

Abstract
Human colon cancers often start as benign adenomas through loss of APC, leading to enhanced βCATENIN (βCAT)/TCF function. These early lesions are efficiently managed but often progress to invasive carcinomas and incurable metastases through additional changes, the nature of which is unclear. We find that epithelial cells of human colon carcinomas (CCs) and their stem cells of all stages harbour an active HH-GLI pathway. Unexpectedly, they acquire a high HEDGEHOG-GLI (HH-GLI) signature coincident with the development of metastases. We show that the growth of CC xenografts, their recurrence and metastases require HH-GLI function, which induces a robust epithelial-to-mesenchymal transition (EMT). Moreover, using a novel tumour cell competition assay we show that the self-renewal of CC stem cells in vivo relies on HH-GLI activity. Our results indicate a key and essential role of the HH-GLI1 pathway in promoting CC growth, stem cell self-renewal and metastatic behavior in advanced cancers. Targeting HH-GLI1, directly or indirectly, is thus predicted to decrease tumour bulk and eradicate CC stem cells and metastases.
Keywords: Hedgehog-GLI1, colon cancer, metastases, cancer stem cells
INTRODUCTION
Colon cancers progress from local adenomas in the intestinal epithelium to invasive carcinomas that usually metastasize to the liver. About half the patients with local CCs will develop metastases. Current therapies show <5% 5-year survival for metastatic disease. Recurrence and metastatic spread have been proposed to depend on cancer stem cells, which express CD133 (AC133; Ieta et al, 2008; O'Brien et al, 2007; Ricci-Vitiani et al, 2007; Todaro et al, 2007; Vermeulen et al, 2008; Zhu et al, 2009).
Whereas CCs are thought to derive from constitutive activation of WNT signalling by mutation of APC of βCAT (e.g. Morin et al, 1997), the involvement of HH-GLI signalling in CC is not clear, even though it is critical for many types of human cancers (reviewed in Ruiz i Altaba et al, 2007). Secreted HH ligands block the function of PATCHED1 (PTCH1) in the responding cell, which normally inhibits SMOOTHENED (SMOH). Briefly, active SMOH then triggers positive function of the GLI zinc finger transcription factors, leading to GLI1 function, and inhibits GLI repressors, mostly GLI3R. The end result is the flipping of the GLI code—the sum of all functions of the three GLI proteins—from a repressive to an activating state, leading to the transcription of GLI1 itself, PTCH1, HIP and other targets that mark of a cell's response to HH signals.
Hh has been suggested to regulate the differentiation of normal intestinal villi (van den Brink et al, 2004), and in mice it affects mostly the surrounding mesenchyme (Alinger et al, 2009; Madison et al, 2005). Moreover, while limited in vitro cell line and expression data suggest a controversial role in epithelial cells (Akiyoshi et al, 2006; Douard et al, 2006; Monzo et al, 2006; Oniscu et al, 2004; Qualtrough et al, 2004), this has been either not observed (Berman et al, 2003) or denied (Chatel et al, 2007; van den Brink et al, 2004). Moreover, recent studies indicate that in mice Hh may act in an exclusive paracrine fashion in pancreatic and gastrointestinal (GI) tumours (Tian et al, 2009; Yauch et al, 2008). This idea has been extended to human CCs without direct data (Yauch et al, 2008; discussed in Ruiz i Altaba, 2008) and it contradicts the presence of active signalling in the tumour cells of human melanomas, prostate and basal cell carcinomas and other human tumours (Dahmane et al, 1997; Sanchez et al, 2004; Stecca et al, 2007; see Ruiz i Altaba, 2008). A role for HH-GLI in human CC has also not been highlighted by genomic approaches (Wood et al, 2007)—although this was also the case of HH-GLI-dependent glioblastomas (Parsons et al, 2008; see also Clement et al, 2007)—and the few mutations in SMOH found in GI tumours are not activating (Guleng et al, 2006). Some of the potential problems with the studies mentioned above in terms of resolving an action of HH-GLI in human CCs include the use of only one or few established human cell lines and the lack of single-cell resolution data on the localization of HH-GLI components. Here we have sought to resolve these discrepancies and test the possible role of HH-GLI function in medically relevant stem cells and metastases of advanced human CCs.
RESULTS
Human CCs display an active HH-GLI pathway in epithelial tumour cells
Analyses of 40 primary local and metastatic CCs as well as CC cell lines chosen by their complementarity, Ls174T (βCAT/KRAS mutant), HT29 (APC/BRAF/p53 mutant) and Caco2 (APC/p53 mutant) (Table S1 of Supporting Information), showed consistent expression of GLI1, PTCH1 and SHH as well as other HH-GLI pathway components, extending previous findings (Bian et al, 2007; Monzo et al, 2006; Oniscu et al, 2004). Expression was detected in epithelial tumour cells of CCs in situ in the colon (Fig 1A) and of metastatic CCs in the liver (Fig 1C), that were positive for carcinoembryonic antigen (CEA), cytokeratins and βCAT (Fig S1 and Table S1 of Supporting Information). No obvious expression was detected in the stroma. Using polyclonal and monoclonal antibodies raised to the same epitope we localized GLI1 protein in the epithelial cells of in situ carcinomas (Fig 1B) and in liver metastases (Fig 1D). While active GLI1 is undetectable by immunocytochemisty in normal development given its very low levels, detectable levels of GLI1 protein in transfected cells or in cancer cells can be found in the nucleus and/or cytoplasm (e.g. Dahmane et al, 1997; Ruiz i Altaba, 1999; Stecca et al, 2007). Here we found GLI1 labelling mostly in the cytoplasm (Fig 1B,D).
Figure 1. HH-GLI pathway expression and localization in human CCs.
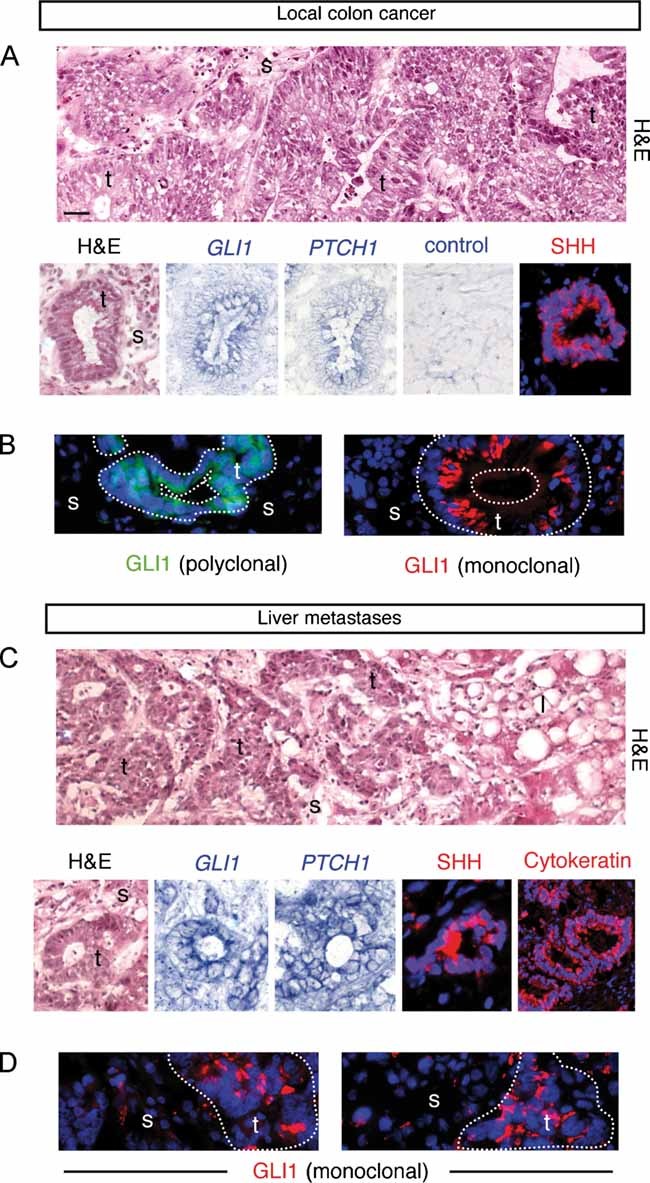
- Analysis of CCs localized in the colon. Hematoxylin and eosin (H&E) staining, in situ hybridization for /GLI1/ and /PTCH1/ mRNAs (blue; CC7) and SHH (red; CC14) in (A) and GLI1 protein (green; CC6) in (B) in CC epithelial cells as indicated.
