

Abstract
The tumour suppressor gene, phosphatase and tensin homolog (PTEN), is one of the most commonly mutated genes in human cancers. Recent evidence suggests that PTEN is important for the maintenance of genome stability. Here, we show that PTEN deficiency causes a homologous recombination (HR) defect in human tumour cells. The HR deficiency caused by PTEN deficiency, sensitizes tumour cells to potent inhibitors of the DNA repair enzyme poly(ADP-ribose) polymerase (PARP), both in vitro and in vivo. PARP inhibitors are now showing considerable promise in the clinic, specifically in patients with mutations in either of the breast cancer susceptibility genes BRCA1 or BRCA2. The data we present here now suggests that the clinical assessment of PARP inhibitors should be extended beyond those with BRCA mutations to a larger group of patients with PTEN mutant tumours.
Keywords: PTEN, PARP inhibitor, homologous recombination
INTRODUCTION
Synthetic lethal approaches to cancer treatment have the potential to deliver relatively large therapeutic windows and significant patient benefit (Kaelin, 2005). We, and others, have previously shown that mutations in either of the tumour suppressor genes, breast cancer 1 (BRCA1) or BRCA2, are synthetically lethal with inhibition of the DNA repair enzyme poly(ADP-ribose) polymerase 1 (PARP1) (Bryant et al, 2005; Farmer et al, 2005). The exquisite sensitivity of BRCA1 or BRCA2 mutant cells to PARP inhibitors (PARPi) forms the rationale behind clinical trials that are now assessing the potential of these agents (Ashworth, 2008). The preliminary results from these clinical trials are promising, with favourable toxicity and sustained tumour responses to the drug (Fong et al, 2009).
Mutations in the phosphatase and tensin homolog (PTEN) gene (OMIM 601728) and loss of PTEN expression have both been associated with a wide range of human tumours (Salmena et al, 2008). PTEN encodes a phosphatase whose role in the control of the phosphoinositide 3 kinase (PI3K) signalling pathway is well established (Salmena et al, 2008). A new functional role for PTEN was recently suggested by the observation that mouse embryonic Pten−/− cells exhibit genomic instability, a phenotype ascribed to either defects in RAD51-mediated DNA double strand break repair (DSBR) (Shen et al, 2007) or defects in cell cycle checkpoints (Gupta et al, 2009). Given that the sensitivity of BRCA mutant cells to PARPi is most likely explained by a defect in RAD51-mediated DSBR by homologous recombination (HR) (Farmer et al, 2005; McCabe et al, 2006), we investigated the possibility that human PTEN mutant cells also display an HR defect and as a consequence, PARPi sensitivity. As PTEN loss of function mutations and loss of PTEN expression are common in a range of hereditary and sporadic cancers (Salmena et al, 2008), we reasoned that such data might significantly extend the utility of this class of drugs.
RESULTS
PTEN involvement in HR repair
To model the effect of PTEN null mutations in human tumour cells, we used isogenically matched wild type and PTEN−/− HCT116 colorectal tumour cell lines (Lee et al, 2007) as well as isogenic wild type and PTEN−/− HEC1A endometroid adenocarcinoma cells (Waldman, unpublished work). PTEN deficiency in both HCT116 and HEC1A lines was achieved by targeting a truncating mutation to both copies of PTEN at exon 2, resulting in an open reading frame encoding only the N-terminal 24 amino acids of the PTEN protein (Lee et al, 2007). First, we confirmed that PTEN−/− human tumour cells express reduced levels of RAD51 (Fig 1A and Fig S1A of Supporting Information), as previously documented in mouse Pten null cells (Shen et al, 2007). Extending this observation, we demonstrated that PTEN mutant tumour cells also had a reduced capacity to form nuclear RAD51 foci in response to DNA damage, a surrogate marker of HR activity (West, 2003) (Fig 1B, Figs S1B and S2 of Supporting Information). To investigate whether these deficiencies in RAD51 expression and recruitment translated into impaired DSBR by HR, we measured HR using a reporter assay. This comprised a previously validated synthetic DNA substrate that bears an inducible double strand DNA break (DSB; Saeki et al, 2006). PTEN deficient human tumour cells exhibited a 5-fold reduction in DSBR by HR when compared to isogenic wild type cells (Fig 1C and Fig S1C of Supporting Information).
Figure 1. PTEN deficiency causes an impairment of DNA repair by HR.
