

Abstract
Mutations in the MSH2 gene predispose to a number of tumourigenic conditions, including hereditary non-polyposis colon cancer (HNPCC). MSH2 encodes a protein in the mismatch repair (MMR) pathway which is involved in the removal of mispairs originating during replication or from damaged DNA. To identify new therapeutic strategies for the treatment of cancer arising from MMR deficiency, we screened a small molecule library encompassing previously utilized drugs and drug-like molecules to identify agents selectively lethal to cells lacking functional MSH2. This approach identified the drug methotrexate as being highly selective for cells with MSH2 deficiency. Methotrexate treatment caused the accumulation of potentially lethal 8-hydroxy-2'-deoxyguanosine (8-OHdG) oxidative DNA lesions in both MSH2 deficient and proficient cells. In MSH2 proficient cells, these lesions were rapidly cleared, while in MSH2 deficient cells 8-OHdG lesions persisted, potentially explaining the selectivity of methotrexate. Short interfering (si)RNA mediated silencing of the target of methotrexate, dihydrofolate reductase (DHFR), was also selective for MSH2 deficiency and also caused an accumulation of 8-OHdG. This suggested that the ability of methotrexate to modulate folate synthesis via inhibition of DHFR, may explain MSH2 selectivity. Consistent with this hypothesis, addition of folic acid to culture media substantially rescued the lethal phenotype caused by methotrexate. While methotrexate has been used for many years as a cancer therapy, our observations suggest that this drug may have particular utility for the treatment of a subset of patients with tumours characterized by MSH2 mutations.
Keywords: colorectal cancer, methotrexate, mismatch DNA repair, oxidative DNA damage
INTRODUCTION
Heterozygous germline mutations in genes such as MSH2 that encode components of the DNA mismatch repair (MMR) pathway predispose individuals to cancer and in particular, hereditary non-polyposis colorectal cancer (HNPCC). As with many tumour suppressor genes, inactivation of the remaining wild-type allele in MSH2 mutant tumours is common and can occur either by somatic mutation (Cunningham et al, 2001; Leach et al, 1993) or loss of heterozygosity (LOH) (Potocnik et al, 2001; Yuen et al, 2002). HNPCC accounts for approximately 5% of all colorectal cancer cases (Jacob & Praz, 2002) and current estimates suggest that just under 40% of HNPCC kindreds bear MSH2 mutations (Peltomaki & Vasen, 1997).
The underlying cause of the association between MSH2 mutation and cancer is thought to be a failure of MMR, a pathway which primarily eliminates base–base mismatches and insertion/deletion loops arising during DNA replication. MSH2 is key to this process, recognizing DNA mismatches as a heterodimer with either MSH3 or MSH6 (Seifert & Reichrath, 2006). In general, the MSH2/MSH6 heterodimer recognizes single base mismatches and short insertion/deletion loops in DNA, whereas the MSH2/MSH3 heterodimer recognizes larger loops (Genschel et al, 1998; Umar et al, 1998). In addition to its role in the repair of replication errors, MMR also repairs mispaired bases that arise during homologous recombination or as a result of oxidative DNA damage (O'Brien & Brown, 2006). Unsurprisingly, MMR deficient cells exhibit a mutator phenotype, characterized by an elevated spontaneous mutation rate alongside microsatellite instability (MSI) (O'Brien & Brown, 2006).
While the cause and effect of the relationship between MMR deficiency and tumourigenesis is clear, this information has not yet been utilized in the development of targeted therapies for tumours characterized by MMR gene defects. Here, we sought to identify agents that could selectively kill MSH2 deficient cells. One of the major limitations to the rapid development of novel drugs is the time, cost and effort that is incurred in developing prototype small molecule inhibitors into potent, drug-like compounds (Collins & Workman, 2006). Given this, one approach is to screen libraries comprised of already approved drugs with known toxicity profiles that could be rapidly progressed into clinical trials (O'Connor & Roth, 2005). Examples of this approach include the use of non-steroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor inhibitors as potential therapeutic agents for Alzheimer's disease (Combs et al, 2001; Yan et al, 2003). Similarly, the generic medication fluphenazine has been identified as a novel therapeutic for multiple myeloma, leading to its progression into clinical trials (Glaser, 2004). Taking a similar approach, we screened a library of off-patent drugs to identify MSH2-selective agents. Notable among the agents we identified was the drug methotrexate. We illustrate that the effects of methotrexate in MSH2 deficient tumour cells can, in part, be explained by an increase in oxidized DNA lesions caused by this drug.
RESULTS
Identification of compounds that are selective for MSH2 deficiency
To clearly identify effects that are selective for genetic differences, the use of isogenic cell lines is essential (Kaelin, 2005). To identify mutS homolog 2 (MSH2)-selective agents, we used the previously characterized human endometrial adenocarcinoma cell line, Hec59, which carries two different loss of function MSH2 nonsense mutations (Boyer et al, 1995). As an MSH2 proficient isogenic comparator, we used the Hec59 + Chr2 cell line (Umar et al, 1997); transfer of human chromosome 2 into Hec59 cells restores wild type MSH2 expression and functional MMR (Umar et al, 1997). To screen these cell lines for agents that are selectively lethal to MSH2 deficient cells, we designed a 96-well plate-based cell viability assay. In this assay, cells were plated in 96-well plates, after which media containing drug was added to each well and replenished every 48 h. After six days continuous culture, cell viability in each well was estimated using a luciferase-based ATP assay (CellTitre Glo). The chemical library used encompassed 1120 small molecules, 90% of which were marketed drugs, the remaining 10% being bioactive alkaloids. The effect of each compound on cell viability was estimated by comparing luciferase readings from drug-treated wells with those from wells treated with the compound vehicle, dimethyl sulphoxide (DMSO). For each cell line, the effect of each compound was represented as a log2-surviving fraction (log2SF), and to identify MSH2-selective effects, we compared log2SFs with Hec59 and Hec59 + Chr2 cells (Fig 1A). As a positive control, we included the methylating agent, N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) in the compound library. MNNG has a well-validated differential drug response in Hec59 and Hec59 + Chr2 cells; Hec59 cells are more resistant to MNNG than Hec59 + Chr2 cells (Umar et al, 1997). Using our screening protocol we were able to reproduce this effect (MNNG log2SF at 1 µM in Hec59 = 0.027 and in Hec59 + Chr2 = −0.519). We also used the effects of MNNG to assess the dynamic range and sensitivity of the screen, as represented by a Z factor (Zhang et al, 1999). Z factors between 0.5–1 predict high-quality screens (Zhang et al, 1999) and the Z-factor for our screen, using MNNG sensitivity, was 0.514. This provided some level of confidence that the screen would produce robust and meaningful data.
Figure 1. Identification of small molecule inhibitors that target MSH2 deficiency.
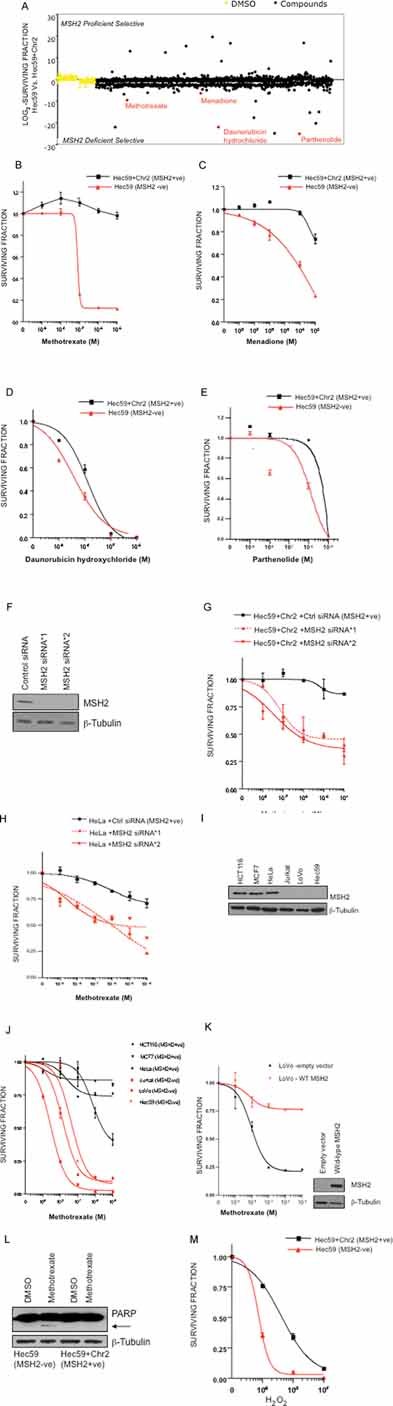
A. High-throughput screen (HTS) identifying MSH2-selective compounds. Scatter plot showing the relative effects of 1120 compounds on Hec59 and Hec59 + Chr2 cells in a high-throughput viability screen. Cells were plated in 96-well plates and exposed to library compounds for five days, using an average compound concentration in media of 0.1 mM. The effect of each compound was represented as a log2 SF, compared to DMSO treated cells. To identify MSH2-selective effects, log2 SFs from the two cell lines were compared.
