptional mechanisms act through the induction of the main target of TNF-α in the inflammatory response, NF-κB.
The link between TNF-α and Snail factors points to the EMT as the molecular and cellular explanation for the association between inflammation, fibrosis and tumour progression. As mentioned above, EMT events are now directly associated with invasion and metastasis in cancer progression. Elegant in vivo imaging studies have shown that carcinoma cells migrate from mouse primary tumours through a process of EMT and that this process is dependent on an inflammatory microenvironment provided by the TAMs and other stromal cells such as the CAFs (Condeelis & Segall, 2003) (Fig 3). TAMs and stromal cells in general are essential for angiogenesis, extracellular matrix remodelling and the inflammatory response associated with cancer (Condeelis & Pollard, 2006). Stromal cells secrete TNF-α which, as in renal cells in the obstructed kidney, induces NF-κB in the tumour cells. The significance of NF-κB activation in cancer cells is evident from the results obtained in inflammation-associated models of cancer (Karin & Greten, 2005; Pikarsky et al, 2004). Selective inactivation of NF-κB in these tumours led to deficient tumour development. Importantly, at least in the liver, activation of NF-κB was not important in the early stages of tumour development, but was crucial for malignant conversion and for the survival of tumour cells. This combined effect is very reminiscent of that produced by the EMT inducers and Snail in particular as, in addition of triggering EMT, Snail-expressing cells are resistant to the cell death induced by the removal of survival factors, by genotoxic stress, chemotherapy and immunotherapy and even by TNF-α (reviewed in Barrallo-Gimeno & Nieto, 2005; see also Kudo-Saito et al, 2009; Vega et al, 2004). In addition to the activation of Snail1 by NF-κB, it should be noted that angiotensin II (Ang II), which behaves as a pro-inflammatory cytokine in the kidney (see below) is able to induce oxidative stress in cancer cells (Uemura et al, 2008), where there is an increase in the production of reactive oxygen species (ROS) which, in turn, also stimulates the transcription of Snail1 and favours EMT (Radisky et al, 2005) (Fig 3). This is in agreement with the finding that Ang II receptor inhibitors have beneficial effects on cancer progression and metastasis (Dehayes & Nahmias, 2005). Needless to say, hypoxia is also present in many regions of the tumour, reinforcing the activation of EMT inducers including Twist, Zeb and Snail factors (see Box 1 and Fig 3).
Back to the kidney, renal obstruction leads to the activation of the intrarenal renin–angiotensin system (RAS) and Ang II (Chevalier, 2006; Ruiz-Ortega et al, 2003) (Fig 3), thus promoting inflammation. Following UUO, and acting as a pro-inflammatory cytokine, Ang II also activates NF-κB (Morrissey & Klahr, 1997), which in turn stimulates the angiotensinogen gene (Klahr & Morrissey, 2002). In addition to NF-κB, Ang II signalling through its AT1 and AT2 receptors augments the expression of other pro-inflammatory genes, including those encoding IL-6, monocyte chemoattractant protein 1 and RANTES (Esteban et al, 2003; Ruiz-Ortega et al, 2001; Wolf et al, 1997). Increased Ang II production and the accumulation of activated phagocytes in the interstitial space following UUO lead to an increase in oxidative stress, concordant with the elevated levels of ROS in the chronically obstructed kidney (Kawada et al, 1999; Moriyama et al, 2001). Simultaneously, the activity of the anti-oxidant enzymes catalase and copper-zinc superoxide dismutase, which prevent free radical damage, is low in the obstructed kidney (Ricardo et al, 1997). The resulting increase in oxidative stress plays an important role in inducing an inflammatory state after obstruction, as oxidative stress is a major activator of the NF-κB pathway (Gloire et al, 2006; Fig 3).
In summary, while some steps of the pathways involved may be tissue or disease specific, it is clear that in both cancer and fibrosis the concerted action of several cytokines as IL-1β and TNF-α plus Ang II together with oxidative stress and hypoxia converge on the activation of NF-κB, a major inducer of the inflammatory response.
Yin and Yang of TGF-β1
In addition to the secretion of inflammatory cytokines, many organs and tissues release TGF-β1 after an injury, as seen in the lung, liver, heart, kidney and bone (Devescovi et al, 2008; Frangogiannis, 2008; Kaneto et al, 1993; Kisseleva & Brenner, 2008; Gharaee-Kermani et al, 2009; Gressner et al, 2008). TGF-β1 is considered a major anti-inflammatory cytokine, as TGF-β1 knockout mice suffer from a lethal multifocal inflammatory disease (Kulkarni et al, 1993; Letterio & Roberts, 1998) and the blockage of TGF-β1 signalling in T cells or in bone marrow results in similar multifocal inflammatory responses (Gorelik & Flavell, 2000; Shull et al, 1992). However, TGF-β1 has been known for many years to display a schizophrenic behaviour in cancer. While it can act as a tumour suppressor at early tumour stages, it can later contribute to the malignant progression by promoting invasion and metastasis (Massague, 2008). In the kidney too, although TGF-β1 acts as an anti-inflammatory cytokine to heal the injured kidney, it also has negative effects on the development of chronic renal disease as it is an important fibrogenic agent. Indeed, the role of TGF-β1 as a promoter of tumour progression and profibrotic agent is associated with its ability to act as a potent activator of EMT inducers (Acloque et al, 2009). The Yin and Yang of TGF-β1 in both cases are explored below.