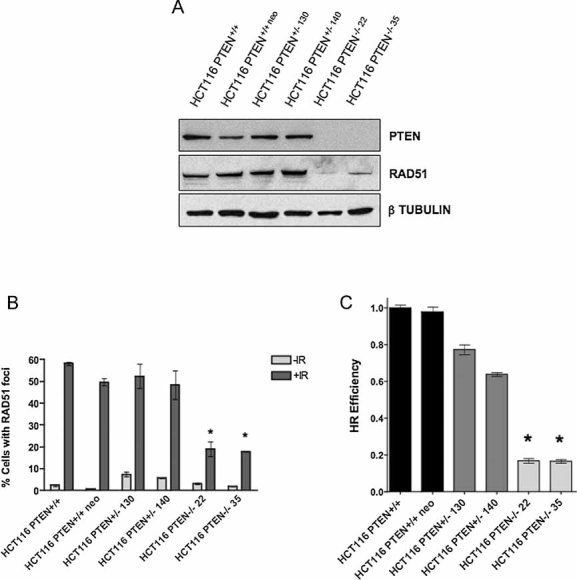
- PTEN deficiency causes a reduction in RAD51 expression. Total cell lysates from isogenic HCT116 colorectal tumour cells were immunoblotted for PTEN and RAD51. Detection of β Tubulin is shown as a loading control. Lysates from parental HCT116 cells were used (HCT116) along with two individually derived PTEN−/− lines (KO35 and KO22) and also a HCT116-derived line bearing a random integration of the PTEN targeting construct (neo124). Lysates from PTEN+/− HCT116 cells are also shown (Lee et al, 2007).
- PTEN deficiency causes a reduction in radiation-induced nuclear RAD51 focus formation. Cells were exposed to 10 Gy γ-irradiation and nuclear RAD51 foci quantified 8 h later by confocal microscopy (Farmer et al, 2005). Bar chart shows the average number of cells with >5 foci per nucleus. Error bars represent three standard deviations of the mean. * p values versus +IR HCT116 PTEN+/+<0.05 (Student's t-test). All other comparisons returned non-significant (>0.05) p values.
- PTEN deficiency causes a reduction in HR as measured using a synthetic HR substrate. As a measure of HR activity, a reporter plasmid-based assay was used comprising two defective copies of GFP, where one serves as a template to restore an induced DSB in the other. HR between the two GFP coding sequences results in an intact GFP coding sequence and cellular fluorescence (Saeki et al, 2006). Error bars represent three standard deviations of the mean. * p values versus HCT116 PTEN+/+<0.05 (Student's t-test).
PTEN deficiency sensitizes to PARP inhibitors
Given the HR repair defect of PTEN deficient cells, we tested the sensitivity of these cells to two DNA-DSB inducers, PARPi and cisplatin. HCT116 PTEN−/− cells were 20 times more sensitive to the PARP inhibitor KU0058948 (Farmer et al, 2005) than their wild type counterparts (compare concentration at which 50% of cells survive (SF50) for HCT116 PTEN+/+ of 1 × 10−5 M with SF50 for HCT116 PTEN−/−22 of 5 × 10−7 M, Fig 2A) and up to 25 times more sensitive to the PARP inhibitor KU0059436/AZD2281/Olaparib (Evers et al, 2008; Fong et al, 2008) (compare SF50 HCT116 PTEN+/+ of 5 × 10−6 M versus SF50 for HCT116 PTEN−/−22 of 2 × 10−7 M, Fig 2B). We also observed PTEN selectivity in the HEC1A model (Fig S1D of Supporting Information). These PTEN/PARP synthetic lethal associations were achievable using sub-micromolar concentrations of PARPi, similar to those required to kill BRCA1 deficient cells (Farmer et al, 2005).
Figure 2. PTEN deficiency sensitizes tumour cells to agents that target HR.
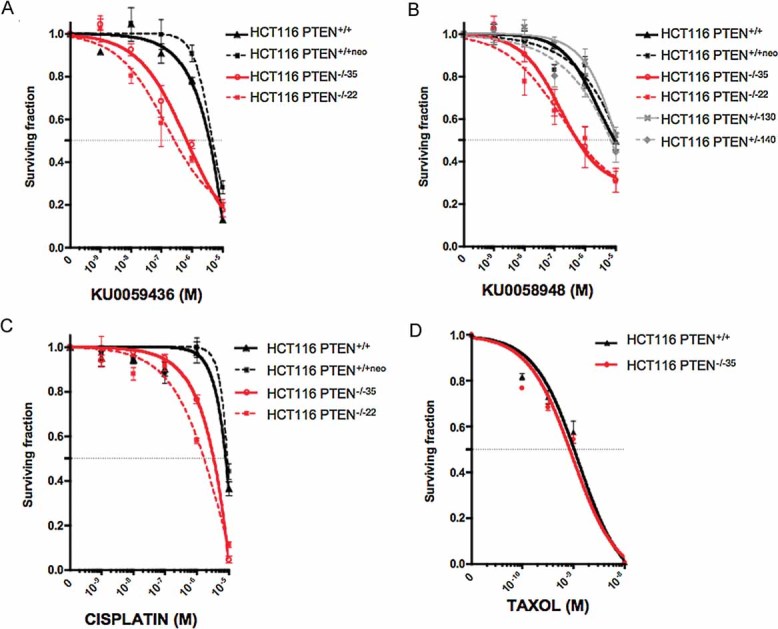
A, B. PTEN deficiency sensitizes cells to drug-like PARP inhibitors. Survival curves for HCT116 cells exposed to the PARP inhibitors KU0058948 (Farmer et al, 2005) or KU0059436 (Evers et al, 2008; Fong et al, 2008). Cells were plated in six-well plates and treated for 15 days, after which SFs were estimated. For both PARP inhibitors, PTEN−/− SFs were significantly different to both PTEN+/+ and PTEN+/− cells (p<0.05 two-way analysis of variance (ANOVA)). No other comparisons returned significant p values.