- Analysis of metastatic CCs localized in the liver. H&E staining, in situ hybridization for /GLI1/ and /PTCH1/ mRNAs (blue; mCC2), SHH (red; mCC11) and cytokeratin (red; mCC11) in (C) and GLI1 protein (green; mCC1) in (D) in CC epithelial cells as indicated.
- The top panels of (A, C) show wider views of the local CC6 and metastatic mCC2 tumors, depicting local colon invasion and high-grade dysplastic morphology (A) and liver invasion (C). Nuclei (blue) are stained with DAPI immunofluorescence images for SHH, cytokeratin and GLI1. l: liver cells, s: stroma; t: tumor. Scale bar = 50 µm for all panels.
Human CCs acquire a high HH-GLI activity signature coincident with the development of metastases
To measure gene expression levels in different CC cell populations and since CC stem cells express CD133 (AC133; O'Brien et al, 2007; Ricci-Vitiani et al, 2007), these were selected from fresh non-metastatic (TNM stages 1,2) and metastatic (TNM3,4) CCs as well as liver metastases (Fig 2A and Fig S2 of Supporting Information). All CCs contained CD133+ cells, which induced tumours in nude mice (CC14: 4/4; mCC11: 4/4; 105 cells injected/site) unlike their CD133− counterparts (CC14: 0/4; mCC11: 0/4; 105 cells injected/site). CD133+ cells were found to be mostly Cytokeratin+ (not shown). Early CCs (TNM1,2) had 7.8-fold larger pools of CD133+ cells on average than normal colon (11.8% ± 1.2 SEM vs. 1.5% ± 0.1 SEM, p < 0.001), and metastatic tumours (TNM3,4) harboured 40% more CD133+ cells than non-metastatic (TNM1,2) CCs (16.6% ± 1.6 SEM vs. 11.8% ± 1.2 SEM, p = 0.04) (Fig 2A).
Figure 2. Gene expression changes in human CC samples.
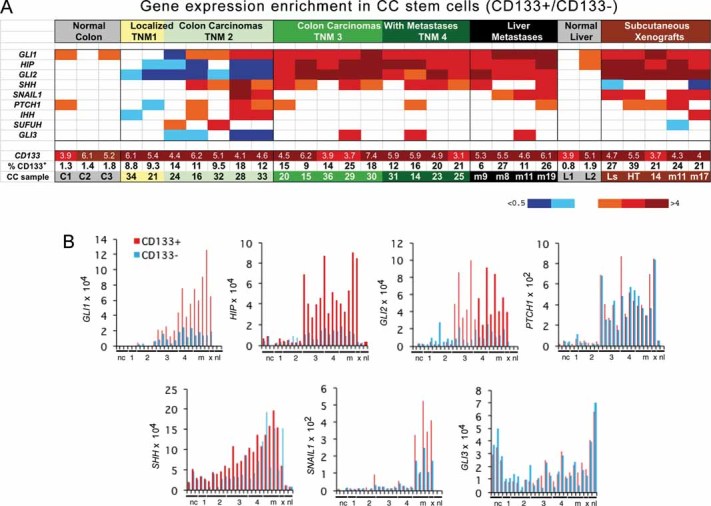
- Heat map of gene expression determined by RT-qPCR shown as CD133+/CD133− expression ratios. CC are TNM staged. Numerical values are given in Fig S2 of Supporting Information. Here and in all figures expression levels were normalized with the geometric mean of the ct values of EEFIa and GAPDH. Ls = LS174T, HT = HT29, m = metastatic tumour in the liver.
- Histograms of individual gene expression changes in CD133+ (red bars) and CD133− (blue bars) cells. The graphs use the same samples as in (A). x = mCC17 xenograft. Numerals refer to TNM stages. nc: normal colon; nl: normal liver; m = metastases.
Gene expression analyses in purified CD133+ and CD133− CC cells, shown as CD133+/CD133− average ratios after normalization (Fig 2A and Fig S2 of Supporting Information) or individually (Fig 2B), revealed ±5-fold enrichment of CD133 mRNA in CD133+ cells. This confirms the selection and its significance since the AC133 epitope used for sorting is displayed by only a fraction of CD133 protein isoforms. Analysis of CD133+ epithelial cells is important as the CD133− epithelial population (and the unfractionated pool) includes also many cell types that form the stroma.
An HH-GLI activity signature (GLI1, PTCH1, GLI2, SHH and HIP) was detected in all CCs (Fig 2A,B and Fig S2 of Supporting Information), consistent with increased SHH expression (Fig 2B; Douard et al, 2006; Oniscu et al, 2004). This signature was not enriched in CD133+ cells of normal colon or liver and it was only slightly enriched in non-metastatic tumours (TNM1,2) (Fig 2A,B). However, there was a large increase in HH-GLI activity signature levels, mostly in CD133+ cells, in metastatic TNM3,4 versus non-metastatic TNM1,2 CCs (CD133+/CD133− averages: GLI1: 1.5 in TNM1.2 vs. 2.8 in TNM3.4, p < 0.01; GLI2: 0.4 vs. 5.5, p < 0.01; HIP: 0.5 vs. 4.4, p < 0.001; Fig 2A,B). PTCH1 increased in both cell populations (0.4 vs. 4.5, p < 0.001 in CD133+ cells; 0.3 vs. 4, p < 0.001 in CD133− cells) (Fig 2A,B and Fig S2 of Supporting Information), suggesting differential regulation of HH-GLI targets. GLI1 itself and SNAIL1, also a GLI target (Li et al, 2006) involved in epithelial-to-mesenchymal transition (EMT; Batlle et al, 2000; Cano et al, 2000), similarly showed an increased CD133+/CD133− average in liver metastases versus TNM3,4 CCs (6 in liver metastases vs. 2.8 in TNM4, p < 0.01; 2 vs. 1, p = 0.01). As CD133+ cells consistently showed the highest increases in the levels of HH-GLI pathway components (Fig 2B), increases in GLI1 levels and targets thus appear to parallel the progression in carcinoma stem cells to metastatic states.
Comparison of advanced TNM4 CCs versus their metastases in the same patients (CC14 vs. mCC9; CC31 vs. mCC19) (Fig 2A and Fig S2 of Supporting Information) showed that GLI1 levels increased only in CD133+ cells (4.4 in TNM4 vs. 6 in liver metastases and 5.5 vs. 12.6 in CD133+ cells; 1.8 vs.1.8 and 2.3 vs.1.4 in CD133− cells, all ×10−4 and normalized).
Human CC cells require HH-GLI function for sustained proliferation and survival in vitro
Primary cultures from patient CC samples contained adherent cells that were >90% pan-Cytokeratin+ (Fig 3A and not shown), demonstrating the epithelial nature of the CC tumour cells used. All primary cultures tested except one from an early stage tumour, and all CC cell lines tested, required sustained activity of HH-GLI signalling for continued proliferation and survival as determined by RNA interference with two independent methods.
Figure 3. Requirement of HH-GLI signalling for CC growth in vitro.