TGF-β1 in cancer: tumour suppressor and promoter of invasion and metastasis
There are paradigmatic examples illustrating how TGF-β1 can impair tumourigenesis while promoting metastasis (Siegel et al, 2003, reviewed in Massague, 2008). In normal cells, TGF-β1 can maintain tissue homeostasis by suppressing cell proliferation and by inducing apoptosis. Tumour cells evade this response by losing the ability to respond to the cytostatic and apoptotic effects through different mechanisms (Shi & Massague, 2003). In addition, TGF-β1 helps cancer cells to acquire the ability to invade and disseminate by inducing the EMT process. Thus, TGF-β1 is a potent EMT inducer in tumours albeit it is also initially secreted to control pathological proliferation and inflammation.
The study of the molecular pathways that drive the differential response to TGF-β1 continues to be a very active field of research and recent findings indicate that both genetic and epigenetic mechanisms are involved. On one hand, the methylation state of the platelet-derived growth factor (PDGF) gene limits the proliferative or cytostatic effect of TGF-β1 in gliomas (Bruna et al, 2007) and the concerted action of TGF-β1 with oncogenic Ras and mutant p53 sequester p63 in a complex that inactivate its tumour suppressor effects (Adorno et al, 2009). This indicates that two molecules frequently mutated in cancer, p53 and Ras, can divert the response to TGF-β1 towards the promotion of metastasis. Indeed, TGF-β1 and oncogenic Ras cooperate in the induction of Snail1 expression and EMT in epithelial cells (Peinado et al, 2003). Interestingly, NF-κB is required for the induction and maintenance of the EMT by TGF-β1 in mammary epithelial cells overexpressing the Ras oncogene (Huber et al, 2004), explaining the role of NF-κB as a modulator of TGF-β1-induced EMT in addition to its role as a direct activator of the EMT inducers.
But what are the sources of TGF-β1 in the tumours? Again, as in the injured organs, TGF-β1 is produced by cancer cells and, particularly, by stromal cells in an attempt to control inflammation. TNF-α can induce TGF-β1 in lung fibroblasts (Sullivan et al, 2009) and TGF-β1 can also activate the resident fibroblasts to convert them into myofibroblasts, called cancer-associated fibroblasts in the contexts of tumours (CAFs) and associated with tumour invasion (Micke & Ostman, 2005). Some of these ‘myofibroblastic’ CAFs are surely the result of the EMT undergone by carcinoma cells, as EMT is now recognized as an important event in carcinoma progression.
TGF-β1 in fibrosis: anti-inflammatory and healing factor versus profibrotic factor
The release of TGF-β1 by renal or by infiltrating cells in the damaged kidney moderates the inflammatory reaction and helps to heal the damaged tissue. Among its anti-inflammatory functions, TGF-β1 antagonizes the pro-inflammatory cytokines IL-1 and TNF-α in glomerular disease, and it is a prominent macrophage deactivator during kidney injury (Kitamura & Suto, 1997). Renal TGF-β1 mRNA expression increases considerably after the onset of obstruction (Kaneto et al, 1993), and its concentration increases in the plasma of patients with obstruction due to ureteral calculi and renal fibrosis (Liu, 2006; Vuruskan et al, 2005). TGF-β1 expression is induced by Ang II (Wolf, 2006), upon increased stretch of tubular epithelial cells (Quinlan et al, 2008), and it is also produced by interstitial fibroblasts and infiltrating macrophages (Diamond et al, 1998; Ding et al, 1993). As already mentioned, in addition to its anti-inflammatory effects, TGF-β1 also acts as a profibrotic agent. The profibrotic effect of TGF-β1 is achieved by increasing the synthesis of matrix proteins and the expression of proteinase inhibitors, including plasminogen activator inhibitor-1 (PAI-1), and by decreasing that of ECM-degrading proteins such as collagenase (Chevalier, 2006). Therefore, sustained aberrant expression of TGF-β1 results in the pathological accumulation of extracellular matrix material in both the glomerulus and interstitial compartments (Bottinger, 2007). TGF-β1 also has some pro-inflammatory properties, as it functions as a chemo-attractant for leukocytes (Wahl et al, 1987) and induces COX-2 in mesangial cells (Rodríguez-Barbero et al, 2006). This profibrotic activity of TGF-β1 reflects its behaviour as a potent inducer of EMT, which contributes to the conversion of renal epithelial cells into myofibroblasts in the context of the injured kidney. In addition, part of the TGF-β1-induced EMT programme promotes the remodelling of the cell contacts with the basal membrane by activating the matrix metalloproteases MMP2 and MMP9 (Li et al, 2003; Strutz et al, 2002), leading to the degradation of the collagen type IV component of basement membrane. Thus, the molecule secreted to control inflammation has an alter ego and it promotes the development of fibrosis.