C. PTEN deficiency sensitizes cells to cisplatin (p<0.05 PTEN−/− lines versus PTEN+/+ cells, two-way ANOVA).
D. PTEN deficiency does not confer sensitization to taxol.
We also assessed the sensitivity of PTEN−/− cells to the platinum salt, cisplatin. Cisplatin, although less selective than PARPi, also targets cells with HR deficiency (Tutt et al, 2005). PTEN−/− cells were five fold more sensitive to cisplatin than their wild type counterparts (compare SF50 HCT116 PTEN+/+ of 1 × 10−5 M versus SF50 for HCT116 PTEN−/−22 of 2 × 10−6 M, Fig 2C), consistent with the hypothesis that PTEN deficiency leads to an HR defect that may be targeted. To eliminate the possibility that these observations were reflective of a general sensitivity to chemotherapeutics caused by PTEN deficiency, we assessed sensitivity to the microtubule poison, paclitaxel (taxol). Paclitaxel was not selectively lethal to PTEN deficient cells (Fig 2D), suggesting that the PARPi and cisplatin sensitivities we observed were more likely reflective of HR deficiency rather than of general chemosensitivity.
Although PTEN haploinsufficiency effects on tumourigenesis have been reported, complete loss of PTEN is observed in many advanced cancers (Salmena et al, 2008). Given this, we thought it pertinent to assess the effects of PTEN gene dosage upon HR deficiency and PARPi sensitivity. Using the HCT116 isogenic system, we demonstrated that PTEN heterozygosity did not cause the same reduction in RAD51 expression as seen in PTEN−/− cells (Fig 1B). Consistent with this observation, the induction of damage induced nuclear RAD51 foci was not abrogated in PTEN+/− cells (Fig 1B) and although a marginal decrease in HR was observed using the synthetic HR assay in heterozygous cells (Fig 1C), this decrease did not translate into PARPi sensitivity (Fig 2B).
To assess the generality of our observations, we examined PARPi sensitivity in an additional panel of tumour cell lines (Fig 3, Fig S3 and Tables S1 and S2 of Supporting Information). Although PTEN mutations are not the only genetic variable within this diverse tumour cell panel, we observed a clear distinction in PARPi sensitivity between tumour lines with wild type PTEN expression, such as DU145 and MCF7, and those with PTEN deficiency, such as HCC70, PC3, RPMI-7951, A172, UM-UC3 and MDA-MB-468 (Fig 3A,B and Fig S3 of Supporting Information). Notably, PARPi sensitivity in PTEN deficient lines was of a scale similar to that observed in human tumour lines with BRCA1 silencing (Farmer et al, 2005). With the exception of the glioma line A172, we also observed consistent cisplatin sensitivity in PTEN deficient lines (Fig 3C).
Figure 3. PARP inhibitor sensitivity in human tumour cells.
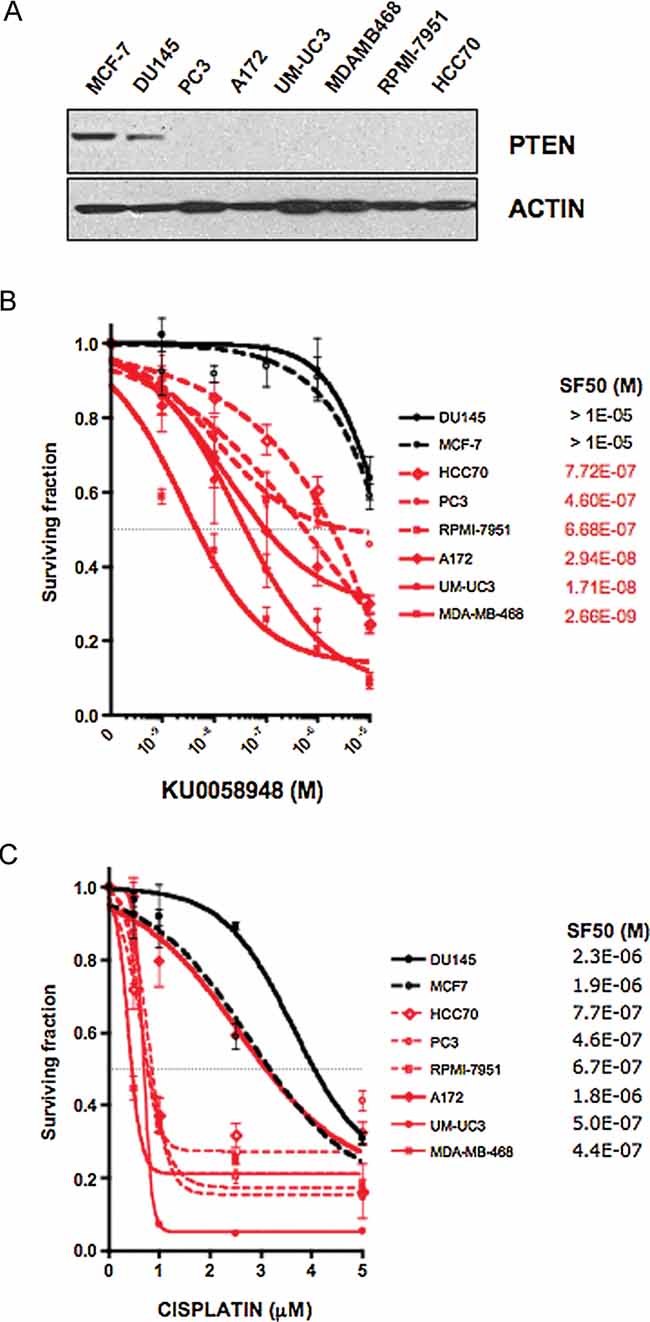