B–E. Validation of MSH2 selective compounds from the HTS. Clonogenic survival curves are shown of Hec59 and Hec59 + Chr2 cells under continuous exposure (14 days) to compounds identified in the HTS. B, methotrexate; C, menadione; D, daunorubicin hydrochloride; E, parthenolide. Error bars represent standard errors of the mean.
F. Western blot analysis of siRNA silencing. Hec59 + Chr2 cells transfected with either control siRNA or two different MSH2 siRNAs. Protein lysates were immunoblotted and probed for MSH2 and β-tubulin (loading control).
G. MSH2 silencing sensitizes Hec59 + Chr2 cells to methotrexate. Survival curves are shown of Hec59 + Chr2 cells transfected with either control siRNA or siRNAs targeting MSH2, and then continuously exposed to methotrexate for 14 days.
H. MSH2 silencing sensitizes HeLa cells to methotrexate. Survival curves are shown of HeLa cells transfected with either control siRNA or siRNAs targeting MSH2, and then continuously exposed to methotrexate for 14 days.
I. Western blot analysis of MSH2 expression. Protein lysates from HeLa, MCF7, HCT116, LoVo, Hec59 and Jurkat cells were immunoblotted and probed for MSH2 and β-tubulin (loading control).
J. Mutation in MSH2 sensitizes a range of tumour cells to methotrexate. Survival curves are shown for the wild-type MSH2 cells HeLa, MCF7 and HCT116 and the MSH2 mutated cell lines, LoVo, Hec59 and Jurkat, continuously exposed to methotrexate for 14 days.
K. Re-expression of wild-type MSH2 restores methotrexate resistance. MSH2 deficient LoVo cells were transfected with either empty or wt-MSH2 pcDNA3 vector and after 48 h were continuously exposed to methotrexate for 14 days. Protein lysates were immunoblotted and probed to show re-expression of MSH2 and β-tubulin as a loading control.
L. Induction of apoptosis in Hec59 cells treated with methotrexate. Western blot of lysates from Hec59 + Chr2 and Hec59 cells, 72 h after treatment with 10 nM methotrexate or DMSO. Protein lysates were immunoblotted and probed for PARP1 and β-tubulin (loading control). Arrow denotes cleavage product of PARP1.
M. H2O2 treatment is selective for MSH2 deficient cells. Survival curves are shown of Hec59 and Hec59 + Chr2 cells under continuous exposure to a range of concentrations of H2O2 for 72 h. Error bars represent standard errors of the mean.
Methotrexate and other oxidative agents are selective for MSH2 deficiency
Using the comparison of log2-SFs from the two cell lines, we identified a number of MSH2 selective agents (Table S1 of Supporting Information). Among these, we noted a number of agents known to cause oxidative damage, namely methotrexate, menadione, parthenolide and daunorubicin hydrochloride (Cojocel et al, 2006; Kurdi et al, 2007; Mizutani et al, 2003; Rajamani et al, 2006). Given a potential link between oxidative agents and MSH2 selectivity, we first validated the selectivity of these agents using the gold standard method for estimating effects on cell viability, colony formation assays (Brown & Attardi, 2005) (Fig 1B–E). Methotrexate (Hec59 SF50 = 70 nM, Hec59 + Chr2 SF50>1 × 10−5 M, over 140 fold difference), menadione (Hec59 SF50 = 400 µM, Hec59 + Chr2 SF50>1 × 10−3 M, over 2.5 fold difference), daunorubicin hydrochloride (Hec59 SF50 = 3 nM, Hec59 + Chr2 SF50 = 13 nM, over 4 fold difference) and parthenolide (Hec59 SF50 = 600 nM, Hec59 + Chr2 SF50 = 6 µM, 10 fold difference) were all able to elicit MSH2 selectivity over a range of doses (Fig 1B–E), validating the results of the initial screen.
We confirmed the MSH2 specificity of the most selective agent, methotrexate. Silencing of MSH2 by short interfering RNA (siRNA) (Fig 1F) sensitized the Hec59 + Chr2 cell line to methotrexate (Fig 1G). MSH2 silencing in HeLa cells also caused sensitivity to methotrexate (Fig 1H), suggesting that the MSH2 selectivity of this drug was not limited to the Hec59 cell model.
To further assess the generality of our observations, we examined methotrexate sensitivity in a panel of MSH2 deficient and proficient tumour cell lines (Fig 1I & J). Although MSH2 mutations are not the only genetic variables within this diverse tumour cell panel, we observed a clear distinction in methotrexate sensitivity between tumour lines with wild-type MSH2 expression, such as HeLa, MCF7 and HCT116, and those with MSH2 deficiency, such as LoVo, Hec59 and Jurkat (Fig 1I & J). Given the role of MSH2 in DNA repair, it was also possible that the methotrexate sensitivity observed in MSH2 deficient cells could be explained by mutations that occur secondary to MSH2 deficiency and not MSH2 deficiency per se. However, expression of wild-type MSH2 in the MSH2 deficient LoVo cell line, restored resistance to methotrexate treatment (Fig 1K), suggesting that this was unlikely to be the case. Finally, stable silencing of MSH2 by short hairpin (sh)RNA in HeLa and MCF7 cells also resulted in methotrexate sensitivity (Fig S1A & B of Supporting Information), confirming our previous results.
We also addressed whether methotrexate sensitivity was particular to MSH2 deficiency or also observed in other cell lines with other MMR defects, such as loss of MutL homologue 1 (MLH1) function. Substantial methotrexate sensitivity was not observed in the MLH1 deficient colorectal cell line HCT116, when compared to the MLH1 proficient comparator cell line HCT116 + Chr3 (Fig S1C of Supporting Information).
We explored whether methotrexate treatment in combination with MSH2 deficiency, results in loss of cellular viability by apoptosis. Cleavage of poly (ADP-ribose) polymerase (PARP) is a recognized marker of apoptosis induction (Boulares et al, 1999) and methotrexate treatment resulted in cleavage of PARP in MSH2 deficient Hec59 cells, but not in the MSH2 proficient Hec59 + Chr2 cells (Fig 1L). This indicated that the loss of viability observed upon MSH2 deficiency combined with methotrexate treatment, is associated with the induction of apoptosis.
The identification of a number of oxidative agents in these studies raised the possibility that, causing oxidative damage could elicit MSH2 selectivity. To directly address this hypothesis, Hec59 and Hec59 + Chr2 cells were treated with increasing concentrations of the oxidizing agent hydrogen peroxide (H2O2), and cell viability was estimated after 72 h. H2O2 was indeed selective for MSH2 deficiency (2-fold difference at 1 nM H2O2, Fig 1M), supporting the suggestion that MSH2 selectivity could be achieved by agents that cause oxidative damage.