TGF-β1, myofibroblasts and EMT
Myofibroblasts play a major role in interstitial fibrosis as they are the main source of matrix proteins. They originate from several sources, including bone marrow cells, resident interstitial fibroblasts, vascular pericytes, as well as endothelial and tubular epithelial cells (reviewed in Grande & Lopez-Novoa, 2009). As mentioned above, TGF-β1 contributes to the formation of myofibroblasts through the activation of resident fibroblasts and by inducing the transition to mesenchyme of epithelial and endothelial cells via EMT or EndMT, respectively (Zavadil & Böttinger, 2005; Zeisberg & Kalluri, 2008; Zeisberg et al, 2008). Indeed, the transformation of tubular and endothelial cells to mesenchymal cells contributes to more than 60% of the myofibroblast population in the obstructed kidney after UUO (Iwano et al, 2002; Zeisberg et al, 2008), and this process plays a major role in tubulo-interstitial fibrosis (Kalluri & Neilson 2003). The induction of EMT by TGF-β1 also explains the fibrotic effect of Ang II, as this anti-inflammatory factor also induces the synthesis of TGF-β1 and its receptors in tubular epithelial cells (Wolf, 2006). Accordingly, the infusion of Ang II into mice subjected to UUO significantly increases their collagen content (Ma et al, 2003).
The induction of EMT by TGF-β1 in the renal cells takes several days and requires the interplay of many pathways, involving SMAD proteins (Li et al, 2002), the integrin-linked kinase (Li et al, 2003) and Rho-family GTPases (Bhowmick et al, 2001; Masszi et al, 2003). β-Catenin is another key element in the myofibroblast transition as it synergizes with TGF-β1 in activating the α-smooth muscle actin (αSMA) gene and protein expression (Masszi et al, 2004). The processes triggered by TGF-β1 converge in the activation of the so-called EMT inducers, which are transcription factors capable of eliciting this dramatic phenotypic change that converts epithelial cells into activated ‘myofibroblast-like’ cells. As in cancer, among the transcription factors involved in the induction of EMT, Snail factors have been associated with renal fibrosis and significantly, TGF-β1 is the most potent inducer of Snail transcription (Fig 3). The expression of these genes increases in the obstructed kidney after UUO (Sato et al, 2003; Tan et al, 2006; Yoshino et al, 2007) and Snail1 activation is sufficient to induce EMT and all the hallmarks of kidney fibrosis in adult mice (Boutet et al, 2006). Snail1 directly represses E-cadherin transcription and that of the renal-specific Cadherin16 gene through the direct repression of HNF-1β gene expression. Snail also activates, albeit indirectly, the transcription of mesenchymal genes, such as vimentin and αSMA, and it promotes collagen I synthesis and deposition (Boutet et al, 2006). Interestingly, Snail factors are strongly upregulated in fibrotic kidneys from patients subjected to nephrectomy due to urinary obstruction and kidney failure (Boutet et al, 2006). Altogether, data from many laboratories indicate that Snail factors are crucial mediators of TGF-β1-induced EMT.
Concluding remarks and perspectives
From all the work carried out in the context of organ fibrosis and cancer we can conclude that the pathways and players identified that lead to pathological EMTs are basically the same. Both in cancer and fibrosis TGF-β1, TNF-α and hypoxia cooperate in the triggering of EMT, converging in the induction of Snail activity through different mechanisms among which the activation of NF-κB seems to play a central role. This can be regarded as the reactivation of a developmental programme designed to endow cells with migratory and invasive properties and with amazing survival properties, crucial for migratory cells to reach their sometimes far destinations within the embryo (Acloque et al, 2009). This developmental programme, firstly reactivated to control the inflammatory response and to heal the injured tissue, is corrupted in the context of chronic inflammation and cancer, favouring the survival of injured or malignant cells and stimulating the production of pro-inflammatory cytokines in the tumour mass or in the organ interstitial space. The cooperation between TGF-β1 and the pro-inflammatory cytokines generates a microenvironment that favours the generation of autoregulatory loops to reinforce the EMT programme (Fig 3). Not only Ang II, TNF-α, ROS and hypoxia converge in the induction of Snail, but also Ang II and TNF-α can induce TGF-β1 at the transcriptional level (Sullivan et al, 2009), the most potent Snail inducer. In addition, Snail1 itself also seems to upregulate the expression of pro-inflammatory mediators such as several ILs (IL-1, IL-6, IL-8; Lyons et al, 2008). It will be interesting to determine whether other EMT inducers are also activated in the context of this inflammatory microenvironment, which seems to be very likely, given that both TGF-β1 and hypoxia can induce the expression of Twist and Zeb transcription factors.