- Western blot showing PTEN expression in a panel of human tumour lines. PTEN mutant lines are shown in red and PTEN wild type lines in black. PTEN genotypes are as follows: DU145 (prostate), PTEN wild type; MCF7 (breast), PTEN wild type; PC3 (prostate) p.R55fs*1 homozygous; MDAMB468 (breast), p.A72fs*5 homozygous; A172 (glioma), p.R55fs*1 homozygous; UM-UC3 (bladder), p.M1*404del homozygous; RPMI-7951 (melanoma) p.null, c.1-79del79 homozygous; HCC70 (breast), p.F90fs*9 homozygous (see Table S1 of Supporting Information for complete genotypes).
- PARPi sensitivity in the same cell line panel. For each PTEN null line, survival was significantly different than in DU145 and MCF7 cells (p<0.05, two-way ANOVA) (see Fig S3 of Supporting Information for analysis of an enlarged cell line panel).
- Cisplatin sensitivity in the same cell line panel. With the exception of A172, survival of each PTEN null line at 1 µM was significantly different than in DU145 and MCF7 cells (p<0.05, two-way ANOVA).
We utilized the PTEN deficient prostate tumour cell line PC3, to assess whether PARPi sensitivity could be abrogated by ectopic expression of PTEN. As expected, infection of PC3 cells with an empty retroviral expression construct did not significantly modify PARPi sensitivity, nor did transfection of an expression construct encoding the p.R55fs*1 PTEN allele normally found in PC3 cells (Fig 4A,B). However, ectopic expression of full length PTEN in PC3 cells elicited PARPi resistance of a scale similar to tumour cells with endogenous wild type PTEN expression (Fig 4A). We also assessed whether PARPi sensitivity by PTEN was independent of the phosphatase activity of PTEN. Ectopic expression of the catalytically inactive C124S PTEN mutant (Maehama & Dixon, 1998; Shen et al, 2007) also caused PARPi resistance in PC3 cells (Fig 4A), suggesting that a non-phosphatase activity of PTEN may determine PARPi sensitivity. We also analysed whether restoring cytoplasmic PTEN expression, while suppressing nuclear PTEN expression could confer PARP inhibitor resistance in a PTEN null cell line, such as PC3. Here we used ectopic expression of a PTEN cDNA that bears a mutation (p.K289E) that reduces the shuttling of PTEN into the nucleus (Song et al, 2008) (Fig 4B). Expression of PTEN p.K289E was able to partially but not completely restore PARP inhibitor resistance (Fig 4A), suggesting that an impairment of the nuclear localization of PTEN also modulates sensitivity to PARP inhibitor. Finally, we sought to assess whether the PARPi sensitivity of a PTEN cell line was RAD51 dependent. Ectopic expression of RAD51 caused PARP inhibitor resistance in PC3 cells (Fig 4A and Fig S4 of Supporting Information), supporting the hypothesis that reduced RAD51 expression underlies the PARP inhibitor sensitivity of PTEN deficient cells.
Figure 4. PARP inhibitor sensitivity in human tumour cells.