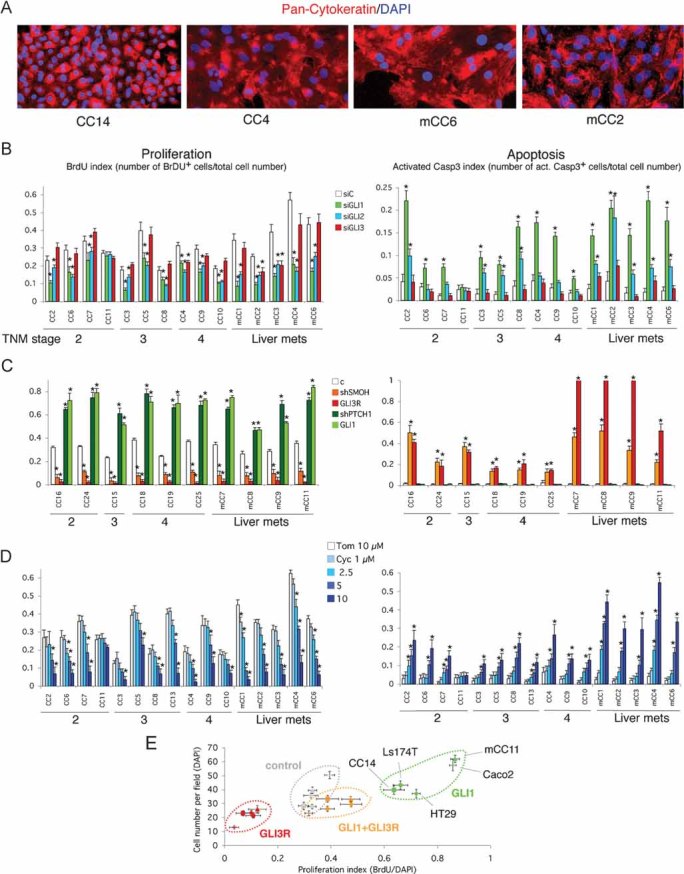
A. Representative CC primary cultures labelled with pan-Cytokeratin antibodies (red) with more than 90% Cytokeratin+ cells (see Fig S1 of Supporting Information), demonstrating their epithelial nature.
B, C. Effects of siRNAs (B) and lentivectors (C) as noted on proliferation (BrdU index: number of BrdU+ cells over total number of cells), and apoptosis (cleaved Caspase-3 index: number of cleaved Caspase-3+ cells over total number of cells). Asterisks in these and all panels denote significant changes (p < 0.05) as compared with controls. mRNA target destruction efficiencies for siGLI1 was 85% on average in seven primary cultures tested; 83.3% for siGLI2; and 83.5% for siGLI3; for shRNAs: 86.9% for shSMOH and 83.9% for shPTCH1 on average in four primary cultures analyzed, measured by qPCR. Primary cultures of normal liver or normal colon did not grow under the conditions used for CCs.
D. Effects of cyclopamine compared with the inactive compound tomatidine on proliferation (right) shown as BrdU index, and apoptosis (left), shown as cleaved Caspase-3 index. Cyclopamine decreases proliferation and increases apoptosis in a concentration-dependent manner. Samples are shown grouped by TNM stage and liver metastases are shown to the right. CC14 proliferation was similarly inhibited by shSMOH, GLI3 and cyclopamine treatment, and enhanced by shPTCH1 and GLI1 (not shown).
E. Graph of the variation in proliferation index (BrdU incorporation/cell number) compared with total cell number (number of DAPI+ cells) of five CC cells as indicated, responding to expression of GLI1, GLI3R or both simultaneously through lentivector transduction. Control cells were transduced with a parental empty lentivector. All cells respond similarly.
Scale bar = 150 µm (A).
First, to knock-down HH-GLI signalling at the level of GLI function we used siRNAs specific for GLI1, GLI2 or GLI3 with ±80–85% targeting efficiencies (Clement et al, 2007; Sanchez et al, 2004; Stecca et al, 2007) (Fig 3B and Fig S3 of Supporting Information). siGLI1 induced a ±2-fold reduction in BrdU incorporation (p < 0.001) and a 5.4-fold increase in apoptosis as assessed by activated Caspase-3 labelling (p < 0.001) on average as compared to a control siRNA (siC). An independent second set of siRNAs (Clement et al, 2007; Sanchez et al, 2004; Stecca et al, 2007) gave the same results (Fig 3B and Fig S3 of Supporting Information). siGLI2 induced a ±1.7-fold decrease in BrdU incorporation on average (p < 0.001) compared with siC, whereas siGLI3 had minor effects in only 3 out of 15 cases. siGLI2 increased apoptosis by 2.5-fold on average (p < 0.01), whereas siGLI3 had no effect (p = 0.6). GLI1, and to a lesser extent GLI2, is therefore essential for metastatic CC cell survival (Fig 3B). Previous chemotherapeutic treatments (Table S1 of Supporting Information) did not affect the response.
Second, to stably and cell-autonomously modulate HH-GLI activity in epithelial cells, we used replication-incompetent lentivectors that modify HH signalling downstream of ligand reception. Expression of an shRNA to SMOOTHENED (shSMOH; Clement et al, 2007) with 85–90% kd targeting efficiency induced a 3.6-fold decrease in BrdU incorporation (p < 0.001) 4–5 days after transduction; and a 26-fold increase in cleaved Caspase-3 labelling (p < 0.001) (Fig 3C). Similar results were obtained by transduction of a lentivector driving the expression of the repressor form of GLI3 (GLI3R), a potent HH-GLI blocker that acts downstream of SMOH, which induced a 13-fold decrease in BrdU incorporation (p < 0.001) and a 42-fold increase in cleaved Caspase-3 labelling (p < 0.01) (Fig 3C). Parental lentivectors at similar multiplicity of infection had no effect, all measured at the same time (Fig S3 of Supporting Information and not shown). Metastastic CCs were more sensitive to GLI3R, the strongest pathway inhibitor, than non-metastatic ones (4.3-fold increase; p < 0.001; Fig 3C), suggesting a near complete dependence on HH-GLI activity for survival.
To complement these studies, we made lentivectors targeting PTCH1 (shPTCH1), an endogenous inhibitor of SMOH, or expressing full-length GLI1 to upregulate HH-GLI signalling. CC cell proliferation and survival were enhanced by shPTCH1, 75–90% targeting efficiency, inducing a two-fold increase in BrdU incorporation (p < 0.001) and a 1.5-fold decrease in cleaved Caspase-3 labelling (p = 0.05) (Fig 3C); or by GLI1, inducing a two-fold increase in BrdU incorporation (p < 0.001) and a 1.6-fold decrease in cleaved Caspase-3 labelling (p = 0.01) (Fig 3C).
Cyclopamine treatment of CC cells in vitro enhances apoptosis and decreases proliferation
As an additional test for the requirement of HH-GLI function, we also inhibited SMOH pharmacologically by treatment with cyclopamine, a plant alkaloid (Keeler, 1970, 1978) that inhibits SMOH (Cooper et al, 1998; Incardona et al, 1998) and that we found to be a specific inhibitor of HH-GLI at effective doses (e.g. Clement et al, 2007; Dahmane et al, 2001; Sanchez et al, 2004; Sanchez & Ruiz i Altaba, 2005; Stecca et al, 2007). Cyclopamine treatment produced 1.9-fold (p < 0.01) and 3.5-fold (p < 0.01) less BrdU incorporation on average at 5 and 10 µM as compared with the control compound tomatidine (10 µM) (Fig 3D). As noted (Qualtrough et al, 2004), cyclopamine induced 1.5-fold (p = 0.02), 3-fold (p < 0.001), 5.4-fold (p < 0.001), and 9-fold (p < 0.001) more cleaved Caspase-3 labelling on average at 1, 2.5, 5 and 10 µM than control (Fig 3D). Similar results were obtained with two CC cell lines (Fig S4 of Supporting Information). An insensitive sample (CC11) and rescue by GLI1 (see below) indicate specificity.
Opposite and compensatory effects of GLI1 and GLI3R in CC cell proliferation
Specificity and rescue analyses also included the expression of GLI1, GLI3R or both into two primary CC samples (CC14 and mCC11) and three established CC cell lines (Ls174T, HT29 and CaCo2) (Fig 3E). GLI1 enhanced and GLI3R repressed cell number and cell proliferation in all cells (Fig 3E). Importantly, these effects were mutually rescued in co-transfected cells (Fig 3E).
Human CC cells require active HH-GLI signalling for growth in vivo
To test for the role of HH-GLI signalling in epithelial tumour cells in vivo we engrafted ∼105 CC cells harbouring GFP+ lentivectors (modulating HH-GLI signalling cell-autonomously) or untransduced CC cells for treatment with cyclopamine, into nude (NMRi-Nude) mice. Gene expression signatures of primary CCs or cell line xenografts (Fig 2A, right) mimicked those of metastases and recapitulated the signatures of the original tumours (CC14 vs. CC14 xenograft and mCC11 vs. mCC11 xenograft), suggesting HH-GLI activity in CD133+ CC cells and validating the xenograft model.
FACS-selected GFP+ CC14, HT29 and Ls174T CC cells expressing shSMOH or GLI3R did not yield tumour growth although these were viable at the time of grafting. In contrast, sorted cells expressing GLI1 or shPTCH1 yielded 6–15 fold larger xenografts than control-GFP+ cells (all p < 0.0001; Fig 4). mCC1 yielded similar results (not shown).