The relationship between inflammation and Snail activity has been established in cancer cells from different origins, indicating that this is a general mechanism in carcinoma progression. While the majority of studies on fibrosis have been carried out in the kidney, the same pathways are likely to operate during other fibrotic processes, including those in the liver and the lung, especially since there is evidence of EMT in hepatic fibrosis (Kim et al, 2006; Zeisberg et al, 2007a). A similar process also occurs when endothelial cells undergo an EMT during cardiac or renal fibrosis (EndMT; Zeisberg et al, 2007b, 2008) as well as during mesothelial fibrosis in patients subjected to peritoneal dialysis where the NF-κB/Snail1 axis has already been described (Strippoli et al, 2008).
In summary, the relationship between inflammation and EMT seems to be a conserved feature in the progression of organ fibrotic diseases and cancer, suggesting that specific anti-EMT drugs should be considered to treat these conditions in combination with anti-inflammatory therapies. With respect to therapies targeting the EMT programme, small molecule inhibitors or antibodies directed against TGFβR have been effective in preclinical and clinical trials, and neutralizing antibodies against TGF-β1 are in Phase 1 clinical trials for renal cell carcinoma and pulmonary fibrosis (Chua et al, 2008). Treatment with BMP7, an inhibitor of TGF-β1 signalling, is a promising strategy to treat fibrosis as it can reverse TGF-β1-induced renal fibrosis in mice (Zeisberg et al, 2003). It should be noted that since signalling molecules such as TGF-β1 or hypoxia trigger complex transduction cascades with deleterious but also beneficial outcomes, inhibiting the EMT inducers rather than the extracellular signalling molecules or their receptors could provide a more specific way to fight both fibrosis and cancer progression. However, EMT inducers are transcription factors, and thus very difficult to target. While RNA interference approaches are promising specific reagents, these are still early days for their wide use due to their low stability and, more importantly, due to the current low efficiency in their cell targeting and intracellular delivery. Finally, as the same signalling pathways can act as promoters or suppressors of fibrosis and cancer, better understanding of the cellular response that depends on the context and stage of the disease is crucial to design more advanced therapies.
Pending issues
The development of additional in vivo models to assess the full significance of EMT in cancer and organ fibrosis.
The characterization of the full complement of EMT inducers activated in organ fibrosis.
The development of optimized antibodies specific for the EMT inducers that can work on histopatological sections.
The development of therapies directed towards the inhibition of the EMT process.
The optimization of methods for the delivery of RNA interference reagents.
Acknowledgments
Work in our laboratories is supported by grants from the Spanish Ministry of Education and Science (SAF2007-63893), from Instituto Carlos III (Red Cooperativa de Investigación Renal, RedinRen Retic 06/0016) and Junta de Castilla y León (SA029/A05 and Excellence Group GR-100), to J. M. L.-N. and from the Spanish Ministry of Education and Science (BFU2008-01042; CONSOLIDER-INGENIO 2010 CSD2007-00017 and CSD2007-00023) and the Generalitat Valenciana (Prometeo 2008/049) to M. A. N. We are very grateful to the Cajal Institute (CSIC) where Cajal's original drawing and slides are kept, to Robert Cardiff for providing Apolant's drawing and to Robert Cardiff and Thomas Brabletz for providing micrographs.
References
- Acloque H, Adams M, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cells' state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acloque H, Thiery JP, Nieto MA. The physiology and pathology of the epithelial to mesenchymal transition. EMBO Rep. 2008;9:322–326. doi: 10.1038/embor.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, et al. A mutnat-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell. 2009;177:87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- Apolant H. Die epithelialen Geschwülste der Maus. Arb Königl Inst Exp Ther. 1906;1:7–62. [Google Scholar]
- Baan C, van Gelder T, Peeters A, Mol W, Niesters H, Weimar W, IJzermans J. Living kidney donors and hypoxia-inducible factor-1alpha. Transplantation. 2003;75:570–571. doi: 10.1097/01.TP.0000034241.55602.AD. [DOI] [PubMed] [Google Scholar]
- Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168:29–33. doi: 10.1083/jcb.200409067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberà MJ, Puig I, Domínguez D, Julien-Grille S, Guaita-Esteruelas S, Peiró S, Baulida J, Francí C, Dedhar S, Larue L, et al. Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene. 2004;23:7345–7354. doi: 10.1038/sj.onc.1207990. [DOI] [PubMed] [Google Scholar]
- Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell TS, Christman JW. The role of nuclear factor-kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol. 1997;17:3–9. doi: 10.1165/ajrcmb.17.1.f132. [DOI] [PubMed] [Google Scholar]
- Bottaro DP, Liotta LA. Out of air is not out of action. Nature. 2003;423:593–595. doi: 10.1038/423593a. [DOI] [PubMed] [Google Scholar]
- Bottinger EP. TGF-beta in renal injury and disease. Semin Nephrol. 2007;27:309–320. doi: 10.1016/j.semnephrol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Boutet A, De Frutos CA, Maxwell PH, Mayol MJ, Romero J, Nieto MA. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006;25:5603–5613. doi: 10.1038/sj.emboj.7601421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA. 2001;98:10356–10361. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna A, Darken RS, Rojo F, Ocana A, Penuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and pomotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11:147–160. doi: 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Bukholm IK, Nesland JM, Børresen-Dale AL. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients. J Pathol. 2000;190:15–19. doi: 10.1002/(SICI)1096-9896(200001)190:1<15::AID-PATH489>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75:1145–1152. doi: 10.1038/ki.2009.86. [DOI] [PubMed] [Google Scholar]
- Chevalier RL. Obstructive nephropathy: towards biomarker discovery and gene therapy. Nat Clin Pract Nephrol. 2006;2:157–168. doi: 10.1038/ncpneph0098. [DOI] [PubMed] [Google Scholar]
- Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–724. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- Chua KN, Ma J, Thiery JP. Targeted therapies in control of EMT in carcinoma and fibrosis. Drug Discov Today. 2008;4:261–267. [Google Scholar]
- Condeelis J, Pollard JW. Macrophages: obligate partners fro tumor cell migration, invasion and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3:921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- Cordon-Cardo C, Prives C. At the crossroads of inflammation and tumorigenesis. J Exp Med. 1999;190:1367–1370. doi: 10.1084/jem.190.10.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehayes F, Nahmias C. Angiotensin receptors: a new role in cancer? Trends Endocrinol Metab. 2005;16:293–299. doi: 10.1016/j.tem.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Devescovi V, Leonardi E, Ciapetti G, Cenni E. Growth factors in bone repair. Chir Organi Mov. 2008;92:161–168. doi: 10.1007/s12306-008-0064-1. [DOI] [PubMed] [Google Scholar]
- Diamond JR. Macrophages and progressive renal disease in experimental hydronephrosis. Am J Kidney Dis. 1995;26:133–140. doi: 10.1016/0272-6386(95)90166-3. [DOI] [PubMed] [Google Scholar]
- Diamond JR, Ricardo SD, Klahr S. Mechanisms of interstitial fibrosis in obstructive nephropathy. Semin Nephrol. 1998;18:594–602. [PubMed] [Google Scholar]
- Ding G, Pesek-Diamond I, Diamond JR. Cholesterol, macrophages, and gene expression of TGF-β1 and fibronectin during nephrosis. Am J Physiol Renal Fluid Electrolyte Physiol. 1993;264:F577–F584. doi: 10.1152/ajprenal.1993.264.4.F577. [DOI] [PubMed] [Google Scholar]
- Dong R, Wang Q, He XL, Chu YK, Lu JG, Ma QJ. Role of nuclear factor kappa B and reactive oxygen species in the tumor necrosis factor-alpha-induced epithelial-mesenchymal transition of MCF-7 cells. Braz J Med Biol Res. 2007;40:1071–1078. doi: 10.1590/s0100-879x2007000800007. [DOI] [PubMed] [Google Scholar]
- Esteban V, Ruperez M, Vita JR, López ES, Mezzano S, Plaza JJ, Egido J, Ruiz-Ortega M. Effect of simultaneous blockade of AT1 and AT2 receptors on the NFkappaB pathway and renal inflammatory response. Kidney Int Suppl. 2003:S33–S38. doi: 10.1046/j.1523-1755.64.s86.7.x. [DOI] [PubMed] [Google Scholar]
- Esteban V, Lorenzo O, Rupérez M, Suzuki Y, Mezzano S, Blanco J, Kretzler M, Sugaya T, Egido J, Ruiz-Ortega M. Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J Am Soc Nephrol. 2004;15:1514–1529. doi: 10.1097/01.asn.0000130564.75008.f5. [DOI] [PubMed] [Google Scholar]
- Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VW, Kim WY, Saravanan A, Maynard MA, Gervais ML, et al. VHL promotes E2 box dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007;27:157–169. doi: 10.1128/MCB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenbach D, Kluth DC, Hughes J. Inflammatory cells in renal injury and repair. Semin Nephrol. 2007;27:250–259. doi: 10.1016/j.semnephrol.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharaee-Kermani M, Hu B, Phan SH, Gyetko MR. Recent advances in molecular targets and treatment of idiopathic pulmonary fibrosis: focus on TGFbeta signaling and the myofibroblast. Curr Med Chem. 2009;16:1400–1417. doi: 10.2174/092986709787846497. [DOI] [PubMed] [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Flavell RA. Abrogation of TGF-ß signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- Grande MT, Lopez-Novoa JM. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat Rev Nephrol. 2009;5:319–328. doi: 10.1038/nrneph.2009.74. [DOI] [PubMed] [Google Scholar]