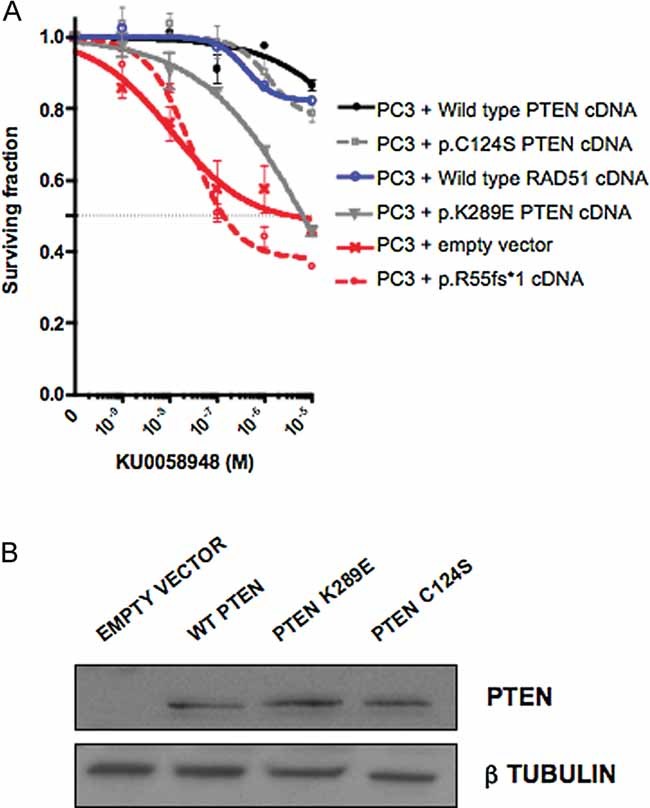
A, B. Wild type and catalytically inactive PTEN, both restore PARP inhibitor resistance, as does expression of RAD51; SFs in PC3 cells infected with PTEN wild type, PTEN p.C124S or RAD51 cDNA expression constructs was significantly different than in PC3 + empty vector (p < 0.05, two-way ANOVA). PC3 cells infected with p.K289E cDNA expression construct did not cause significant resistance. PTEN null PC3 cells were infected with cDNA expression constructs as shown and exposed to PARP inhibitor. Western blot and survival curves are shown.
To assess PARPi selective efficacy in vivo, we xenografted PTEN deficient HCT116 cells into mice and treated animals with the PARPi KU0059436 (Olaparib) (Evers et al, 2008) which is currently undergoing clinical assessment (Fong et al, 2009). Although xenograft growth in this model was extremely rapid, growth from PTEN deficient xenografts was suppressed by KU0059436 when compared to vehicle-treated xenografts (Fig 5A). Conversely PARPi did not have a similar effect on the growth of PTEN wild type xenografts (Fig 5B). This provides preliminary evidence to suggest that our in vitro observations may extrapolate to an in vivo setting.
Figure 5. In vivo efficacy of Olaparib in PTEN deficient xenografts.
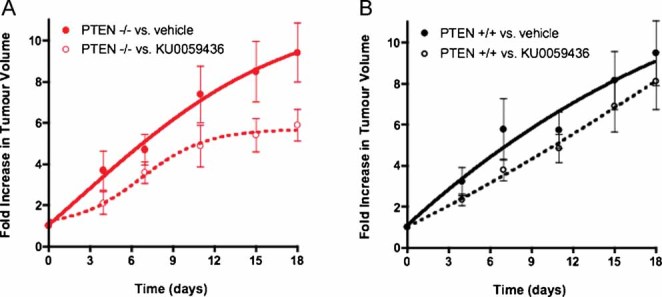
A, B. The clinical PARP inhibitor KU0059436/Olaparib (Evers et al, 2008; Fong et al, 2008) suppresses growth in PTEN−/− subcutaneous xenografts (A), but not in PTEN+/+ xenografts (B). PTEN deficient or proficient HCT116 cells were each mixed 2:1 in matrigel and then injected into either bilateral flank of 6–8 week old female athymic nude mice. After two days recovery, mice were randomized into treatment and control groups, (10 animals per cohort, 20 in total) and drug dosing initiated. A treatment regime similar to that known to elicit Brca2 selectivity (Farmer et al, 2005) was used. For five consecutive days, single daily intraperitoneal doses of KU0059436 (or vehicle) were administered at a dose of 15 mg/kg in HBC (Farmer et al, 2005). This was followed by two consecutive days of no treatment after which the same treatment cycle was continued until the end of the study. Tumour volumes were measured every four days from the initiation of drug dosing and the results expressed as fold increase in tumour volume relative to that at the first drug administration. Tumour volume vehicle treated vs. drug treated, p = 0.04 for PTEN−/− xenografts and p = 0.44 for PTEN+/+ xenografts (two-way repeated measures ANOVA).
DISCUSSION
PARP inhibitors are now showing considerable promise in the treatment of cancers characterized by genetic deficiencies that lead to an impairment of HR. For example, observations from a phase I trial of the PARP inhibitor KU0059436/AZD2281/Olaparib (AstraZeneca) suggest that significant tumour responses can be achieved in ovarian cancer patients carrying BRCA mutations (Fong et al, 2009). These tumour responses, demonstrated by both radiological estimation of tumour size and by the use of biochemical markers of tumour burden, are sustained and can be achieved without the significant toxicity normally associated with standard chemotherapeutic regimes (Fong et al, 2009). These promising results have led to phase II trials of KU0059436/AZD2281/Olaparib for breast and ovarian cancers in BRCA mutation carriers (Tuma, 2007). Here, we show that PARP inhibitors may have utility outside the relatively small proportion of cancer patients carrying BRCA mutations. PTEN mutations are common in both endometrial cancers and glioblastomas and are also found in a significant proportion of malignant melanomas, prostate, breast, lung and colorectal tumours (Keniry & Parsons, 2008). In addition, transcriptional or translational repression of PTEN expression in the absence of PTEN mutation is also a common characteristic of multiple tumour types (Wiencke et al, 2007; Yang et al, 2008). The profound sensitivity of PTEN deficient tumour cells to PARP inhibitors demonstrated here suggests that clinical assessment of these agents in a wider context is warranted and perhaps even in combination with PI3K/AKT inhibitors that have been already been proposed as a means to target PTEN deficient tumours (Knight & Shokat, 2007).