Figure 4. Requirement of HH-GLI signalling for CC growth in vivo.
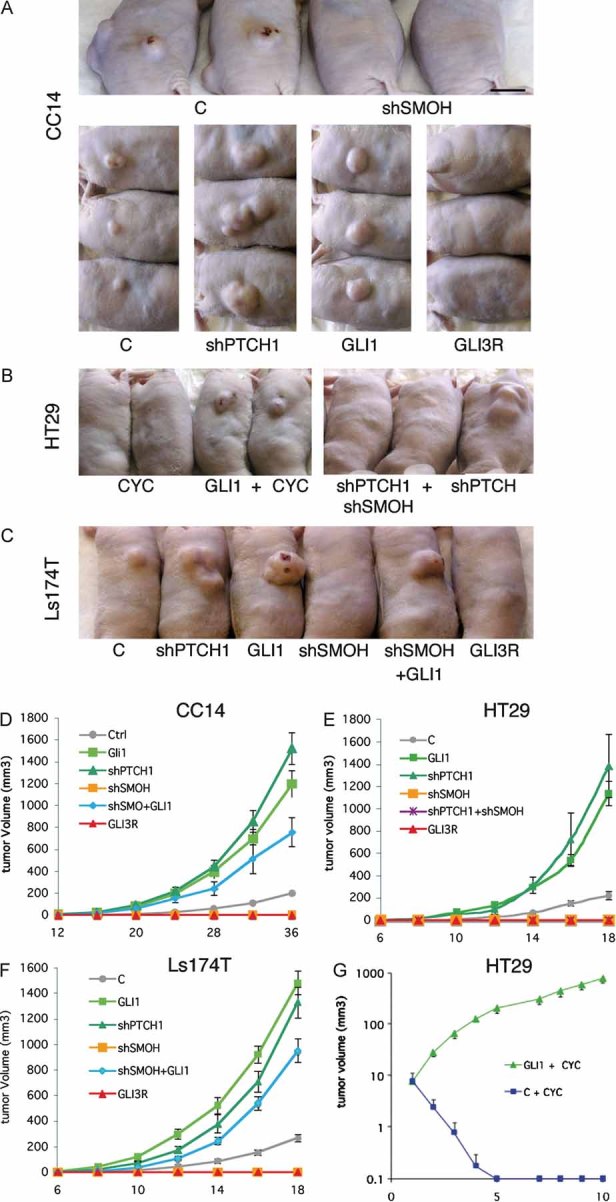
A-C. Representative CC subcutaneous xenografts in nude mice taken at the same time for each group (10–20 days) as indicated with cells expressing shRNAs or cDNAs noted. Control cells were transduced with a GFP-only parental lentivector.
D-G. CC xenograft growth curves transduced or treated as shown. In (G), the graph starts at the beginning of treatment with cyclopamine rather than at the time of cell grafting (D–F). For each curve at least six independent tumours were scored. All graphs show tumour volume over days.
Scale bar = 1 cm for (A–C).
The growth of HT29 and CC14 xenografts was also abolished by intratumoural treatment with cyclopamine (10 mg/kg/twice daily cyclodextrin-complexed), whereas no response was observed by injection of carrier alone (cyclodextrin at same doses) (Fig 4B,G and not shown). Importantly, the blockade of tumour growth by cyclopamine or by shSMOH were both fully rescued by GLI1 and the overgrowth induced by shPTCH1 was fully rescued by shSMOH (Fig 4A–G) (p < 0.001 for both), consistent with their linear epistatic relationship and demonstrating specificity.
CC recurrence requires HH-GLI function
Recurrence is a major problem in the treatment of human CC. Even after apparent complete resection of the primary tumour, recurrence at nearby or distal locations has poor prognosis. To analyze CC recurrence, xenografts of HT29 CC cells were allowed to grow until the tumour was visible and then treated intratumourally with cyclopamine until their disappearance. Cessation of the treatment, however, resulted in recurrence in 100% of cases (not shown). Cyclopamine treatment was then continued for an additional 5, 10 or 20 days into the site where the tumour had been. In all cases, the tumours recurred (Fig 5) except for those treated for 20 days, which prevented all recurrences. Such mice kept up to 1 year after the end of treatment were healthy and tumour-free (Fig 5).
Figure 5. Interference with HH-GLI prevents CC recurrence.
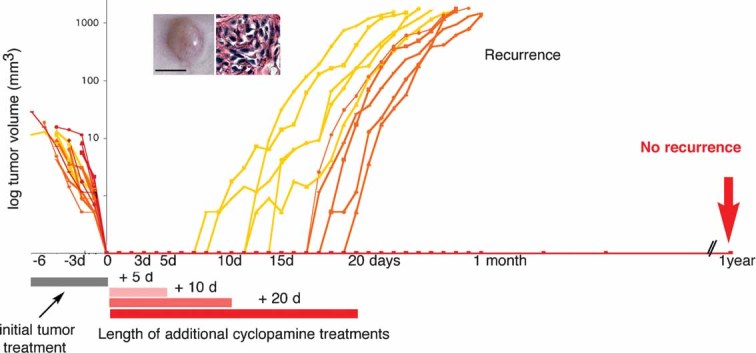
Suppression of HT29 xenografts by cyclopamine treatment and their subsequent recurrence after additional treatments of 5 (yellow) or 10 (orange) days, but not after an additional 20 day treatment (red). n = 5 for each cohort. Insets show the morphological appearance of a recurrence (left) and its histology (right) showing the presence of XGal+ LacZ-HT29 cells (blue) surrounded by host stroma (pink). Scale bar = 0.5 cm for (left inset), 50µm (right inset).
CC metastastic growth requires HH-GLI function
In addition to recurrence, CC growth in distal organs following metastasis leads to an incurable disease. To analyze if metastatic growth is HH-GLI-dependent, we injected LacZ-transduced CC cells into the mouse tail vein, which leads to the formation of metastatic lesions in the lungs of nude mice. These can then be easily visualized by XGal staining yielding blue metastatic cells (Stecca et al, 2007).
Expression of shSMOH in injected LacZ-HT29 CC cells, which were viable at the time of injection, completely inhibited CC metastatic growth detected after ±1.5 months (Fig 6A–C). In contrast, enhanced GLI1 function increased by ±2-fold the number and ±2–5-fold the size of metastatic colonies (Fig 6A–C). GLI1 rescued the inhibition by shSMOH (Fig 6A,C), proving specificity and epistasis. Similarly, LacZ-CC14 and LacZ-Ls174T cells, which normally produced few lung metastases after ±1.5 months, yielded ±3–7-fold more metastatic colonies with shPTCH1 (Fig 6D,E).
Figure 6. Interference with HH-GLI prevents CC metastatic growth and GLI1 induces EMT in CC Cells.
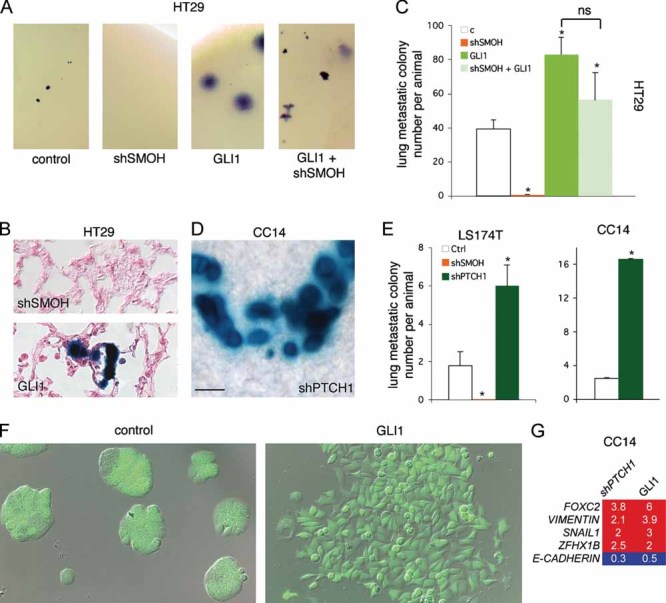
A, B. Wholemount (A) and histological sections (B) of LacZ-HT29 lung metastases after HH-GLI pathway modulation. The development of large metastases is contrary to local ethical rules. Sections were counterstained with eosin (pink).