- Gressner OA, Rizk MS, Kovalenko E, Weiskirchen R, Gressner AM. Changing the pathogenetic roadmap of liver fibrosis? Where did it start, where will it go? J Gastroenterol Hepatol. 2008;23:1024–1035. doi: 10.1111/j.1440-1746.2008.05345.x. [DOI] [PubMed] [Google Scholar]
- Hay ED. Organization and fine structure of epithelium and mesenchyme in the developing chick embryo. In: Fleischmajer R, Billingham RE, editors. Epithelial-Mesenchymal Interactions. Baltimore, MD, USA: Williams & Wilkins Co; 1968. pp. 31–55. [Google Scholar]
- Hay ED. Role of cell-matrix contacts in cell migration and epithelial-mesenchymal transformation. Cell Differ Dev. 1990;32:367–375. doi: 10.1016/0922-3371(90)90052-x. [DOI] [PubMed] [Google Scholar]
- Hay ED. Collagen and other matrix glycoproteins in embryogenesis. In: Hay ED, editor. Cell Biology of Extracellular Matrix. New York: Plenum Press; 1991. pp. 419–462. [Google Scholar]
- Hegarty NJ, Young LS, Kirwan CN, O'Neill AJ, Bouchier-Hayes DM, Sweeney P, Watson RW, Fitzpatrick JM. Nitric oxide in unilateral ureteral obstruction: effect on regional renal blood flow. Kidney Int. 2001;59:1059–1065. doi: 10.1046/j.1523-1755.2001.0590031059.x. [DOI] [PubMed] [Google Scholar]
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber MA, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinde K, Nikolic-Paterson DJ, Huang XR, Sakai H, Kurokawa K, Atkins RC, Lan HY. Tubular phenotypic change in progressive tubulointerstitial fibrosis in human glomerulonephritis. Am J Kidney Dis. 2001;38:761–769. doi: 10.1053/ajkd.2001.27693. [DOI] [PubMed] [Google Scholar]
- Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A, Larue L. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26:7445–7456. doi: 10.1038/sj.onc.1210546. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg R. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneto H, Morrissey J, Klahr S. Increased expression of TGF-beta 1 mRNA in the obstructed kidney of rats with unilateral ureteral ligation. Kidney Int. 1993;44:313–321. doi: 10.1038/ki.1993.246. [DOI] [PubMed] [Google Scholar]
- Kawada N, Moriyama T, Ando A, Fukunaga M, Miyata T, Kurokawa K, Imai E, Hori M. Increased oxidative stress in mouse kidneys with unilateral ureteral obstruction. Kidney Int. 1999;56:1004–1013. doi: 10.1046/j.1523-1755.1999.00612.x. [DOI] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB. Linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivoduring pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, Lewis MT, Gottardis MM, Wong TW, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol. 2007;27:3165–3175. doi: 10.1128/MCB.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva T, Brenner DA. Fibrogenesis of parenchymal organs. Proc Am Thorac Soc. 2008;5:338–342. doi: 10.1513/pats.200711-168DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura M, Suto TS. TGF-beta and glomerulonephritis: anti-inflammatory versus prosclerotic actions. Nephrol Dial Transplant. 1997;12:669–679. doi: 10.1093/ndt/12.4.669. [DOI] [PubMed] [Google Scholar]
- Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol. 2002;283:F861–F875. doi: 10.1152/ajprenal.00362.2001. [DOI] [PubMed] [Google Scholar]
- Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH, Semenza GL. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66:2725–2731. doi: 10.1158/0008-5472.CAN-05-3719. [DOI] [PubMed] [Google Scholar]
- Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009;15:195–206. doi: 10.1016/j.ccr.2009.01.023. [DOI] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor ß1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara N, Tamada S, Iwai T, Teramoto K, Kaneda N, Yukimura T, Nakatani T, Miura K. Attenuation of renal fibrosis by curcumin in rat obstructive nephropathy. Urology. 2006;67:440–446. doi: 10.1016/j.urology.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Lange-Sperandio B, Trautmann A, Eickelberg O, Jayachandran A, Oberle S, Schmidutz F, Rodenbeck B, Hömme M, Horuk R, Schaefer F. Leukocytes induce epithelial to mesenchymal transition after unilateral ureteral obstruction in neonatal mice. Am J Pathol. 2007;171:861–871. doi: 10.2353/ajpath.2007.061199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB. Regulation of immune responses by TGF-ß. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Li JH, Zhu HJ, Huang XR, Lai KN, Johnson RJ, Lan HY. Smad7 inhibits fibrotic effect of TGF-beta on renal tubular epithelial cells by blocking smad2 activation. J Am Soc Nephrol. 2002;13:1464–1472. doi: 10.1097/01.asn.0000014252.37680.e4. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest. 2003;112:503–516. doi: 10.1172/JCI17913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 2006;69:213–217. doi: 10.1038/sj.ki.5000054. [DOI] [PubMed] [Google Scholar]