Our initial studies suggest that homozygous PTEN mutations that severely truncate the open reading frame sensitize tumour cells to PARP inhibitors. For example, glioblastoma, breast and prostate tumour cell lines bearing homozygous PTEN mutations that truncate the open reading frame before residue c.279 (amino acid residue 93) are as sensitive to KU0058948, as human tumour cells with BRCA1 or BRCA2 deficiencies (Farmer et al, 2005; Turner et al, 2008). Notably, reconstitution of PC3 cells (p.R55fs*1 homozygous) with ectopic expression of a catalytically inactive PTEN allele (p.C124S) was sufficient to restore PARPi resistance (Fig 4). Therefore, it seems likely that tumour-associated PTEN missense mutations that only modify catalytic activity without ostensibly compromising PTEN expression or non-phosphatase activities may not sensitize tumour cells to PARP. Conversely, PTEN mutations that severely truncate the open reading frame or cause instability of the PTEN protein are more likely to cause PARPi sensitivity. Mutations that truncate the open reading frame of PTEN before residue 93, as in the PARPi sensitive HCC70 cell line (p.F90fs*9 homozygous) are found in a wide variety of tumour types (Table S2 of Supporting Information), suggesting at least one patient subgroup that should be rapidly assessed for response to PARP inhibitors in clinical trials. Tumours that have PTEN deficiency brought about by PTEN mutations in the C-terminus that compromise protein stability (Leslie et al, 2008) or non-genetic alterations that cause changes in PTEN transcription or translation, may also respond to PARP inhibitors, although considerable work is required to establish a threshold of PTEN expression below which PARPi sensitivity occurs. Furthermore, we envisage that the role PTEN plays in determining response to PARP inhibitors, is mediated by a nuclear activity of the protein, suggesting that a complete absence of nuclear PTEN expression, determined by immunohistochemical (IHC) detection in biopsy material, could predict response. This raises the possibility that PARP inhibitors could specifically be assessed in tumourigenic conditions such as the more aggressive forms of oesophageal squamous cell carcinoma (Tachibana et al, 2002), cutaneous melanoma (Whiteman et al, 2002) and colorectal cancer (Trotman et al, 2007; Zhou et al, 2002), where nuclear PTEN expression is absent. Defining thresholds of IHC detection that are predictive of PARPi response will be a key in assessing this possibility, and we propose that combining PTEN IHC with surrogate markers of PARPi sensitivity, such as the inability to form nuclear RAD51 foci (Farmer et al, 2005), may allow further refinement of the use of PARP inhibitors in cancer treatment.
MATERIALS AND METHODS
Materials
PARP inhibitors KU0058948 (Farmer et al, 2005) and KU0059436 (Evers et al, 2008; Fong et al, 2008) (KuDOS Pharmaceuticals/Astra Zeneca), cisplatin and paclitaxel (Sigma–Aldrich) were used as described previously (Edwards et al, 2008; Farmer et al, 2005).
Cell culture
Wild type, PTEN+/− and PTEN−/− HCT116 or HEC1A cells (Kim et al, 2007; Lee et al, 2007) were maintained in McCoy's 5A medium (Invitrogen, Glasgow, UK) supplemented with 10% v/v foetal bovine serum (FBS) (PAA gold, GIBCO). In the case of PTEN−/− cells, two individually derived lines were used, (KO22 and KO35 for HCT116 and KO22 and KO16 for HEC1A). For the HCT116 experiments, parental cells and a HCT116 derived line bearing a random integration of the PTEN targeting construct (neo124) were used as PTEN wild type comparators. Two HCT116 PTEN+/− cell lines bearing one targeted allele (130 and 140) were also analysed. All other cells lines were obtained from the ECACC (Teddington, UK) and cultured according to the supplier's instructions.
Plasmids
The full length coding sequence of PTEN was PCR amplified from a cDNA IMAGE clone (Geneservice, Cambridge, UK), using a 5′ primer harbouring a Kozak consensus sequence (GCCACC prior to initiating ATG). The resultant PCR product was cloned into a tag-free pBabe-puro retroviral expression vector. Mutation of the PTEN cDNA sequence was achieved using the Quick Change site-directed mutagenesis kit (Stratagene). Twenty-four hours after infection, cells were selected in 1 mg/l puromycin (InvivoGen) for three days to remove non-infected cells, then seeded in six-well plates (at 1500 cells/well) and treated with drug the following day, as previously described (Edwards et al, 2008; Farmer et al, 2005).