C. Quantification of metastastic colony number from injected cells expressing shSMOH, GLI1 or both. ns = not significative. n = 8 mice each for control (c), shSMOH and GLI1; n = 5 for shSMOH + GLI1.
D, E. Whole-mount image (D) and quantification (E) of the metastatic behavior of LacZ-Ls174T and LacZ-CC14 and the modulation by shSMOH or shPTCH1.
F. Images of transduced CC14 cells with control (left) or GLI1 (right) GFP+ lentivectors showing EMT induction by GLI1 (right) with elongated dispersing cells (right) that disintegrate the compact epithelial islands characteristic of CC cultures (left).
G. RT-qPCR analyses of CC14 cells as in (F) showing the regulation of EMT genes by enhanced HH-GLI activity through shPTCH1 or GLI1 in relation to control (set at 1). Note the increased expression of EMT genes and the repression of E-CADHERIN.
Scale bar = 5 mm for (A), 80 µm for (B), 30 µm for (D) and 60 µm for (F).
The increase of metastatic growth of cells with high HH-GLI activity could be due to a growth advantage and/or to an induction of EMT (Li et al, 2006). To test the possibility that GLI1 directly drives EMT advanced CC cells, GLI1 was expressed at higher levels in CC14 cells, which derive from an advanced tumour in the colon (Table S1 of Supporting Information). Transduced CC14 GFP+ cells expressing shPTCH1 or GLI1 displayed changes in cell shape, loss of epithelial morphology and increased dispersion (Fig 6F). Consistent with EMT induction, they expressed higher levels of the EMT markers FOXC2, VIMENTN, SNAIL1 and ZFHX1B while repressing E-CADHERIN (Fig 6G), suggesting that GLI1 drives EMT and metastatic behavior in human CCs.
Development of a novel ‘red/green’ competition assay to test stem cell self-renewal in vivo
The involvement of HH-GLI signalling in recurrence and metastasis suggested a possible role in CC stem cells. To test for stem cell behavior in vivo, we designed a novel assay in which freshly purified CD133+ CC cells from primary tumours were transduced with lentivectors expressing RFP (TomatoRED) alone as internal control, GFP alone, GFP plus shSMOH, or GFP plus shPTCH1. RFP+ cells and one of the GFP+ variants above are then mixed with untransduced CC cells and ∼105 cells injected subcutaneously into nude mice (Fig 7A). The untransduced and RFP+ control populations always promote tumour development, in which the RFP+ and GFP+ populations compete. Altered self-renewal of CD133+/GFP+ cells in vivo in a tumour context is predicted to result in a change in the size of the GFP+ population comprising CD133+ cells as well as derived CD133− cells. Relative population size changes of sorted CD133+ cells are measured by FACS following tumour dissociation ∼2–3 weeks post-grafting when the tumours were clearly visible. Serial passages in mice of such sorted CD133+ cells under the same conditions allow for the assessment of survival, tumour contribution and cancer stem cell self-renewal in vivo.
Figure 7. Requirement of HH-GLI function for CC stem cell survival and modulation of self-renewal in vivo.
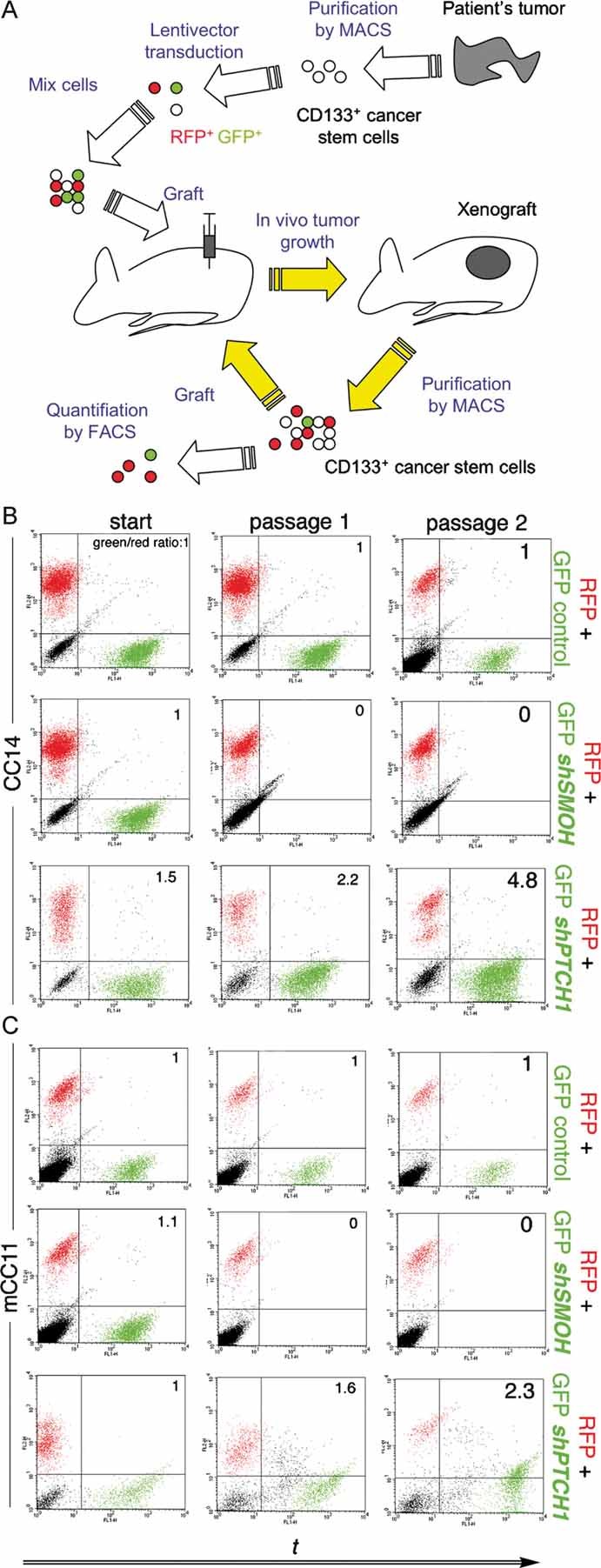
A. Diagram of the novel red/green in vivo protocol described in the text.
B, C. FACS plots of CC14 (B) or mCC11 (C) cell populations with untransduced, TomatoRed+ (RFP+) and GFP+ cells. The GFP/RFP ratio is given for each case.
Advanced and metastatic CC stem cell survival and expansion in vivo depend on HH-GLI activity
Using our novel ‘red/green’ assay, we found that CD133+ CC14 and mCC11 cells expressing RFP or GFP alone were maintained over serial passes, and served as controls (Fig 7B,C). In contrast, those expressing GFP plus shSMOH disappeared in the first round (Fig 7B,C). No GFP+ plus shSMOH CD133− cells were observed (not shown). Conversely, the population size of CD133+ (and CD133−) cells expressing GFP plus shPTCH1 increased over serial passages whereas the RFP+ population did not (Fig 7B,C). HH-GLI pathway activity is thus required for the survival of CC stem cells in vivo and its levels modulate the extent or rate of CD133+ stem cell self-renewal.
Bulk tumour growth in vitro and in vivo is driven by HH-GLI1 activity in the red/green competition assay
The results presented above indicate a key role of HH-GLI in tumour maintenance and expansion through a direct action of cancer stem cells. However, the data on whole tumour growth in vitro and in xenografts additionally indicates an action of HH-GLI on the tumour bulk, possibly paralleling the requirement of HH-GLI in both neural stem cells and derived precursors (Palma & Ruiz i Altaba, 2004; Stecca & Ruiz i Altaba, 2009).
Thus, to extend the results of the new red/green assay to whole CC cell populations, we tested the behavior of GLI1 in vitro and the requirement of HH-GLI in vivo in unfractionated CC14 cells.
GLI1 transduction led to the progressive expansion of GLI1/GFP+ cells over control/RFP+ CC14 cells, reaching a ∼200-fold enrichment after four passages in vitro (Fig 8A). In contrast, control GFP cells maintained a ±1:1 ratio with RFP+ cells over the same four passages (Fig 8A). This result confirms the ability of GLI1 to drive the expansion of a CC cell subpopulation in vitro.
