

Abstract
Various age-related neurodegenerative diseases, including Parkinson's disease, polyglutamine expansion diseases and Alzheimer's disease, are associated with the accumulation of misfolded proteins in aggregates in the brain. How and why these proteins form aggregates and cause disease is still poorly understood. Small model organisms—the baker's yeast Saccharomyces cerevisiae, the nematode worm Caenorhabditis elegans and the fruit fly Drosophila melanogaster—have been used to model these diseases and high-throughput genetic screens using these models have led to the identification of a large number of genes that modify aggregation and toxicity of the disease proteins. In this review, we revisit these models and provide a comprehensive comparison of the genetic screens performed so far. Our integrative analysis highlights alterations of a wide variety of basic cellular processes. Not all disease proteins are influenced by alterations in the same cellular processes and despite the unifying theme of protein misfolding and aggregation, the pathology of each of the age-related misfolding disorders can be induced or influenced by a disease-protein-specific subset of molecular processes.
Keywords: neurodegeneration, protein aggregation, genetic modifiers, small model organisms, meta-analysis
Introduction
Several age-related neurodegenerative diseases, including Parkinson's disease, Alzheimer's disease and Huntington's disease are associated with the accumulation of misfolded proteins into microscopically visible aggregates in the brain (see Fig 1). These aggregates contain fibrillar structures that are mainly composed of disease-specific misfolded proteins, such as α-synuclein in Parkinson's disease, amyloid-β and tau in Alzheimer's disease, superoxide dismutase (SOD) in Amyotrophic lateral sclerosis (ALS), and mutant huntingtin in Huntington's disease (Fig 1) (Masters et al, 1985; Scherzinger et al, 1997; Spillantini et al, 1998 Durham et al., 1997). There is still controversy about the role of the aggregates but the prevalent hypothesis is that they represent a cellular protection mechanism against toxic aggregation intermediates (Arrasate et al, 2004; Kaganovich et al, 2008; Lansbury & Lashuel, 2006; Sanchez et al, 2003; Saudou et al, 1998).
Figure 1. Protein aggregation in neurodegenerative disease.

Examples of aggregation in brains of patients with neurodegenerative disorders (disorders in dark blue). Typically, these aggregates contain amyloid-like fibrils composed of specific disease proteins (disease proteins in brown). All photographs provided by Wilfred den Dunnen, UMCG, Groningen, The Netherlands.
Familial forms of these misfolding diseases often involve toxic gain-of-function mutations that increase misfolding and aggregation properties as well as the toxicity of the disease proteins. This is well illustrated by Huntington's disease, in which expansion of a polyglutamine stretch within the huntingtin protein causes disease. In this and several other polyglutamine diseases, there is a direct correlation between the length of the polyglutamine expansion and the aggregation kinetics and toxicity of disease proteins (Scherzinger et al, 1997; Chen et al, 2002). Additionally, there is an inverse correlation between the length of the polyglutamine stretch and the age-at-onset of disease (Gusella & MacDonald, 2006). Apart from the misfolding properties of the disease proteins, other inherited factors are predicted to modify the age-at-onset of these familial misfolding diseases (Rosenblatt et al, 2001).
Non-familial forms of neurodegenerative diseases, which include the majority of cases of Parkinson's and Alzheimer's disease, typically develop in old age. In these sporadic cases the characteristic aggregates in the brain are primarily composed of misfolded, but wild type, disease proteins. The cause of protein misfolding and pathogenesis in these non-familial forms remains elusive. One can speculate that several phenomena associated with old age, for example cell shrinkage and a decline in protein quality control, could cause or contribute to protein misfolding and aggregation and thereby lead to disease (Gaczynska et al, 2001; Reznick & Gershon, 1979; Shtilerman et al, 2002). These phenomena do not explain, however, why different people develop different protein-misfolding diseases in old age. Other disease-modifying factors are likely to play a role as well. Finding the genetic modifiers that influence protein misfolding and toxicity is, therefore, expected to expand our understanding of the molecular cause(s) of protein-misfolding diseases and could provide important cues for therapeutic strategies.
So far, human genetic screens and pathological studies have been able to provide limited mechanistic insight into the molecular processes that determine disease susceptibility or age-at onset of disease (Carrasquillo et al, 2009; Lesage & Brice, 2009; Metzger et al, 2006; Mougeot et al, 2009). Small genetic model organisms, transgenically expressing human misfolding disease-related proteins, together with large-scale genetic screens, have therefore been exploited to generate additional hypotheses about protein-misfolding disease mechanisms (Bilen & Bonini, 2007; Fernandez-Funez et al, 2000; Ghosh & Feany, 2004; Giorgini et al, 2005; Hamamichi et al, 2008; Kaltenbach et al, 2007; Kazemi-Esfarjani & Benzer, 2000; Kraemer et al, 2006; Nollen et al, 2004; Outeiro & Lindquist, 2003; Van Ham et al, 2008; Wang et al, 2009a; Willingham et al, 2003). Here we describe these models and provide a first integrative and comprehensive comparison of the results of these large-scale screens. We aimed to pinpoint evolutionary conserved processes with a role in misfolding and to extract common and disease-specific processes, which will provide a focus for future mechanistic studies and for development of therapeutic strategies.
Glossary
- Age-at-onset of disease
The mean age at which a disease is revealed by its characteristic symptoms.