- Lyons JG, Patel V, Roue NC, Fok SY, Soon LL, Halliday GM, Gutkind JS. Snail up-regulates proinflammatory mediators and inhibits differentiation in oral keratinocytes. Cancer Res. 2008;68:4525–4530. doi: 10.1158/1078-0432.CCR-07-6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LJ, Yang H, Gaspert A, et al. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6 (7/7) mice. Am J Pathol. 2003;163:1261–1273. doi: 10.1016/s0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Massague J. TGFβ in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masszi A, Di Ciano C, Sirokmany G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol. 2003;284:F911–F924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- Masszi A, Fan L, Rosivall L, McCulloch CA, Rotstein OD, Mucsi I, Kapus A. Integrity of cell-cell contacts is a critical regulator of TGF-beta 1-induced epithelial-to myofibroblast transition: role for beta-catenin. Am J Pathol. 2004;165:1955–1967. doi: 10.1016/s0002-9440(10)63247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum KK, Metcalfe P, Leslie JA, Misseri R, Hile KL, Meldrum DR. TNF-alpha neutralization decreases nuclear factor-kappaB activation and apoptosis during renal obstruction. J Surg Res. 2006;131:182–188. doi: 10.1016/j.jss.2005.11.581. [DOI] [PubMed] [Google Scholar]
- Metcalfe PD, Leslie JA, Campbell MT, Meldrum DR, Hile KL, Meldrum KK. Testosterone exacerbates obstructive renal injury by stimulating TNF-alpha production and increasing proapoptotic and profibrotic signaling. Am J Physiol Endocrinol Metab. 2008;294:E435–E443. doi: 10.1152/ajpendo.00704.2006. [DOI] [PubMed] [Google Scholar]
- Misseri R, Meldrum DR, Dinarello CA, Dagher P, Hile KL, Rink RC, Meldrum KK. TNF-alpha mediates obstruction-induced renal tubular cell apoptosis and proapoptotic signaling. Am J Physiol Renal Physiol. 2005;288:F406–F411. doi: 10.1152/ajprenal.00099.2004. [DOI] [PubMed] [Google Scholar]
- Micke P, Ostman A. Exploring the tumour environment: cancer-associated fibroblasts as targets in cancer therapy. Expert Opin Ther Targets. 2005;9:1217–1233. doi: 10.1517/14728222.9.6.1217. [DOI] [PubMed] [Google Scholar]
- Miyajima A, Kosaka T, Seta K, Asano T, Umezawa K, Hayakawa M. Novel nuclear factor kappa B activation inhibitor prevents inflammatory injury in unilateral ureteral obstruction. J Urol. 2003;169:1559–1563. doi: 10.1097/01.ju.0000045686.21766.c1. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Kawada N, Nagatoya K, Takeji M, Horio M, Ando A, Imai E, Hori M. Fluvastatin suppresses oxidative stress and fibrosis in the interstitium of mouse kidneys with unilateral ureteral obstruction. Kidney Int. 2001;59:2095–2103. doi: 10.1046/j.1523-1755.2001.00724.x. [DOI] [PubMed] [Google Scholar]
- Morrissey JJ, Klahr S. Enalapril decreases nuclear factor kappa B activation in the kidney with ureteral obstruction. Kidney Int. 1997;52:926–933. doi: 10.1038/ki.1997.414. [DOI] [PubMed] [Google Scholar]
- Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113–21123. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Quinlan MR, Docherty NG, Watson RW, Fitzpatrick JM. Exploring mechanisms involved in renal tubular sensing of mechanical stretch following ureteric obstruction. Am J Physiol Renal Physiol. 2008;295:F1–F11. doi: 10.1152/ajprenal.00576.2007. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon y Cajal S. Manual de Anatomía Patológica General. Barcelona: Imprenta de la Casa Provincial de la Caridad; 1890. [Google Scholar]
- Rastaldi MP, Ferrario F, Giardino L, Dell'Antonio G, Grillo C, Grillo P, Strutz F, Muller GA, Colasanti G, D'Amico G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002;62:137–146. doi: 10.1046/j.1523-1755.2002.00430.x. [DOI] [PubMed] [Google Scholar]
- Ricardo SD, Ding G, Eufemio M, Diamond JR. Antioxidant expression in experimental hydronephrosis: role of mechanical stretch and growth factors. Am J Physiol. 1997;272:F789–F798. doi: 10.1152/ajprenal.1997.272.6.F789. [DOI] [PubMed] [Google Scholar]
- Rius J, Guma M, Schachtrup C, Akassaglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Barbero A, Dorado F, Velasco S, Pandiella A, Banas B, López-Novoa JM. TGF-beta1 induces COX-2 expression and PGE2 synthesis through MAPK and PI3K pathways in human mesangial cells. Kidney Int. 2006;70:901–909. doi: 10.1038/sj.ki.5001626. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ortega M, Lorenzo O, Rupérez M, Blanco J, Egido J. Systemic infusion of angiotensin II into normal rats activates nuclear factor-kappaB and AP-1 in the kidney: role of AT(1) and AT(2) receptors. Am J Pathol. 2001;158:1743–1756. doi: 10.1016/s0002-9440(10)64130-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ortega M, Rupérez M, Esteban V, Rodriguez-Vita J, Sanchez-Lopez E, Egido J. Modulation of angiotensin II effects, a potential novel approach to inflammatory and immune diseases. Curr Med Chem. 2003;2:379–394. [Google Scholar]
- Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA. 2008;105:6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signalling from cell membrane to the nucleus Cell. 2003;113:685–690. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-ß gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel PM, Shu W, Cardiff RD, Muller WJ, Massagué J. Transforming growth factor β signalling impairs Neu-induced tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci USA. 2003;100:8430–8435. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein DM, Travis BR, Thornhill BA, Schurr JS, Kolls JK, Leung JC, Chevalier RL. Altered expression of immune modulator and structural genes in neonatal unilateral ureteral obstruction. Kidney Int. 2003;64:25–35. doi: 10.1046/j.1523-1755.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- Smith JM, Stablein DM, Munoz R, Hebert D, McDonald RA. Contributions of the transplant registry: the 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Pediatr Transplant. 2007;11:366–373. doi: 10.1111/j.1399-3046.2007.00704.x. [DOI] [PubMed] [Google Scholar]
- Strippoli R, Benedicto I, Pérez Lozano ML, Cerezo A, López-Cabrera M, del Pozo MA. Epithelial-to-mesenchymal transition of peritoneal mesothelial cells is regulated by an ERK/NF-kappaB/Snail1 pathway. Dis Model Mech. 2008;1:264–274. doi: 10.1242/dmm.001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Muller GA, Neilson EG. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002;61:1714–1728. doi: 10.1046/j.1523-1755.2002.00333.x. [DOI] [PubMed] [Google Scholar]
- Sullivan DE, Ferris M, Nguyen H, Abboud E, Brody AR. TNF-alpha induces TGF-beta(1) expression in lung fibroblasts at the transcriptional level via AP-1 activation. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2008.00647.x. DOI: 10.1111/j.1582-4934.2008.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Ning X, Zhang Y, Lu Y, Nie Y, Han S, Liu L, Du R, Xia L, He L, et al. Hypoxia-inducible factor-1alpha induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 2009;75:1278–1287. doi: 10.1038/ki.2009.62. [DOI] [PubMed] [Google Scholar]
- Tan R, Zhang J, Tan X, Zhang X, Yang J, Liu Y. Downregulation of SnoN expression in obstructive nephropathy is mediated by an enhanced ubiquitin-dependent degradation. J Am Soc Nephrol. 2006;17:2781–2791. doi: 10.1681/ASN.2005101055. [DOI] [PubMed] [Google Scholar]
- Tashiro K, Tamada S, Kuwabara N, Komiya T, Takekida K, Asai T, Iwao H, Sugimura K, Matsumura Y, Takaoka M, et al. Attenuation of renal fibrosis by proteasome inhibition in rat obstructive nephropathy: possible role of nuclear factor kappaB. Int J Mol Med. 2003;12:587–592. [PubMed] [Google Scholar]
- Uemura H, Ishiguro H, Ishiguro Y, Hoshino K, Takhashi S, Kubota Y. Angiotensin II induces oxidative stress in prostate cancer. Mol Cancer Res. 2008;6:250–258. doi: 10.1158/1541-7786.MCR-07-0289. [DOI] [PubMed] [Google Scholar]
- Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vongwiwatana A, Tasanarong A, Rayner DC, Melk A, Halloran PF. Epithelial to mesenchymal transition during late deterioration of human kidney transplants: the role of tubular cells in fibrogenesis. Am J Transplant. 2005;5:1367–1374. doi: 10.1111/j.1600-6143.2005.00843.x. [DOI] [PubMed] [Google Scholar]
- Vuruskan H, Caliskan Z, Kordan Y, Ozakin C, Yavascaoglu I, Oktay B. Elevated plasma concentrations of transforming growth factor-beta 1 in patients with unilateral ureteral obstruction. Urol Res. 2005;33:465–469. doi: 10.1007/s00240-005-0509-z. [DOI] [PubMed] [Google Scholar]
- Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, Sporn MB. Transforming growth factor type β induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci USA. 1987;84:5788–5792. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G. Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int. 2006;70:1914–1919. doi: 10.1038/sj.ki.5001846. [DOI] [PubMed] [Google Scholar]
- Wolf G, Ziyadeh FN, Thaiss F, Tomaszewski J, Caron RJ, Wenzel U, Zahner G, Helmchen U, Stahl RA. Angiotensin II stimulates expression of the chemokine RANTES in rat glomerular endothelial cells. Role of the angiotensin type 2 receptor. J Clin Invest. 1997;100:1047–1058. doi: 10.1172/JCI119615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of Snail by NF-κB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi H, Yokoo T, Imasawa T, Mitarai T, Kawamura T, Utsunomiya Y. Genetically modified bone marrow-derived vehicle cells site specifically deliver an anti-inflammatory cytokine to inflamed interstitium of obstructive nephropathy. J Immunol. 2001;166:609–616. doi: 10.4049/jimmunol.166.1.609. [DOI] [PubMed] [Google Scholar]
- Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Monkawa T, Tsuji M, Inukai M, Itoh H, Hayashi M. Snail1 is involved in the renal epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2007;362:63–68. doi: 10.1016/j.bbrc.2007.07.146. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Kalluri R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci. 2008;13:6991–6998. doi: 10.2741/3204. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007a;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007b;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]