Survival assays
Long-term survival assays were performed as previously described (Edwards et al, 2008; Farmer et al, 2005). For measurement of sensitivity to PARP inhibitor, cisplatin or paclitaxel (taxol), exponentially growing cells were seeded in six-well plates at a concentration of 1000–2000 cells per well. For PARP inhibitor and taxol treatment, cells were continuously exposed to the drug with media and drug replaced every 60 h. For cisplatin treatment, cells were exposed to the drug at the indicated concentrations for 24 h, washed twice with phosphate buffered saline (PBS), then normal media replaced every 3-4 days. After 15 days, cells were fixed and stained with sulphorhodamine-B (Sigma, St. Louis, USA) and a colorimetric assay performed as described previously (Edwards et al, 2008). Surviving fractions (SFs) were calculated and drug sensitivity curves plotted as previously described (Farmer et al, 2005).
Protein analysis
Whole-cell protein extracts were prepared from cells lysed with radioimmunoprecipitation (RIPA) buffer (Upstate) supplemented with protease inhibitor cocktail tablets (Roche, Burgess Hill, UK). Protein concentrations were measured using BioRad Protein Assay Reagent (BioRad, Hemel Hempstead, UK). For Western blot analysis, lysates were electrophoresed on Novex 4–12% gradient bis–tris pre-cast gels (Invitrogen) and immunoblotted overnight at 4°C with antibodies targeting the following epitopes: PTEN C-terminus (Cell Signalling, Danvers, USA), RAD51 (H-92, Santa Cruz Biotech, Santa Cruz, USA), ACTIN (Santa Cruz Biotech) or β-Tubulin (T4026, Sigma). Incubation with primary antibody was followed by incubation with a horseradish peroxidase-conjugated secondary antibody and chemiluminescent detection of proteins (Amersham Pharmacia, Cardiff, UK).
DSB Repair Assay
DSBR was quantified using a synthetic repair substrate as previously described (Saeki et al, 2006). Briefly, the DR-green fluorescent protein (GFP) HR reporter plasmid, containing two differentially mutated GFP-encoding cassettes, one of which contains a unique ISceI restriction site, was co-transfected into cells alongside a ISceI expression vector, pcBASce (Saeki et al, 2006). Seventy-two hours after transfection, HR competence was estimated by quantifying the number of GFP positive cells from a population of 50,000 cells, using flow cytometry (Saeki et al, 2006).
The paper explained
PROBLEM
The tumour suppressor gene, PTEN, is one of the most commonly mutated genes in human cancers. Selectively targeting cancerous PTEN deficient cells would likely make for an efficient and relatively safe cancer therapy applicable to the large number of PTEN deficient cancers. Selective targeting of BRCA1 or BRCA2 deficient tumours has recently been demonstrated with Olaparib, a high-potency PARP inhibitor. This drug can elicit sustained responses in BRCA deficient cancers without causing some of the negative side effects caused by standard chemotherapies. Tumour cells in BRCA mutant patients have a defect in the DNA repair process of HR that repairs DSBs and it is thought that PARP inhibitors target this HR defect by increasing the formation of DSBs.
RESULTS
Recent evidence suggests that, like BRCA mutant cells, PTEN mutant cells are also characterized by an increase in DSBs, suggesting that drugs that target BRCA mutation could also be used to target PTEN mutation. We demonstrate that PTEN deficiency in human tumour cells also leads to suppression of HR. Using in vitro cell culture and in vivo tumour modelling in mice, we show that, as in BRCA deficient tumours, the HR deficiency in PTEN mutant cells can be exploited therapeutically by Olaparib.
IMPACT
Our work suggests that this relatively non-toxic anti-cancer drug can be used to target the large number of PTEN deficient tumours, significantly extending its previously anticipated limited spectrum.
Analysis of RAD51 foci formation
Nuclear RAD51 foci were visualized and quantified by confocal microscopy as previously described (Farmer et al, 2005). Detection of gamma (γ)-H2AX foci was used a control marker for the formation of DSBs (data not shown). In brief, cells were grown onto l-lysine coated cover slips (BD Biosciences, Oxford, UK) and exposed to 10 Gy ionizing radiation. Eight hours later, cells were fixed, permeabilized and then co-immunostained with primary antibodies targeting RAD51 (rabbit, Santa Cruz Biotech) and γH2AX (mouse, Upstate), detected with fluorescein isothiocyanate (FITC) and Texas red conjugated secondary antibodies respectively. 4′-6-Diamidino-2-phenylindole (DAPI) staining was used to detect nuclei. Nuclear foci were visualized by confocal microscopy and a minimum of 100 fields of view used to estimate RAD51 focus formation frequency.
Xenografts
HCT116 cells were mixed 2:1 in matrigel (BD Biosciences) and then injected into the bilateral flanks of 6–8 week old female athymic nude mice (2 × 106 cells per animal). After two days recovery, mice were randomized into treatment and control groups, (10 animals per cohort, 20 in total) and drug dosing initiated. We used a treatment regime similar to that known to elicit Brca2 selectivity (Farmer et al, 2005). For five consecutive days, single daily intraperitoneal doses of KU0059436 (or vehicle) were administered at a dose of 15 mg/kg in 2-hydroxypropyl-b-cyclodextrin (HBC, Farmer et al, 2005). This was followed by two consecutive days of no treatment after which the same treatment cycle was continued until the end of the study. Tumour volumes were measured every four days from the initiation of drug dosing and the results expressed as fold increase in tumour volume, relative to that at the first drug administration.