Figure 8. GLI1 drives in vitro expansion of CC cells and HH-GLI function is required for the growth of the bulk of CC tumours.
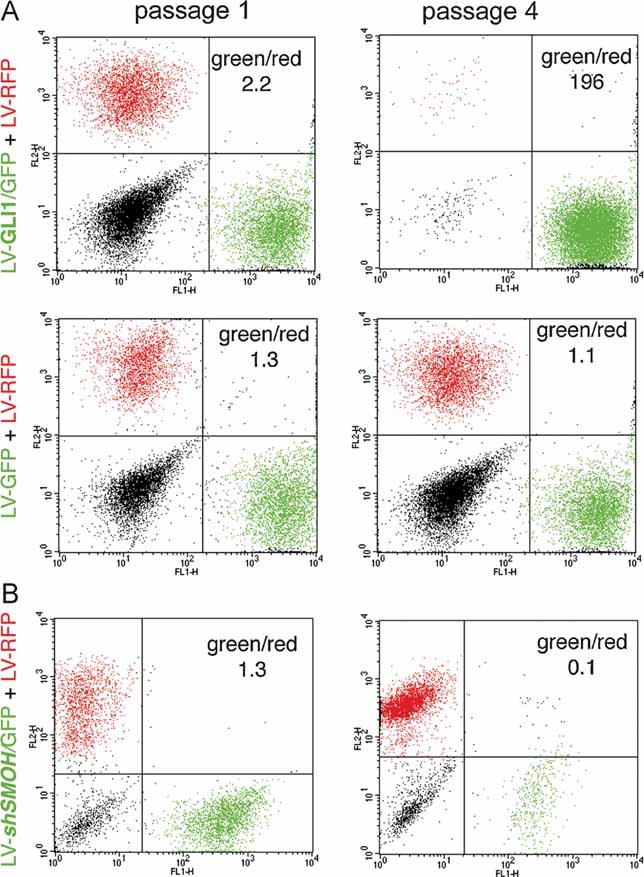
A, B. In vitro (A) and in vivo (B) red/green assay, showing the dominance of GLI1-expressing cells (A) and the requirement of SMOH through the use of shSMOH (B) in CC14 cells. Assays were with untransduced CC14 cells (black in FACS plots), or CC14 cells transduced with RFP+ control (red) or GFP+ GLI1-expressing (green) lentivectors, or RFP plus GFP-only control lentivectors as indicated. FACS plots of cells at passage 1 and 4 are shown in all cases. Control GFP-only transduced CC14 cells are maintained and the green/red cell ratio around 1 is preserved. 106 cells were injected in (B).
In vivo expression of shSMOH in the whole CC14 cell population led to the progressive disappearance of expressing cells from the red/green tumours over time. After four passages from mouse to mouse as subcutaneous xenografts, the population of shSMOH expressing cells became almost undetectable (Fig 8B). Taken together, all in vitro and in vivo results support the critical requirement for HH-GLI activity in the bulk population of CC cells and in their cancer stem cells.
DISCUSSION
Essential role of HH-GLI1 signalling in sporadic human CC
A possible role of HH-GLI in human CC had been proposed based on limited in vitro data (e.g. Douard et al, 2006; Qualtrough et al, 2004). This, however, has been questioned and denied (e.g. Berman et al, 2003; Chatel et al, 2007; van den Brink et al, 2004). Our extensive analyses with cell lines and primary tumours utilizing multiple in vitro and in vivo assays resolves the existing controversy in favor of an essential role of HH-GLI in human sporadic CCs.
Importantly, we find that HH-GLI activity is required by both early and advanced CC in vitro and in vivo. Moreover, the strong upregulation of the HH-GLI activity signature in metastatic patient samples, together with higher apoptosis levels in metastatic tumours as compared with earlier cancers upon HH-GLI pathway blockade, indicate that metastatic human CCs show an enhanced dependence on HH-GLI function.
Human CC epithelial cells display and require HH-GLI activity
We have previously mapped the site of action of HH-GLI signalling in several human cancers by in situ RNA hybridization to the epithelial tumour cells, as these express GLI1 and PTCH1 (Clement et al, 2007; Dahmane et al, 2001; Stecca et al, 2007). However, this conclusion has recently been challenged for colon and other cancers, with suggestions of purely paracrine signalling—from SHH-expressing epithelium to GLI1-expressing responding stroma—largely based on extrapolation of data from mice to the human situation (Tian et al, 2009; Yauch et al, 2008; discussed in Ruiz i Altaba, 2008).
Here we have directly localized the expression of SHH, PTCH1 and GLI1 mRNAs and GLI1 protein in patient-derived human CCs of different stages. We demonstrate the clear presence of active HH-GLI signalling in SHH+/PTCH1+/GLI1+ tumour epithelial cells, but not the stroma, of early and late sporadic human CCs. Differences between the mode of action of HH-GLI in humans versus mice may explain the different results. For instance, in rodents there is convincing data that signalling occurs from epithelium to mesenchyme in the prostate and colon (e.g. Madison et al, 2005; Pu et al, 2004) but from epithelium to epithelium in humans (Sanchez et al, 2004; this work).
The paper explained
PROBLEM
Early human colon tumors often progress to invasive colon carcinomas (CCs) and produce distant metastases, commonly in the liver. Whereas early and localized colon tumors can be effectively treated by surgery, metastatic CC remains largely incurable. Understanding the molecular mechanisms that drive advanced CC is thus essential to design novel rational therapies.
RESULTS
We demonstrate the presence of active HH-GLI signalling in the epithelial tumour cells of patient-derived primary CCs and liver metastases. Morever, HH-GLI activity in epithelial tumour cells is essential for tumour growth, recurrence and metastatic growth. Finally, we also show that HH-GLI regulates the behavior of human CC stem cells in vivo.
IMPACT
The results presented resolve the controversies on the role and mode of action of HH-GLI in CCs, demonstrate a key role in CC stem cells and point to players of this pathway, and GLI1 in particular, as validated targets for novel anti-cancer therapies, specially for those targeting so far intractable metastatic cancer.
Functionally, the cell-autonomous modulation of HH-GLI signalling at the level of PTCH1, SMOH and GLI in vitro and in vivo by cell-autonomous RNA interference defines tumour epithelial and CC stem cells as primary targets of HH-GLI signalling. In CC, as in stomach (Berman et al, 2003) and prostate (Sanchez et al, 2004) cancers, ligand-driven autocrine signalling in SHH+/GLI1+ epithelial cells may be active. The rare GLI1-dependent, SMOH-independent cases we observe (see also Sanchez et al, 2004) could have alterations in components downstream of SMOH.
Finally, the promotion of EMT by HH-GLI1 activity further supports the role of GLI1 as a central nexus regulating cancer cell behavior (Ruiz i Altaba et al, 2007), including the promotion of metastases.
HH-GLI signalling drives CC stem cell survival and self-renewal
Several studies have established the human CD133+ (AC133+) CC fraction as a population of cells highly enriched in CC cancer stem cells (Ieta et al, 2008; O'Brien et al, 2007; Ricci-Vitiani et al, 2007; Todaro et al, 2007; Vermeulen et al, 2008; Zhu et al, 2009). Moreover, CD133+ (Prominin1+) mouse intestinal cells are stem cells that can induce tumourigenesis (Snippert et al, 2009; Zhu et al, 2009). Our results support these findings and not the suggestion that all epithelial cells of local human CCs are CD133+ and that CD133+ cells are not tumourigenic (Shmelkov et al, 2008). We find only ±9–18% CD133+ cells in local CCs, that CD133+ cells are tumourigenic and that their population size is modulated by HH-GLI activity in vivo.
The results with our novel red/green in vivo competition assay tracking CD133+ cells allow us to conclude that human CC stem cells require active HH-GLI signalling for survival and self-renewal, with increased signalling driving a population expansion in advanced cancers and metastases.
Targeting HH-GLI signalling and GLI1 in human metastatic cancers
Human CC metastases remain incurable. The data we present suggest that early, advanced and metastatic CCs have a complete dependence on HH-GLI1. This suggests that inhibitors of GLI1 or HH-GLI signalling will be beneficial to treat local CCs of all stages, but importantly, also metastatic lesions, which remain as an incurable and prevalent disease worldwide.