Oxidative damage caused by methotrexate persists in MSH2 deficient cells
To further investigate the possibility that MSH2 selectivity of agents such as methotrexate and menadione could be explained by oxidative damage, we measured levels of the most prevalent product of DNA oxidation, the oxidized base 8-hydroxy-2'-deoxyguanosine (8-OHdG). MSH2 is thought to play a role in the repair of 8-OHdG lesions and has been shown to be involved in reducing the levels of oxidized bases in DNA (Macpherson et al, 2005). Inactivation of Msh2 has also been associated with increased oxidized lesions in several organs of the mouse (Russo et al, 2007). Msh2−/− mouse embryo fibroblasts (MEFs) accumulate approximately two-fold more oxidized guanine than wild-type MEFs (Colussi et al, 2002). Hec59 and Hec59 + Chr2 cells were treated with sub-lethal concentrations of either methotrexate (10 nM) or menadione (100 nM) and DNA was extracted for analysis 72 h later. We quantified 8-OHdG levels in DNA using a highly sensitive 8-OHdG competitive ELISA assay (Shen et al, 2007). In MSH2 deficient Hec59 cells, methotrexate and menadione treatment caused significant increases in the amount of cellular 8-OHdG, compared to DMSO-treated cells (Fig 2A). However, neither methotrexate nor menadione elicited the same increase in 8-OHdG in MSH2 proficient Hec59 + Chr2 cells 72 h after the initiation of treatment, demonstrating that the level of oxidative DNA damage correlated with the MSH2 selectivity of methotrexate and menadione (Fig 2A). Silencing of MSH2 by siRNA (Fig S2A of Supporting Information) in the Hec59 + Chr2 cell line also resulted in an accumulation of 8-OHdG lesions following treatment of methotrexate. Similarly, in MCF7 cells carrying a doxycycline-inducible shRNA targeting MSH2 (shMSH2), methotrexate treatment caused an increase in 8-OHdG in comparison to MCF7 cells carrying a doxycycline-inducible non-silencing shRNA control (shCtrl; Fig S2B of Supporting Information).
Figure 2. Oxidative damage inducing compounds are selectively lethal for MSH2 deficiency.
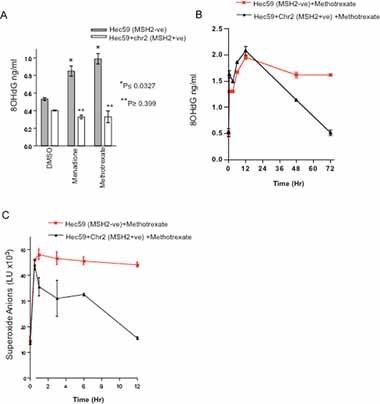
- Methotrexate and menadione induce oxidative DNA damage in MSH2 deficient cells. Hec59 and Hec59 + Chr2 cells were treated with 10 nM methotrexate or 100 nM menadione. After 72 h, DNA was isolated and analysed for 8-OHdG content using a specific 8-OHdG ELISA assay. Oxidized lesions were quantified according to an 8-OHdG standard curve. Assays were performed in triplicate and bar chart showing relative levels of 8-OHdG in each cell line is shown *p ≤ 0.0327 compared to the similarly treated MSH2 proficient Hec59 + Chr2 cells (Student's t-test). **p ≥ 0.399 compared to the DMSO treated Hec59 + Chr2 cells. Error bars represent standard errors of the mean.
- Methotrexate causes an accumulation of 8-OHdG lesions over time in MSH2 deficient cells. Hec59 and Hec59 + Chr2 cells were treated with 10nM methotrexate and DNA analysed for 8-OHdG content over time, as before. A line graph showing relative levels of 8-OHdG content over time is shown. Assays were performed in triplicate. Error bars represent standard errors of the mean.
- Methotrexate causes a sustained increase in superoxide anions over time in MSH2 deficient cells. Hec59 and Hec59 + Chr2 cells were treated with 10 nM methotrexate and analysed for levels of superoxide anions over time. A line graph showing relative levels of superoxide anions over time is shown. Assays were performed in triplicate. Error bars represent standard errors of the mean.
To further investigate this rise in 8-OHdG levels, we assessed the presence of this oxidized base over time (Fig 2B; Fig S2C of Supporting Information). In both MSH2 proficient and deficient cells, methotrexate initially caused an increase in 8-OHdG levels that peaked 12 h after the initial exposure to the drug. In MSH2 proficient Hec59 + Chr2 cells and in MCF7 cells carrying a doxycycline-inducible shCtrl, levels of 8-OHdG rapidly declined after 12 h, falling to basal levels by 72 h, supporting our previous observation (Fig 2A & B; Fig S2B & C of Supporting Information). However, in MSH2 deficient Hec59 cells and MCF7 cells in which MSH2 is silenced with a doxycycline-inducible shMSH2, 8-OHdG levels remained elevated after 12 h and failed to significantly diminish, even after 72 h (Fig 2B; Fig S2C of Supporting Information). Taken together, these data support the hypothesis that in the presence of functional MSH2, agents such as methotrexate cause oxidative damage, which is rapidly cleared from DNA. Conversely, in the absence of wild-type MSH2, this damage persists and perhaps limits survival or at least fitness of cells, potentially explaining the MSH2 selectivity of methotrexate. In support of this hypothesis, levels of superoxide anions were also elevated after methotrexate treatment (Fig 2C); in MSH2 proficient cells, levels of superoxide anions fell to a basal level 12 hours after initial methotrexate exposure, while in MSH2 deficient cells, levels of superoxide anions remain elevated. Taken together, this data further suggested that the differential selectivity of MSH2 deficiency and methotrexate treatment could be due to a sustained accumulation of oxidative damage in the cell.
We also analysed the expression of the oxidative damage enzymes MutYH (MYH), 8-oxoguanine glycosylase (OGG1) and MTH1 in MSH2 deficient and proficient cell lines (Fig S2D of Supporting Information). Hec59 and Hec59 + Chr2 cells were treated with either DMSO or methotrexate (100 nM) and after 72 h, protein lysates were isolated. The protein levels of MTH1 or OGG1 did neither appear to be grossly changed by MSH2 deficiency nor methotrexate exposure. However, MYH levels were considerably higher in MSH2 deficient cells than in MSH2 competent cells. One of the known effects of 8-OHdG accumulation is an increase in GC>TA transversions as 8-OHdG lesions are replicated. MYH excises adenine residues inserted opposite 8-OHdG at replication, providing a mechanism by which the transversion frequency can be suppressed (Oka et al, 2008). It is possible that the elevated levels of MYH expression in MSH2 deficient cells represent the response to a reduced ability to process 8-OHdG lesions prior to replication.
Anti-oxidants and folate rescue the effects of methotrexate on MSH2 deficient cells
While the persistence of oxidative DNA damage represented a potential mechanism, explaining the selectivity of methotrexate for MSH2 deficient cells, it was still possible that the elevated levels of 8-OHdG observed were merely coincidental and not the cause of methotrexate selectivity. To address this issue, we attempted to abrogate the lethal effect of methotrexate in MSH2 deficient cells by counteracting the oxidative damage caused by this drug. Selenium is a cofactor of the enzyme glutathione peroxidase which aids the elimination of free radicals and reactive oxygen species from cells (Rotruck et al, 1973). Supplementation of tissue culture media with selenomethionine has been shown to significantly reduce 8-OHdG levels caused by ultraviolet radiation (Rafferty et al, 2003). We treated MSH2 deficient and proficient cells with methotrexate alone, or in combination with 1 µM selenomethionine and then estimated cell viability and 8-OHdG levels. Selenomethionine treatment rescued the lethal phenotype caused by methotrexate in MSH2 deficient cells (Fig 3A; Fig S3A of Supporting Information). Furthermore, selenomethionine substantially reduced 8-OHdG accumulation in MSH2 deficient cells treated with methotrexate (Fig 3B; Fig S3B of Supporting Information). Taken together, this suggested that oxidative damage most likely underlies the MSH2 selectivity of methotrexate.
Figure 3. Treatment with selenomethionine rescues the oxidative damage induced selectivity with MSH2 deficiency.
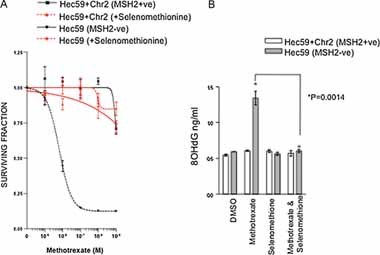
- Selenomethionine rescues methotrexate selectivity in MSH2 deficient cells. Survival curves are shown from Hec59 and Hec59 + Chr2 cells under continuous exposure to a range of concentrations of Methotrexate ± 1 µM selenomethionine for 14 days. Error bars represent standard errors of the mean.