Acknowledgments
We thank Drs. Graeme Smith and Niall Martin of KuDOS Pharmaceuticals for the provision of PARP inhibitors. This work was funded by Breakthrough Breast Cancer and Cancer Research UK. We acknowledge NHS funding to the NIHR Biomedical Research Centre.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author Contributions
AMP, CJL and AA conceived and designed the experiments. AMP, SAM, RB, AM and JT performed the experiments. AMP, SAM, RB, AM, JT, CJL and AA analysed the experiments. TW and JSK provided reagents. AMP, CJL and AA wrote the manuscript. CJL and AA devised the project and obtained funding.
For more information
The Breakthrough Research Center:
http://www.breakthroughresearch.org.uk/breakthrough_research_centre/index.html
Clinical trials (Olaparib):
http://clinicaltrials.gov/ct2/results?term=olaparib
Cancer Research UK: Cancer Help UK (Olaparib):
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- Evers B, Drost R, Schut E, de Bruin M, van der Burg E, Derksen PW, Holstege H, Liu X, van Drunen E, Beverloo HB, et al. Selective Inhibition of BRCA2-Deficient Mammary Tumor Cell Growth by AZD2281 and Cisplatin. Clin Cancer Res. 2008;14:3916–3925. doi: 10.1158/1078-0432.CCR-07-4953. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Carden CP, Roelvink M, De Greve J, Gourley CM, Carmichael J, De Bono JS, Schellens JH, Kaye SB. AZD2281 (KU-0059436), a PARP (poly ADP-ribose polymerase) inhibitor with single agent anti-cancer activity in patients with BRCA deficient ovarian cancer. Results from a phase I study. J Clin Oncol. 2008;26 abstr 5510. [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Gupta A, Yang Q, Pandita RK, Hunt CR, Xiang T, Misri S, Zeng S, Pagan J, Jeffery J, Puc J, et al. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle. 2009;8 doi: 10.4161/cc.8.14.8947. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27:5477–5485. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2007;27:662–677. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Shokat KM. Chemically targeting the PI3K family. Biochem Soc Trans. 2007;35:245–249. doi: 10.1042/BST0350245. [DOI] [PubMed] [Google Scholar]
- Lee C, Kim JS, Waldman T. Activated PI3K signaling as an endogenous inducer of p53 in human cancer. Cell Cycle. 2007;6:394–396. doi: 10.4161/cc.6.4.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene. 2008;27:5464–5476. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- Saeki H, Siaud N, Christ N, Wiegant WW, van Buul PP, Han M, Zdzienicka MZ, Stark JM, Jasin M. Suppression of the DNA repair defects of BRCA2-deficient cells with heterologous protein fusions. Proc Natl Acad Sci USA. 2006;103:8768–8773. doi: 10.1073/pnas.0600298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature. 2008;455:813–817. doi: 10.1038/nature07290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Shibakita M, Ohno S, Kinugasa S, Yoshimura H, Ueda S, Fujii T, Rahman MA, Dhar DK, Nagasue N. Expression and prognostic significance of PTEN product protein in patients with esophageal squamous cell carcinoma. Cancer. 2002;94:1955–1960. doi: 10.1002/cncr.0678. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma RS. Combining carefully selected drug, patient genetics may lead to total tumor death. J Natl Cancer Inst. 2007;99:1505–1506. doi: 10.1093/jnci/djm194. 1509. [DOI] [PubMed] [Google Scholar]
- Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. Embo J. 2008;27:1368–1377. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutt AN, Lord CJ, McCabe N, Farmer H, Turner N, Martin NM, Jackson SP, Smith GC, Ashworth A. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb Symp Quant Biol. 2005;70:139–148. doi: 10.1101/sqb.2005.70.012. [DOI] [PubMed] [Google Scholar]
- West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- Whiteman DC, Zhou XP, Cummings MC, Pavey S, Hayward NK, Eng C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int J Cancer. 2002;99:63–67. doi: 10.1002/ijc.10294. [DOI] [PubMed] [Google Scholar]
- Wiencke JK, Zheng S, Jelluma N, Tihan T, Vandenberg S, Tamguney T, Baumber R, Parsons R, Lamborn KR, Berger MS, et al. Methylation of the PTEN promoter defines low-grade gliomas and secondary glioblastoma. Neuro Oncol. 2007;9:271–279. doi: 10.1215/15228517-2007-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Kong W, He L, Zhao JJ, O'Donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008;68:425–433. doi: 10.1158/0008-5472.CAN-07-2488. [DOI] [PubMed] [Google Scholar]
- Zhou XP, Loukola A, Salovaara R, Nystrom-Lahti M, Peltomaki P, de la Chapelle A, Aaltonen LA, Eng C. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am J Pathol. 2002;161:439–447. doi: 10.1016/S0002-9440(10)64200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.