MATERIALS AND METHODS
Human CC specimens, primary cells and cell lines
The fresh human CC tumours were obtained under approved ethical protocols from the University Hospital of Geneva. Tumours were histopathologically evaluated and classified by the Tumour-Node-Mestastasis (TNM) staging system. Excisions obtained straight after surgery were finely chopped and enzymatically dissociated for 45 minutes in Collagenase A (300 units/ml; Worthington), Dnase I (Roche) and Hyaluronidase (100 units/ml; Sigma) in 1× HBSS. Samples were then mechanically dissociated by pipetting and filtered with a 70 µm strainer, washed twice in 1× PBS and used for culture, sorting, transductions and xenografts. Culture of primary cells was performed using DMEM/F12 supplemented with either 20% BIT 9500 (Stem Cell Technologies) or 10% heat-inactivated FCS, plus 10 ng/ml EGF. HT29 and Caco2 cell lines were purchased from ATCC and Ls174T cells were a gift from E. Batlle (Barcelona) and were routinely cultured with 10% FBS in DMEM/F12. Primary cells were treated with cyclopamine (Toronto Research Chemicals) or tomatidine (Sigma) at 1–10 µM in 1% serum.
In situ hybridization, immunohistochemistry and immunocytochemistry
In situ hybridizations with frozen sections were performed with digoxygenin-labelled antisense RNA probes for GLI1 and PTCH1 as described previously (Dahmane et al, 2001; Stecca et al, 2007). Immunohistotochemistry of frozen sections used mouse anti-CEA (DAKO) mouse anti-Ki67 (Cell Signalling), mouse anti-SHH (University of Iowa Hybridoma Bank), mouse anti-PanCytokeratin (Sigma), anti-GLI1 monoclonal and polyclonal (Cell Signalling) antibodies. Cell proliferation was measured by BrdU incorporation for 48 h followed by staining with anti-BrdU antibodies (BD Biosciences). Apoptosis was measured after 48 h by assessing the number of cells expressing cleaved Caspase-3 with specific antibodies (Cell Signalling). All were followed by FITC- or RITC-conjugated secondary antibodies (Molecular Probes). Labelled cells were imaged with a Zeiss Axiocam optical microscope. Frozen sections were pretreated by bleaching with hydrogen peroxide and boiling for detection of βCAT with horseradish peroxidase-coupled secondary antibodies and development with diaminobenzidine as described previously (van de Wetering et al, 2002).
Plasmids and transfections
Full-length GLI1, GLI3 and GLI3R plasmids were described previously (Ruiz i Altaba, 1999). 106 of the total or CD133+ selected cell populations of HT29, Ls174T, CC14 and mCC11 were mixed with 5 µg of total DNA and transfected with a Nucleofector (Amaxa) according to the manufacturer's instructions. After 16 h, transfection efficiency was 70–90% and cells were collected in 500 µl of Trizol (Invitrogen).
siRNAs
21nt-long, double-stranded, siRNAs were purified and desalted (Dharmacon Inc). The sequences for the siRNAs were 5′–3′: First set (Clement et al, 2007; Sanchez et al, 2004; Stecca et al, 2007) – GLI1, AACUCCACAGGCAUACAGGAU; GLI2, AAGAUCUGGACAGGGAUGACU; GLI3, AAUGAGGAUGAAAGUCCUGGA. Second set (Ambion) – GLI1, GCCCAGAUGAAUCACCAAA; GLI2, CCCUGUCGCCAUUCACAAG; GLI3, GGGCCGUUACCAUUACGAU. Control siRNA: ACGUACGCGGAAUACAACGA. siRNA transfections (0.2 µM) were performed with Oligofectamine (Invitrogen) for 48 h in DMEM/F12 with 2.5% heat-inactivated FBS.
Lentivectors
LV-shSMOH was as in Clement et al (2007). LV-shPTCH1 was made in the same LV-CTH vector as LV-shSMOH with the target sequence 5′-GCACTATGCTCCTTTCCTC-3′. GLI3R cDNA Flag tagged was amplified by PCR using Phusion highfidelity DNA-polymerase (Finnzymes) with primers 5′ to 3′: forward – ATATTCTAGACCATGGACTACAAAGACGATGA and reverse – ATATGATATCCTAATCGATGGCACTGAGGTCTCC, containing the XbaI and EcoRV sites respectively. The PCR product was subcloned in the pRRL-CMV-PGK-GFP-WPRE (TWEEN) lentivector. GLI1 cDNA: a 3370 bp cDNA fragment encompassing the complete ORF was amplified using high fidelity polymerase (Phusion) and the following primers 5′ to 3′: GLI1-forward – ATAGTTTAAACATGGACTACAAAGACGATG A; GLI1-reverse – ATATGTTTAACTTAGGCACTAGAGTTGAGGAA, which introduced PmeI restriction enzyme sites. The resulting PCR product was inserted into an appropriately restricted TWEEN lentivector. All the constructs were checked by restriction enzyme analysis and DNA sequencing. These shRNA and cDNA lentivectors also expressed GFP. A TomatoRed expressing lentiviral vector was a gift from Dr Patrick Salmon (Geneva).
CD133+ cell selection
CD133+ CC cells (O'Brien et al, 2007; Ricci-Vitiani et al, 2007) were sorted from human resections, mouse xenografts of cultured cells using magnetic cell sorting (MACS) as described by the manufacturer (MiltenyiBiotech). Single cell suspensions were incubated with a biotinylated anti-CD133 monoclonal antibody. After the washing steps cells were incubated with streptavidin coupled with magnetic beads. Magnetic sorting was performed using the possel-s program on an autoMACS machine. CD133+ and CD133− fractions were collected and analyzed.
Mouse xenografts of human CC cells and metastasis assays
Human CC cells were infected with appropriate lentiviral vectors (GFP-, TomatoRED- or LacZ-expressing) to allow tracing in vivo. Cells were transduced 48–72 h after infection, GFP and TomatoRED-expressing cells were purified by FACS and resuspended in HBSS. For subcutaneous xenografts, each 6–8 weeks-old female nude (NMRI) mouse received two injections of 105–106 cells each, on the back above each hind leg. As soon as the tumour was palpable (1–2 mm), cyclodextrin-conjugated cyclopamine or cyclodextrin alone (Sigma) was injected intra- and/or peritumourally at 10 mg/kg once daily. Tumour growth was periodically monitored with a caliper. For metastatic growth (Stecca et al, 2007), 1 × 106 HT29-LacZ and CC14-LacZ infected with different lentivectors were injected into the tail vein (i.v.) of adult (6–8 weeks) nude mice.
In vivo self-renewal competition ‘red/green’ assay
Fresh tumours were dissociated as described previously, CD133+ cells isolated by MACS and subsequently infected with either TomatoRed (RFP alone), CTH (GFP alone) or shSMOH (also expressing GFP) lentivectors. Uninfected cells were mixed with red and green cells and FACS analysis was performed to estimate the proportion of each cell population (start). Mixed cells were then grafted under nude mice skin and developing tumours were collected about 14 days post-grafting. The xenograft was then dissociated, CD133+ cells were isolated, an FACS profile established and cells reinjected into mice for additional passages in vivo.
Quantitative RT-PCR
Real-time quantitative PCR amplifications with different primers were carried out at 60 °C on an Opticon machine (MJ Research), by using iQTm SYBR green supermix (Bio-Rad) and values calculated by using the standard curve method. Primers were as described in Dahmane et al (2001), Sanchez et al (2004), Stecca et al (2007) and Clement et al (2007). Additional primer sets were 5′ to 3′: KLF4-Forward – AGAAGGATCTCGGCCAATTT; and KLF4-R, GTGGAGAAAGATGGGAGCAG; VIMENTIN-F, TGCCCTTAAAGGAACCAATG and VIMENTIN-R: TCCAGCAGCTTCCTGTAGGT; IHH -F: CACCCCCAATTACAATCCAG and IHH-R: CGGTCTGATGTGGTGATGTC; CD133-F: GCCACCGCTCTAGATACTGC and CD133-R: TGTTGTGATGGGCTTGTCAT; FOXC2-F: AGTTCATCATGGACCGCTTC and FOXC2-R: GCTCCTCCTTCTCCTTGGAC; ZFHX1B-F:TCCTAATATTCCGCCTGTCG and ZFHX1B-R: GGCATGAAAATGGAGTGGAT; E-CADHERIN-F: GGATGTGCTGGATGTGAATG and E-CADHERIN-R: CTCAAAATCCTCCCTGTCCA.