- Selenomethionine reduces 8-OHdG accumulation in MSH2 deficient cells. Cells were treated as in (A) and DNA analysed for 8-OHdG content as before. A bar chart showing relative levels of 8-OHdG in each cell line is shown. Assays were performed in triplicate. *p ≤ 0.0014 compared to the similarly methotrexate treated Hec59 cells in combination with selenomethionine (Student's t-test). Error bars represent standard errors of the mean.
Methotrexate inhibits dihydrofolate reductase (DHFR), a critical component of the folate metabolic pathway (Goodsell, 1999). To assess whether the effects of methotrexate on MSH2 deficient cells could be explained by inhibition of DHFR, we silenced DHFR expression by RNA interference (Fig 4A). Silencing of DHFR in MSH2 deficient cells resulted in a significant reduction in survival compared to MSH2 proficient cells (Fig 4B; Fig S3C of Supporting Information). To validate this observation, we attempted to rescue the effects of methotrexate using folic acid, the natural substrate of DHFR. MSH2 deficient and proficient cells were treated with methotrexate and, in addition, folic acid (100 µM) (Fig 4C; Fig S3D of Supporting Information). Folic acid treatment also rescued the lethal phenotype caused by methotrexate in MSH2 deficient cells (Fig 4C; Fig S3D of Supporting Information), suggesting that the observed selectivity of methotrexate upon MSH2 deficient cells is due to the inhibition of DHFR.
Figure 4. Methotrexate treatment selection for MSH2 deficient cells is via inhibition of folate synthesis.
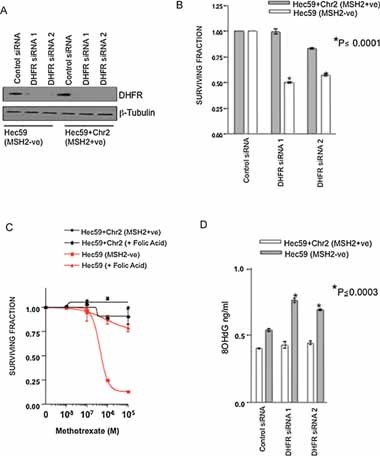
- Western blot analysis of DHFR siRNA silencing. Cells were transfected with either control siRNA or two different DHFR siRNAs. Protein lysates were immunoblotted and probed for DHFR and β-tubulin (loading control).
- Silencing of DHFR is selective for MSH2 deficient cells. Survival bar chart is shown of Hec59 + Chr2 and Hec59 transfected with siRNA oligonucleotides targeting DHFR. *p ≤ 0.0001 compared to the similarly transfected MSH2 proficient Hec59 + Chr2 cells (Student's t-test). Error bars represent standard errors of the mean.
- Folic acid rescues methotrexate selectivity in MSH2 deficient cells. Survival curves are shown of Hec59 and Hec59 + Chr2 cells under continuous exposure to a range of concentrations of methotrexate ± 100 µM folic acid for 14 days. Error bars represent standard errors of the mean.
- DHFR silencing causes an accumulation of 8-OHdG accumulation. Hec59 and Hec59 + Chr2 cells were transfected with control or DHFR siRNA and DNA analysed for 8-OHdG content as before. A bar chart showing relative levels of 8-OHdG in each cell line is shown. Assays were performed in triplicate. *p ≤ 0.0003 compared to the similarly transfected MSH2 proficient Hec59 + Chr2 cells (Student's t-test). Error bars represent standard errors of the mean.
To extend this finding, we investigated whether inhibition of DHFR could also result in an accumulation of 8-OHdG lesions in MSH2 deficient cells. DHFR silencing by RNA interference caused a significant increase (p ≤ 0.0003) in 8-OHdG levels in MSH2 deficient Hec59 cells, whereas no increase was observed in the similarly treated MSH2 proficient cell line, Hec59 + Chr2 (Fig 4D). This data supported the hypothesis that in MSH2 deficient cells, inhibition of DHFR via treatment with methotrexate, induces the accumulation of 8-OHdG oxidative lesions beyond a level that is incompatible with viability.
Methotrexate is selective for MSH2 deficiency in vivo
To assess methotrexate selective efficacy in vivo, we xenografted MSH2 deficient Hec59 cells into mice and treated animals with methotrexate. Once established, tumour growth from these xenografts was extremely rapid. Nevertheless, we observed that xenograft growth from MSH2 deficient xenografts was significantly reduced (p = 0.049) by methotrexate treatment when compared to vehicle-treated MSH2−/− xenografts (Fig 5A). No difference in tumour growth (p = 0.33) was observed in the methotrexate treated MSH2+/+ xenograft tumours when compared to the vehicle-treated MSH2+/+ tumours (Fig 5B). The efficacy of methotrexate was also suggested by a high level of apoptosis in MSH2−/− xenografts treated with methotrexate (p = 0.0001), as assessed by TUNEL staining (Fig 5C; Fig S4A of Supporting Information). Significantly no increase in TUNEL stained cells was observed in the MSH2+/+ xenografts upon methotrexate treatment. An increased level of necrosis was also observed in methotrexate treated MSH2−/− xenografts, as defined by nuclear fragmentation, loss of cell borders, presence of cell debris, karyorexis and cystification (Fig S4B of Supporting Information). To further validate our in vitro observations in vivo, 8-OHdG accumulation was quantified in DNA extracted from xenograft tumours (Fig 5D). Methotrexate treatment caused a significant increase in the amount of 8-OHdG in MSH2−/− xenografts, when compared to vehicle-treated MSH2−/− xenografts. No such increase in 8-OHdG lesions was observed in the MSH2+/+ xenograft tumours upon treatment with methotrexate. Taken together, these in vivo observations provide preliminary evidence that methotrexate, a drug already in clinical use, might show utility in patients with MSH2-deficient tumours.
Figure 5. Methotrexate is selective for MSH2 deficiency in vivo.
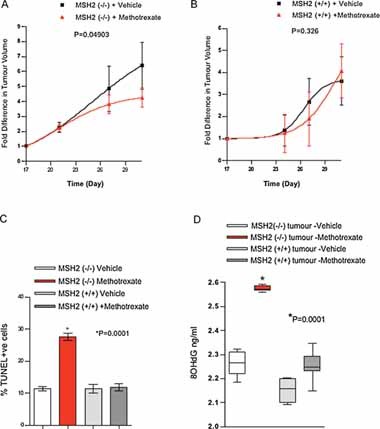
- A, B. Methotrexate suppresses growth in MSH2−/− subcutaneous xenografts. Hec59 (A) or Hec59 + Chr2 (B) cells were mixed 1:1 in matrigel and then injected subcutaneously into the lateral flank of 6–8 week old female athymic nude mice. The tumours were allowed to develop to a mean tumour diameter between 4 and 8 mm and randomized into treatment and control groups (10 animals per cohort, 20 in total) and drug dosing initiated. Methotrexate (or vehicle) was administered by intrapertineal injection at a dose of 30 mg/kg in PBS for five consecutive days followed by two days of no treatment after which the same treatment cycle was continued until the end of the study. Tumour volumes were measured every four days from the initiation of drug dosing and the results expressed as fold increase in tumour volume relative to that at the first drug administration. (A) p = 0.04903, compared to the vehicle treated MSH2(−/−) xenograft tumours (Two-way ANOVA test). (B) p = 0.326 compared to the vehicle treated MSH2(+/+) xenograft tumours (Two-way ANOVA test).
- C. Methotrexate increases the number of apoptotic cells in MSH2−/− subcutaneous xenografts. TUNEL-positive cells were measured in frozen tumour sections from Hec59 and Hec59 + Chr2 xenograft tumours treated with either vehicle or methotrexate, using an ApopTag Plus In Situ Apoptosis Detection Kit (Chemicon International, Temecula, CA). The total number of TUNEL-positive cells was quantified in four randomly selected microscopic fields consisting of approximately 800 cells/field at ×200 magnification within cell nuclei. *p = 0.0001 compared to the vehicle treated Hec59 xenograft tumours (Student's t-test). Error bars represent standard errors of the mean.
- D. 8-OHdG accumulation in methotrexate treated MSH2−/− subcutaneous xenografts. DNA extracted from Hec59 and Hec59 + Chr2 xenograft tumours treated with either vehicle or methotrexate were analysed for 8-OHdG content as before. Assays were performed in triplicate. *p = 0.0001 compared to the vehicle treated Hec59 xenograft tumours (Student's t-test). Error bars represent standard errors of the mean.
DISCUSSION
The potential of molecularly directed therapy, based on targeting the underlying genetic defects in tumour cells, is that it may cause highly selective killing of tumour cells, while sparing normal cells. However, where loss of a tumour suppressor gene function is a known determinant of the disease, it is difficult to identify a direct therapeutic target as it is often technically complex to efficiently recapitulate tumour suppressor function. Given these considerations, the concept of synthetic lethality/sickness has been proposed as a means to functionally target susceptibilities in cancer cells, especially where loss of a tumour suppressor gene is evident (Ashworth, 2008; Kaelin, 2005). Here we describe a synthetic lethality/sickness approach to targeting a defect in a specific DNA repair pathway, MMR. We demonstrate a novel selective lethality resulting from treatment with oxidative damage-inducing drugs including methotrexate, in an MSH2-deficient background. We describe how in the absence of MSH2, this selectivity results in an increase in 8-OHdG oxidative lesions upon inhibition of DHFR, potentially identifying a molecular targeted therapy for MSH2 deficient tumours. It seems most likely that in tumour cells with a MSH2 deficiency, this rise in 8-OHdG is incompatible with cellular viability; conversely, in MSH2 proficient cells, 8-OHdG lesions caused by methotrexate are rapidly cleared, hence the genotypic selectivity. We do, of course, acknowledge the possibility that drugs that cause DNA damage could also, by increasing the mutational load in tumour cells with DNA repair defects, increase malignancy. This is indeed a common critique of therapeutic strategies that target DNA repair deficiencies. However, we believe that if used for limited periods and in the correct patient group, the therapeutic effect would far outweigh the potential mutagenic effects. When used in such a context, we believe that MSH2 deficient tumour cells would more probably die in response to methotrexate, rather than survive and become more malignant. Nevertheless, limited treatment regimens are appropriate as they would also limit the potential for the development of drug resistance.
The standard adjuvant treatments for colorectal cancer, involving 5-fluorouracil (5-FU)-based chemotherapy regimens (Chau & Cunningham, 2006) are less effective in patients with MMR deficient tumours. For example, the presence of MSI, a sign of MMR deficiency, can predict a poor response to adjuvant 5-FU chemotherapy, as measured by progression free survival or overall survival (Jover et al, 2006; Ribic et al, 2003; Sargent et al, 2008). In addition, in vitro studies suggest that MMR defective colon cancer cells show increased resistance to 5-FU compared with MMR proficient cells (Arnold et al, 2003). Taken together, this suggests that there is a clinical need for therapies that specifically target MMR gene defects particularly in cases at high risk of relapse, such as Dukes C colorectal cancer. The work that we have presented here, suggests that methotrexate may have some utility in this area. Methotrexate has been frequently used in the treatment of cancer and its low toxicity profile has led to efforts to derive further antifolates for cancer treatment (McGuire, 2003). In fact, methotrexate has already been trialed in colorectal cancer, in a sequential regimen with 5-FU (Sobrero et al, 2005). While this study determined that adjuvant sequential use of 5-FU and methotrexate results in similar outcome as the standard combination of 5-FU and leucovorin, the patient group studied was not stratified by MMR or MSH2 status and thus any selectivity of methotrexate for MSH2 deficient tumours would have gone unnoticed. It should also be noted that patients with HNPCC are also at increased risk of tumours of the endometrium, stomach and transitional cell carcinoma of the urinary tract (Lynch & Lynch, 1995; Muller & Fishel, 2002), suggesting the potential utility of our approach outside of colorectal cancer.
It is also possible that other agents could be used for the treatment of MMR-defective cancers. Berry et al describe how MMR deficient cells can be targeted for radiosensitization by the halogenated thymidine analogues iododeoxyuridine (IdUrd) and bromodeoxyuridine (BrdUrd) (Berry et al, 2000). This approach relies on the reduced removal of the DNA adducts caused by these drugs in MMR deficient cells. Gemcitabine has also been proposed as an MMR selective agent (Robinson et al, 2003). For example, radiosensitization is increased in MLH1 deficient HCT116 cells exposed to Gemcitabine. This effect is most likely mediated by the inhibition of ribonucleotide reductase by Gemcitabine, the resultant reduction in dATP levels and the subsequent misincorporation of nucleotides into DNA; in MMR deficient cells, the removal of these misincorporated bases is reduced, which most likely impairs the fitness of cells. MMR defective mouse and human cell lines have also been reported to be sensitive to drugs inducing interstrand-cross-links (ICLs) including CCNU and MMC (Aquilina et al, 1998; Wu et al, 2005). We should also note that a number of compounds in the screening set that are thought to cause oxidative damage but were not identified as selective agents (Table S1 of Supporting Information). This may be for a number of reasons, including (i) these non-selective compounds could have other (non oxidative damage) effects in both MSH2 competent and deficient cells. These additional effects could mask the potentially selective effects of DNA damage (ii) like the majority of drug screens, the compound collection was screened at a single concentration to enable the high-throughput approach to be taken. It is possible that some of the non-selective agents only cause sufficient oxidative damage at concentrations not assessed in the screen.
In addition to methotrexate, inhibition of DHFR by RNA interference also elicited MSH2 selectivity and the accumulation of 8-OHdG lesions in MSH2 deficient cells (Fig 4). This suggests that folate metabolism most likely underlies the mechanism of lethality in MSH2 deficient cells. A number of reports have also suggested that oxidative stress can be induced by folate deficiency (Balaghi et al, 1993; Lan et al, 2007; Mato & Lu, 2005; Varela-Moreiras & Selhub, 1992). While the exact mechanisms underlying the antioxidant function of folates are still unclear, folic acids produced by DHFR control the catabolism of the oxidizing agent homocysteine (Hcy); folic acids donate methyl groups to Hcy as part of the remethylation pathway, producing methionine (Brosnan et al, 2004). In the absence of DHFR activity and methyl group donation, Hcy is known to auto-oxidize, producing disulphides such as Hcy as well as reactive oxygen species such as superoxide and H2O2 (Brosnan et al, 2004) that may well cause the formation of DNA lesions such as 8-OHdG. It should be noted that reports have been made suggesting that MMR-deficient cells grown in folate-deprived media are more resistant to apoptosis (Gu et al, 2002; Li et al, 2003). However, these latter studies focussed upon MLH1 deficient cell lines that express the MSH2 protein. Here, we demonstrate that methotrexate or antifolate treatment may be appropriate for cases of MSH2 deficiency but not MLH1 deficiency. In a similar vein, a number of studies have suggested that MMR deficient cells, including MLH1 and PMS2, produce more methotrexate resistant colonies than MMR proficient cells (Frouin et al, 2001; Lin et al, 2001; Snijders et al, 2008). Lin et al (2001) report that the MSH2 and MSH6 mutated Hec59 and DLD1 cells, respectively, gave rise to increased methotrexate-resistant colonies compared to their MMR proficient paired cell lines. Frouin et al (2001) describe how the MLH1 deficient cell line HCT116 and a HeLa cell-clone12 strain deficient in PMS2 are less sensitive to methotrexate treatment when compared to their MMR-competent cell lines. Here, we show that methotrexate is not an appropriate agent to be used in patients with MLH1 deficient tumours as no differential selectivity is observed (Fig S1C of Supporting Information).
The availability of a biomarker for MSH2 deficiency should enable the identification of patients who would possibly benefit from methotrexate treatment. HNPCC patients may exhibit a distinct clinicopathological phenotype, presenting at a younger age, with tumours being predominantly right-sided, often mucinous, poorly differentiated, and sometimes distinguished by peritumoural lymphocytic reaction (Half & Bresalier, 2004). Additionally, both immunohistochemical detection of MSH2 and assessment of MSI status from primary tumour specimens are well-validated markers already in use in the clinical setting to screen for cases at high risk of HNPCC. In combination, these clinical and pathological features may serve as suitable markers for patients who may benefit from treatment with methotrexate.