Acknowledgments
We thank N. Dahmane and all Ruiz i Altaba lab members for discussion and P. Morel for support in obtaining samples. We are grateful to P. Salmon and E. Batlle for the reagents. This work was supported by grants from the Jeantet, Oncosuisse Foundation and Swissbridge Foundations, FNS and NIH to ARA.
Supporting information is available at EMBO Molecular Medicine online.
ARA is a consultant for and holds stocks in Phistem.
Author contributions
FV performed all experiments in collaboration with AD for the red-green and EMT assays, and MM and ARA for metastases assays. MZ and CM provided GLI3 and GLI1 lentivectors, and PG provided tumour samples, diagnoses and medical insights. FV, AD, MM and ARA designed and analyzed experiments and ARA wrote the paper with FV, AD and MM.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Akiyoshi T, Nakamura M, Koga K, Nakashima H, Yao T, Tsuneyoshi M, Tanaka M, Katano M. Gli1, downregulated in colorectal cancers, inhibits proliferation of colon cancer cells involving Wnt signalling activation. Gut. 2006;55:991–999. doi: 10.1136/gut.2005.080333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alinger B, Kiesslich T, Datz C, Aberger F, Strasser F, Berr F, Dietze O, Kaserer K, Hauser-Kronberger C. Hedgehog signaling is involved in differentiation of normal colonic tissue rather than in tumor proliferation. Virchows Arch. 2009;454:369–379. doi: 10.1007/s00428-009-0753-7. [DOI] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- Bian YH, Huang SH, Yang L, Ma XL, Xie JW, Zhang HW. Sonic hedgehog-Gli1 pathway in colorectal adenocarcinomas. World J Gastroenterol. 2007;13:1659–1665. doi: 10.3748/wjg.v13.i11.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Chatel G, Ganeff C, Boussif N, Delacroix L, Briquet A, Nolens G, Winkler R. Hedgehog signaling pathway is inactive in colorectal cancer cell lines. Int J Canc. 2007;121:2622–2627. doi: 10.1002/ijc.22998. [DOI] [PubMed] [Google Scholar]
- Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Lee J, Robins P, Heller P, Ruiz i Altaba A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389:876–881. doi: 10.1038/39918. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Sánchez P, Gitton Y, Palma V, Sun T, Beyna M, Weiner H, Ruiz i Altaba A. The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development. 2001;128:5201–5212. doi: 10.1242/dev.128.24.5201. [DOI] [PubMed] [Google Scholar]
- Douard R, Moutereau S, Pernet P, Chimingqi M, Allory Y, Manivet P, Conti M, Vaubourdolle M, Cugnenc PH, Loric S. Sonic Hedgehog-dependent proliferation in a series of patients with colorectal cancer. Surgery. 2006;139:665–670. doi: 10.1016/j.surg.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Guleng B, Tateishi K, Ohta M, Asaoka Y, Jazag A, Lin LJ, Tanaka Y, Tada M, Seto M, Kanai F, et al. Smoothened gene mutations found in digestive cancer have no aberrant Hedgehog signaling activity. J Gastroenterol. 2006;41:1238–1239. doi: 10.1007/s00535-006-1955-2. [DOI] [PubMed] [Google Scholar]
- Ieta K, Tanaka F, Haraguchi N, Kita Y, Sakashita H, Mimori K, Matsumoto T, Inoue H, Kuwano H, Mori M. Biological and genetic characteristics of tumor-initiating cells in colon cancer. Ann Surg Oncol. 2008;15:638–648. doi: 10.1245/s10434-007-9605-3. [DOI] [PubMed] [Google Scholar]
- Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development. 1998;125:3553–3562. doi: 10.1242/dev.125.18.3553. [DOI] [PubMed] [Google Scholar]
- Keeler RF. Teratogenic compounds of Veratrum californicum (Durand) X. Cyclopia in rabbits produced by cyclopamine. Teratology. 1970;3:175–180. doi: 10.1002/tera.1420030210. [DOI] [PubMed] [Google Scholar]
- Keeler RF. Cyclopamine and related steroidal alkaloid teratogens: their occurrence, structural relationship, and biologic effects. Lipids. 1978;13:708–715. doi: 10.1007/BF02533750. [DOI] [PubMed] [Google Scholar]
- Li X, Deng W, Nail CD, Bailey SK, Kraus MH, Ruppert JM, Lobo-Ruppert SM. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene. 2006;25:609–621. doi: 10.1038/sj.onc.1209077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Braunstein K, Kuizon E, Portman K, Qiao XT, Gumucio DL. Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development. 2005;132:279–289. doi: 10.1242/dev.01576. [DOI] [PubMed] [Google Scholar]
- Monzo M, Moreno I, Artells R, Ibeas R, Navarro A, Moreno J, Hernandez R, Granell M, Pie J. Sonic hedgehog mRNA expression by real-time quantitative PCR in normal and tumor tissues from colorectal cancer patients. Canc Lett. 2006;233:117–123. doi: 10.1016/j.canlet.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Oniscu A, James RM, Morris RG, Bader S, Malcomson RD, Harrison DJ. Expression of Sonic hedgehog pathway genes is altered in colonic neoplasia. J Pathol. 2004;203:909–917. doi: 10.1002/path.1591. [DOI] [PubMed] [Google Scholar]
- Palma V, Ruiz i Altaba A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development. 2004;131:337–345. doi: 10.1242/dev.00930. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu Y, Huang L, Prins GS. Sonic hedgehog-patched Gli signaling in the developing rat prostate gland: lobe-specific suppression by neonatal estrogens reduces ductal growth and branching. Dev Biol. 2004;273:257–275. doi: 10.1016/j.ydbio.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualtrough D, Buda A, Gaffield W, Williams AC, Paraskeva C. Hedgehog signaling in colorectal tumour cells: induction of apoptosis with cyclopamine treatment. Int J Canc. 2004;110:831–837. doi: 10.1002/ijc.20227. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development. 1999;126:3205–3216. doi: 10.1242/dev.126.14.3205. [DOI] [PubMed] [Google Scholar]
- Ruiz I Altaba A. Therapeutic inhibition of Hedgeog-GLI signaling in cancer: Epithelial, stromal or stem cell targets? Canc Cell. 2008;14:281–283. doi: 10.1016/j.ccr.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007;17:438–447. doi: 10.1016/j.tcb.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez P, Hernández AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, Beyna M, Datta MW, Datta S, Ruiz i Altaba A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci USA. 2004;101:12561–12566. doi: 10.1073/pnas.0404956101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez P, Ruiz i Altaba A. In vivo inhibition of endogenous brain tumors through systemic interference with Hedgehog signaling in mice. Mech Dev. 2005;122:223–230. doi: 10.1016/j.mod.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, St Clair R, Baljevic M, White I, Jin DK, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J Clin Invest. 2008;118:2111–2120. doi: 10.1172/JCI34401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van Es JH, van den Born M, Begthel H, Stange DE, Barker N, Clevers H. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology. 2009;136:2187–2194. doi: 10.1053/j.gastro.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, Beermann F, Ruiz i Altaba A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci USA. 2007;104:5895–5900. doi: 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecca B, Ruiz i Altaba A. A GLI1-p53 inhibitory loop regulates neural stem cell and tumor cell numbers. EMBO J. 2009;28:663–679. doi: 10.1038/emboj.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ, de Sauvage FJ. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci USA. 2009;106:4254–4259. doi: 10.1073/pnas.0813203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G, Medema JP, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- van den Brink GR, Bleuming SA, Hardwick JC, Schepman BL, Offerhaus GJ, Keller JJ, Nielsen C, Gaffield W, van Deventer SJ, Roberts DJ, et al. Indian Hedgehog is an antagonist of Wnt signaling in colonic epithelial cell differentiation. Nat Genet. 2004;36:277–282. doi: 10.1038/ng1304. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, Richel DJ, Stassi G, Medema JP. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci USA. 2008;105:13427–13432. doi: 10.1073/pnas.0805706105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–410. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603–607. doi: 10.1038/nature07589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.