In summary, our data supports the hypothesis that MSH2 deficiency can be targeted by therapies that cause oxidative DNA damage. Resistance of MMR deficient tumours to standard treatments such as 5-FU further argues for the testing of oxidative damaging agents, in particular methotrexate, as a specific therapeutic agent in the treatment of MSH2 deficient cancers.
METHODS
Cell lines and reagents
The human endometrial cell lines Hec59 and Hec59 + Chr2 were the kind gift of Dr. T. Kunkel (NIEHS, NC, USA). Hec59 + Chr2 and Hec59 cells were grown in DMEM F12 supplemented with FCS (10% v/v), glutamine and antibiotics. Cells containing human chromosome 2 were cultured under selective pressure of 400 µg/mL geneticin (G418 sulphate). The human colon cancer cell line HCT116 and HCT116 + chr3 were the kind gift of Dr. A. Clark (NIEHS, NC, USA) and were grown in McCoys 5A supplemented with FCS (10% v/v), glutamine, and antibiotics. Cells containing human chromosome 3 were cultured under selective pressure of 400 µg/mL geneticin (G418 sulphate). Jurkat cells were were grown in Rosewell Park Memorial Institute (RPMI-1640) medium supplemented with FCS (10% v/v), glutamine and antibiotics. LoVo, HeLa and MCF7 cells were grown in Dulbecco's Modified Eagle Medium (DMEM), supplemented with FCS (10% v/v), glutamine and antibiotics. shRNA expressing cells were established by infecting HeLa or MCF7 cells with shRNA expressing empty or hMSH2 vectors, which were generated by PCR amplification of 97mer DNA oligonucleotides as described (Paddison et al, 2004) and cloned into either the LMP vector (Dickins et al, 2005) or the doxycycline-inducible TRIPZ lentiviral vector. shRNA sequences were as follows:
shMsh2: TGCTGTTGACAGTGAGCGCCTCAGTGAATTAAGAGAAATATAGTGAAGCCACAGATGTATATTTCTCTTAATTCACTGAGATGCCTACTGCCTCGGA
The pcDNA3-wt-MSH2 vector was a kind gift from Dr. M. Meuth (Sheffield, UK). Selenomethionine, folic acid and H2O2 were purchased from Sigma.
Compound inhibitor screen
Compound libraries were purchased from Prestwick Chemicals (Saffron Walden, Essex, UK). Cells were plated in 96-well plates. After 12 h incubation, cells were exposed to compound or equimolar DMSO and media and compounds replenished every 48 h. The library was screened using an average compound concentration in media of 0.1 mM. Cell viability was measured six days later, using the CellTitre Glo assay (Promega) according to the manufacturer's instructions. Luminescence measurements were used to determine growth in the presence of the inhibitors. The effect of each of the compounds on viability in a particular cell line, was estimated by comparing luminescence readings from drug-treated cells with those from cells treated with the compound vehicle, DMSO. The effect of each compound was expressed as a log2 SF, compared to DMSO-treated cells and to identify selective effects, the log2 SFs were compared from the two cell lines.
Validation experiments were carried out with Methotrexate (Biomol International L.P.), Menadione, Parthenolide and Daunorubicin hydrochloride (Prestwick Chemicals). Validation of hit compounds was performed by clonogenic assays. Exponentially growing cells were seeded at various densities in six-well plates. Cells were treated with increasing concentrations of the compound. Cell medium was replaced every four days. After ten to fourteen days, cells were fixed in methanol, stained with crystal violet and counted. The plating efficiencies were calculated as the number of colonies divided by the number of cells plated for each compound treatment. The SF for a given sample was calculated as the plating efficiencies for each compound treated cells divided by the plating efficiencies of DMSO treated cells.
Measurement of 8-OHdG
Genomic DNA was extracted using the QIamp DNA isolation kit (Qiagen) and digested with nuclease P1. A commercially available ELISA kit from Cell Biolabs (UK) was used to determine levels of 8-OHdG in isolated DNA. The assays were performed according to the manufacturer's instructions. The 8-OHdG standard (0.078–20 ng/ml) or 10 µg DNA from cells was incubated with a 8-OHdG monoclonal antibody in a microtiter plate precoated with 8-OHdG. The assay was normalized by an equal amount of DNA used for each sample. Addition of 3,35,5-tetramethylbenzidine to replicate samples was followed by measurement of absorbance at 450 nm. Standard curves were calculated for all reactions with serial dilutions of 8-OHdG standard to calculate reaction efficiency. Samples were assayed in triplicate.
Measurement of superoxide anions
We used the superoxide anion cell-based assay kit (Sigma) according to the manufacturer's instructions. This assay is based on the oxidation of luminol by superoxide anions resulting in the formation of chemiluminescence light.
siRNA and plasmid transfections
Cells were transfected with siRNA (Qiagen, UK) targeting the following genes (target sequences shown):
DHFR*1, 5′- TACGGAGAAACTGAACTGAGA-3′
DHFR*2, 5′- AACCTCCACAAGGAGCTCATT-3′
MSH2*1, 5′-CCCATGGGCTATCAACTTAAT-3′
MSH2*2, 5′-TCCAGGCATGCTTGTGTTGAA-3′
Control, 5′-CATGCCTGATCCGCTAGTC-3′
For clonogenic assays, exponentially growing cells were seeded at various densities in six-well plates. For siRNA transfections, cells were transfected with individual siRNA using Lipofectamine 2000 (Invitrogen, UK) according to manufacturer's instructions. As a control for each experiment, cells were left un-transfected or transfected with a non-targeting control siRNA and concurrently analysed. pcDNA3 plasmids were transfected into cells using Fugene 6 (Roche, Burgess Hill, UK) according to manufacturer's instructions. Twenty-four hours after transfection, cells were seeded in six-well plates (at 1500 cells/well) and treated with drug or in the case of DHFR siRNA transfections, media was replaced the following day. After ten to fourteen days, cells were fixed in methanol, stained with crystal violet and counted. The plating efficiencies were calculated as the number of colonies divided by the number of cells plated for each siRNA transfection. The SF for a given sample was calculated as the plating efficiency of each siRNA transfection divided by the plating efficiencies of control siRNA transfected cells. All transfections were carried out in triplicate.
The paper explained
PROBLEM
Defects in components of the DNA MMR pathway, such as those caused by mutation of the MSH2 gene, predispose to a range of cancers and, in particular, HNPCC. In patients with cancers characterized by MSH2 mutations, tumour cells are normally MSH2 deficient, whereas normal cells are not—leading to efforts to target this tumour-specific difference as a therapeutic approach.
RESULTS
We screened MSH2 proficient and deficient human tumour cells with a library of commonly used drugs and drug-like compounds and show here that MSH2 deficient tumour cells are particularly sensitive to methotrexate. Methotrexate treatment of MSH2 deficient tumour cells causes an accumulation of potentially lethal oxidative DNA damage; in cells with functional MSH2, this damage is efficiently repaired but in MSH2 deficient tumour cells, repair of oxidative DNA damage is impaired and this eventually limits the survival of tumour cells.
IMPACT
The data demonstrate that a drug already used in cancer therapy, methotrexate, is MSH2-selective and could be used to treat cancer in specific subgroups of the patient population. A clinical trial aimed at assessing this possibility is now underway (see web link).
Protein analysis
Cell pellets were lysed in 20 mmol/l Tris (pH 8), 200 mmol/l NaCl, 1 mmol/l EDTA, 0.5% (v/v) NP40, 10% (v/v) glycerol and protease inhibitors. Lysates were electrophoresed on Novex precast gels (Invitrogen, UK) and immunoblotted using the following antibodies: anti-MSH2 (Ab-1, Calbiochem, UK), anti-DHFR (ab49881, Abcam, UK), anti-PARP (Cell Signalling), anti-MYH (ab55551, Abcam), anti-OGG1 (Novus), anti-MTH1 (ab98–230, Abcam) and anti-β-tubulin (T4026, Sigma, UK). This was followed by incubation with anti-IgG-horseradish peroxidase and chemiluminescent detection (SuperSignal West Pico Chemiluminescent Substrate, Pierce, UK). Immunoblotting for β-tubulin was used as a loading control.
Xenograft analysis
Hec59 and Hec59 + Chr2 cells were mixed 1:1 in matrigel and then injected subcutaneously into the lateral flank of 6–8 week old female athymic nude mice. Tumours were allowed to develop to a mean tumour diameter between 4 and 8 mm and randomized into treatment and control groups (10 animals per cohort, 20 in total) and drug dosing initiated. Methotrexate (or vehicle) was administered by intrapertineal injection at a dose of 30mg/kg in phosphate buffered saline (PBS) for five consecutive days followed by two days of no treatment after which the same treatment cycle was continued until the end of the study. Tumour volumes were measured every four days from the initiation of drug dosing and the results expressed as fold increase in tumour volume relative to that at the first drug administration.
TUNEL staining
Apoptosis was measured using an ApopTag Plus In Situ Apoptosis Detection Kit (Chemicon International, Temecula, CA) according to the manufacturer's instructions. The total number of TUNEL-positive cells was quantified in four randomly selected microscopic fields containing approximately 800 cells/field at ×200 magnification within cell nuclei.
Acknowledgments
We thank Dr. T. Kunkel and Dr. A. Clark for the provision of cell lines and Dr. M. Meuth for providing the MSH2 expression construct. We also thank Dr. M. Hewish for helpful discussions. This work was supported by grants from Cancer Research UK and Breakthrough Breast Cancer. We acknowledge NHS funding to the NIHR Biomedical Research Centre.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
Sarah A. Martin: Experimental design, all in vitro experiments, manuscript preparation. Afshan McCarthy and Suzanne Parry: Tumour xenograft analysis. Louise J. Barber and Darren J. Burgess: Reagent contribution. Christopher J. Lord and Alan Ashworth: Experimental design & manuscript preparation.
For more information
Clinical trial of Methotrexate in treating patients with MSH2 deficient advanced bowel cancer:
http://clinicaltrials.gov/ct2/show/NCT00952016
Breakthrough Research Center:
http://www.breakthroughresearch.org.uk/breakthrough_research_centre/index.html
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Aquilina G, Ceccotti S, Martinelli S, Hampson R, Bignami M. N-(2-chloroethyl)-N′-cyclohexyl-N-nitrosourea sensitivity in mismatch repair-defective human cells. Cancer Res. 1998;58:135–141. [PubMed] [Google Scholar]
- Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int J Cancer. 2003;106:66–73. doi: 10.1002/ijc.11176. [DOI] [PubMed] [Google Scholar]
- Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- Balaghi M, Horne DW, Woodward SC, Wagner C. Pancreatic one-carbon metabolism in early folate deficiency in rats. Am J Clin Nutr. 1993;58:198–203. doi: 10.1093/ajcn/58.2.198. [DOI] [PubMed] [Google Scholar]
- Berry SE, Davis TW, Schupp JE, Hwang HS, de Wind N, Kinsella TJ. Selective radiosensitization of drug-resistant MutS homologue-2 (MSH2) mismatch repair-deficient cells by halogenated thymidine (dThd) analogues: Msh2 mediates dThd analogue DNA levels and the differential cytotoxicity and cell cycle effects of the dThd analogues and 6-thioguanine. Cancer Res. 2000;60:5773–5780. [PubMed] [Google Scholar]
- Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274:22932–22940. doi: 10.1074/jbc.274.33.22932. [DOI] [PubMed] [Google Scholar]
- Boyer JC, Umar A, Risinger JI, Lipford JR, Kane M, Yin S, Barrett JC, Kolodner RD, Kunkel TA. Microsatellite instability, mismatch repair deficiency, and genetic defects in human cancer cell lines. Cancer Res. 1995;55:6063–6070. [PubMed] [Google Scholar]
- Brosnan JT, Jacobs RL, Stead LM, Brosnan ME. Methylation demand: a key determinant of homocysteine metabolism. Acta Biochim Pol. 2004;51:405–413. [PubMed] [Google Scholar]
- Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–237. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- Chau I, Cunningham D. Adjuvant therapy in colon cancer–what, when and how? Ann Oncol. 2006;17:1347–1359. doi: 10.1093/annonc/mdl029. [DOI] [PubMed] [Google Scholar]
- Cojocel C, Novotny L, Vachalkova A. Mutagenic and carcinogenic potential of menadione. Neoplasma. 2006;53:316–323. [PubMed] [Google Scholar]
- Collins I, Workman P. New approaches to molecular cancer therapeutics. Nat Chem Biol. 2006;2:689–700. doi: 10.1038/nchembio840. [DOI] [PubMed] [Google Scholar]
- Colussi C, Parlanti E, Degan P, Aquilina G, Barnes D, Macpherson P, Karran P, Crescenzi M, Dogliotti E, Bignami M. The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr Biol. 2002;12:912–918. doi: 10.1016/s0960-9822(02)00863-1. [DOI] [PubMed] [Google Scholar]
- Combs CK, Bates P, Karlo JC, Landreth GE. Regulation of beta-amyloid stimulated proinflammatory responses by peroxisome proliferator-activated receptor alpha. Neurochem Int. 2001;39:449–457. doi: 10.1016/s0197-0186(01)00052-3. [DOI] [PubMed] [Google Scholar]
- Cunningham JM, Kim CY, Christensen ER, Tester DJ, Parc Y, Burgart LJ, Halling KC, McDonnell SK, Schaid DJ, Walsh Vockley C, et al. The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am J Hum Genet. 2001;69:780–790. doi: 10.1086/323658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, Lowe SW. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37:1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- Frouin I, Prosperi E, Denegri M, Negri C, Donzelli M, Rossi L, Riva F, Stefanini M, Scovassi AI. Different effects of methotrexate on DNA mismatch repair proficient and deficient cells. Eur J Cancer. 2001;37:1173–1180. doi: 10.1016/s0959-8049(01)00095-8. [DOI] [PubMed] [Google Scholar]
- Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem. 1998;273:19895–19901. doi: 10.1074/jbc.273.31.19895. [DOI] [PubMed] [Google Scholar]
- Glaser V. Immune Control pursues serotonin antagonists. Genet Eng News. 2004;24:15. [Google Scholar]
- Goodsell DS. The molecular perspective: methotrexate. Stem Cells. 1999;17:314–315. doi: 10.1002/stem.170314. [DOI] [PubMed] [Google Scholar]
- Gu L, Wu J, Qiu L, Jennings CD, Li GM. Involvement of DNA mismatch repair in folate deficiency-induced apoptosis small star, filled. J Nutr Biochem. 2002;13:355–363. doi: 10.1016/s0955-2863(02)00178-x. [DOI] [PubMed] [Google Scholar]
- Half EE, Bresalier RS. Clinical management of hereditary colorectal cancer syndromes. Curr Opin Gastroenterol. 2004;20:32–42. doi: 10.1097/00001574-200401000-00008. [DOI] [PubMed] [Google Scholar]
- Jacob S, Praz F. DNA mismatch repair defects: role in colorectal carcinogenesis. Biochimie. 2002;84:27–47. doi: 10.1016/s0300-9084(01)01362-1. [DOI] [PubMed] [Google Scholar]
- Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, Pinol V, Xicola RM, Bujanda L, Rene JM, et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut. 2006;55:848–855. doi: 10.1136/gut.2005.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Kurdi M, Bowers MC, Dado J, Booz GW. Parthenolide induces a distinct pattern of oxidative stress in cardiac myocytes. Free Radic Biol Med. 2007;42:474–481. doi: 10.1016/j.freeradbiomed.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Lan W, Guhaniyogi J, Horn MJ, Xia JQ, Graham B. A density-based proteomics sample fractionation technology: folate deficiency induced oxidative stress response in liver and brain. J Biomol Tech. 2007;18:213–225. [PMC free article] [PubMed] [Google Scholar]
- Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki P, Sistonen P, Aaltonen LA, Nystrom-Lahti M, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- Li GM, Presnell SR, Gu L. Folate deficiency, mismatch repair-dependent apoptosis, and human disease. J Nutr Biochem. 2003;14:568–575. doi: 10.1016/s0955-2863(03)00115-3. [DOI] [PubMed] [Google Scholar]
- Lin CT, Lyu YL, Xiao H, Lin WH, Whang-Peng J. Suppression of gene amplification and chromosomal DNA integration by the DNA mismatch repair system. Nucleic Acids Res. 2001;29:3304–3310. doi: 10.1093/nar/29.16.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, Lynch J. Genetics, natural history, surveillance, management, and gene mapping in the Lynch syndrome. Pathol Biol (Paris) 1995;43:151–158. [PubMed] [Google Scholar]
- Macpherson P, Barone F, Maga G, Mazzei F, Karran P, Bignami M. 8-oxoguanine incorporation into DNA repeats in vitro and mismatch recognition by MutSalpha. Nucleic Acids Res. 2005;33:5094–5105. doi: 10.1093/nar/gki813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato JM, Lu SC. Homocysteine, the bad thiol. Hepatology. 2005;41:976–979. doi: 10.1002/hep.20708. [DOI] [PubMed] [Google Scholar]
- McGuire JJ. Anticancer antifolates: current status and future directions. Curr Pharm Des. 2003;9:2593–2613. doi: 10.2174/1381612033453712. [DOI] [PubMed] [Google Scholar]
- Mizutani H, Oikawa S, Hiraku Y, Murata M, Kojima M, Kawanishi S. Distinct mechanisms of site-specific oxidative DNA damage by doxorubicin in the presence of copper(II) and NADPH-cytochrome P450 reductase. Cancer Sci. 2003;94:686–691. doi: 10.1111/j.1349-7006.2003.tb01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A, Fishel R. Mismatch repair and the hereditary non-polyposis colorectal cancer syndrome (HNPCC) Cancer Invest. 2002;20:102–109. doi: 10.1081/cnv-120000371. [DOI] [PubMed] [Google Scholar]
- O'Brien V, Brown R. Signalling cell cycle arrest and cell death through the MMR System. Carcinogenesis. 2006;27:682–692. doi: 10.1093/carcin/bgi298. [DOI] [PubMed] [Google Scholar]
- O'Connor KA, Roth BL. Finding new tricks for old drugs: an efficient route for public-sector drug discovery. Nat Rev Drug Discov. 2005;4:1005–1014. doi: 10.1038/nrd1900. [DOI] [PubMed] [Google Scholar]
- Oka S, Ohno M, Tsuchimoto D, Sakumi K, Furuichi M, Nakabeppu Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008;27:421–432. doi: 10.1038/sj.emboj.7601975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison PJ, Cleary M, Silva JM, Chang K, Sheth N, Sachidanandam R, Hannon GJ. Cloning of short hairpin RNAs for gene knockdown in mammalian cells. Nat Methods. 2004;1:163–167. doi: 10.1038/nmeth1104-163. [DOI] [PubMed] [Google Scholar]
- Peltomaki P, Vasen HF. Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology. 1997;113:1146–1158. doi: 10.1053/gast.1997.v113.pm9322509. [DOI] [PubMed] [Google Scholar]
- Potocnik U, Glavac D, Golouh R, Ravnik-Glavac M. Causes of microsatellite instability in colorectal tumors: implications for hereditary non-polyposis colorectal cancer screening. Cancer Genet Cytogenet. 2001;126:85–96. doi: 10.1016/s0165-4608(00)00399-x. [DOI] [PubMed] [Google Scholar]
- Rafferty TS, Green MH, Lowe JE, Arlett C, Hunter JA, Beckett GJ, McKenzie RC. Effects of selenium compounds on induction of DNA damage by broadband ultraviolet radiation in human keratinocytes. Br J Dermatol. 2003;148:1001–1009. doi: 10.1046/j.1365-2133.2003.05267.x. [DOI] [PubMed] [Google Scholar]
- Rajamani R, Muthuvel A, Senthilvelan M, Sheeladevi R. Oxidative stress induced by methotrexate alone and in the presence of methanol in discrete regions of the rodent brain, retina and optic nerve. Toxicol Lett. 2006;165:265–273. doi: 10.1016/j.toxlet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BW, Im MM, Ljungman M, Praz F, Shewach DS. Enhanced radiosensitization with gemcitabine in mismatch repair-deficient HCT116 cells. Cancer Res. 2003;63:6935–6941. [PubMed] [Google Scholar]
- Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Selenium: biochemical role as a component of glutathione peroxidase. Science. 1973;179:588–590. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- Russo MT, De Luca G, Degan P, Bignami M. Different DNA repair strategies to combat the threat from 8-oxoguanine. Mutat Res. 2007;614:69–76. doi: 10.1016/j.mrfmmm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Sargent D, Marsoni S, Thibodeau S, Labianca R, Hamilton S, Torri V, Monges G, Ribic CM, Grothey A, Gallinger S. Confirmation of deficient mismatch repair as a predictive marker for lack of benefit from 5-FU based chemotherapy in stage II and III colon cancer. J Clin Oncol. 2008;26 abr. 4008. [Google Scholar]
- Seifert M, Reichrath J. The role of the human DNA mismatch repair gene hMSH2 in DNA repair, cell cycle control and apoptosis: implications for pathogenesis, progression and therapy of cancer. J Mol Histol. 2006;37:301–307. doi: 10.1007/s10735-006-9062-5. [DOI] [PubMed] [Google Scholar]
- Shen J, Deininger P, Hunt JD, Zhao H. 8-Hydroxy-2′-deoxyguanosine (8-OH-dG) as a potential survival biomarker in patients with nonsmall-cell lung cancer. Cancer. 2007;109:574–580. doi: 10.1002/cncr.22417. [DOI] [PubMed] [Google Scholar]
- Snijders AM, Hermsen MA, Baughman J, Buffart TE, Huey B, Gajduskova P, Roydasgupta R, Tokuyasu T, Meijer GA, Fridlyand J, et al. Acquired genomic aberrations associated with methotrexate resistance vary with background genomic instability. Genes Chromosomes Cancer. 2008;47:71–83. doi: 10.1002/gcc.20509. [DOI] [PubMed] [Google Scholar]
- Sobrero A, Frassineti G, Falcone A, Dogliotti L, Rosso R, Di Costanzo F, Bruzzi P. Adjuvant sequential methotrexate → 5-fluorouracil vs 5-fluorouracil plus leucovorin in radically resected stage III and high-risk stage II colon cancer. Br J Cancer. 2005;92:24–29. doi: 10.1038/sj.bjc.6602276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar A, Koi M, Risinger JI, Glaab WE, Tindall KR, Kolodner RD, Boland CR, Barrett JC, Kunkel TA. Correction of hypermutability, N-methyl-N′-nitro-N-nitrosoguanidine resistance, and defective DNA mismatch repair by introducing chromosome 2 into human tumor cells with mutations in MSH2 and MSH6. Cancer Res. 1997;57:3949–3955. [PubMed] [Google Scholar]
- Umar A, Risinger JI, Glaab WE, Tindall KR, Barrett JC, Kunkel TA. Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics. 1998;148:1637–1646. doi: 10.1093/genetics/148.4.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela-Moreiras G, Selhub J. Long-term folate deficiency alters folate content and distribution differentially in rat tissues. J Nutr. 1992;122:986–991. doi: 10.1093/jn/122.4.986. [DOI] [PubMed] [Google Scholar]
- Wu Q, Christensen LA, Legerski RJ, Vasquez KM. Mismatch repair participates in error-free processing of DNA interstrand crosslinks in human cells. EMBO Rep. 2005;6:551–557. doi: 10.1038/sj.embor.7400418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23:7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen ST, Chan TL, Ho JW, Chan AS, Chung LP, Lam PW, Tse CW, Wyllie AH, Leung SY. Germline, somatic and epigenetic events underlying mismatch repair deficiency in colorectal and HNPCC-related cancers. Oncogene. 2002;21:7585–7592. doi: 10.1038/sj.onc.1205968. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.