- Alzheimer's disease
Age-related neurodegenerative disease, characterized pathologically by protein aggregates known as plaques and tangles consisting mainly of the proteins amyloid beta and phosphorylated tau. The main symptoms are memory loss and reduced cognitive functioning.
- Amyotrophic lateral sclerosis
Progressive, fatal, neurodegenerative disease caused by the degeneration of motor neurons resulting in loss of ability to initiate and control all voluntary movement.
- Essential genes
Genes that are required for (early) embryonic development, and loss of which is lethal at an early stage.
- Huntington's disease
Heritable neurodegenerative disease, characterized pathologically by loss of neurons and protein aggregates consisting mainly of mutant Huntingtin protein. The main symptoms are involuntary movements (chorea), declined mental abilities and behavioural and psychiatric problems.
- Lipid metabolism
The synthesis, breakdown and transport of various lipid-containing molecules.
- miRNA
microRNAs are small RNA molecules that regulate gene expression.
- Molecular chaperone
Proteins that assist in the correct folding of other proteins and protect against misfolding.
- Parkinson's disease
Age-related neurodegenerative disease, characterized pathologically by protein aggregates in the brain, mainly composed of alpha-synuclein known as Lewy bodies. The main symptoms are reduced motor skills and movement defects.
- Proteasomal degradation
Proteolytic breakdown of damaged or superfluous proteins into short peptides. In mammalian cells this is affected by the proteasome, a large ∼2000 kDa enzymatic complex.
- Protein quality control
The recognition and disposal of misfolded, damaged and no longer required proteins.
- RNA metabolism
The synthesis (transcription), breakdown and transport of ribonucleic acids.
- Sarcopenia
Degenerative loss of skeletal muscle tissue and skeletal muscle strength as a consequence of aging.
- Spinocerebellar ataxias
Progressive neurodegenerative diseases of the cerebellum, resulting in motor coordination loss.
- Superoxide dismutase
Important antioxidant defence enzymes that catalyse the dismutation of superoxide into oxygen and hydrogen peroxide.
- Synaptic vesicles
Small membrane-bounded compartments containing neurotransmitters at the synapse of neurons.
- Tau
Microtubule-associated protein abundant in neurons in the central nervous system. Hyperphosphorylation of the tau protein can result in self-assembly of fibrillary tangles involved in Alzheimer's disease and other tauopathies.
- Vesicle trafficking
The movement of small membrane-bounded compartments (vesicles) between organelles, such as from the ER to the Golgi system.
Yeast, worm and fly models of protein-misfolding disease
Small model organisms—baker's yeast (Saccharomyces cerevisiae), the nematode worm (Caenorhabditis elegans) and the fruit fly (Drosophila melanogaster)—have been used to model several aspects of neurodegenerative diseases, including aggregation and toxicity of misfolding disease-related proteins (Fernandez-Funez et al, 2000; Ghosh and Feany, 2004; Hamamichi et al, 2008; Kazemi-Esfarjani and Benzer, 2000; Kraemer et al, 2006; Kuwahara et al, 2008; Morley et al, 2002; Outeiro and Lindquist, 2003; Van Ham et al, 2008; Wang et al, 2009a). These organisms share at least 30% of their genes with humans and show a strong evolutionary conservation of many cellular pathways (Table 1). In fact, several regulatory pathways that play a major role in human embryonic development and physiology were first elucidated in these organisms, including genetic pathways that regulate programmed cell death and neuronal function (Ellis & Horvitz, 1986; McIntire et al, 1993). In addition, molecular pathways that regulate aging have been resolved in yeast, worms and flies, which makes these models excellently suited to studying aging-associated neurodegenerative diseases (Clancy et al, 2001; Kaeberlein et al, 1999; Kenyon et al, 1993).
Table 1.
Conservation of genetics of human disease in popular model organisms for protein aggregation research
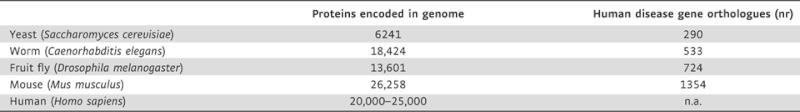 |
‘Proteins encoded in genome’ indicates the estimated number of proteins encoded by the genome; for yeast, C. elegans and Drosophila the estimations from Rubin et al (2000) were used, for mouse, the Mouse Genome Informatics from Jackson Laboratory (http://jax.bioinformatics.org; September 2009), and for human the estimation from the International Genome Sequencing Consortium. The human disease homology indicates the number of genetic orthologue clusters to human disease genes, relating to the number of orthologues in that model organism of human disease genes, a measure for the genetic conservation of human disease genes (O'Brien et al, 2004, Human Mutation; http://orthodisease.cgb.ki.se).
The misfolded protein pathology of human neurodegenerative diseases has been modelled in these small model organisms by transgenic expression of human disease proteins (Table 2, Fig 2). As an example, expression of human α-synuclein in yeast is toxic in a dose-dependent manner, resembling the situation in humans in which multiplication of the α-synuclein locus causes early-onset Parkinson's disease (Ibanez et al, 2004; Outeiro and Lindquist, 2003; Singleton et al, 2003). Similarly, worms expressing fluorescently labelled polyglutamine stretches, show a repeat-length dependent- and age-dependent aggregation and toxicity, which captures the inverse correlation between the onset-age of disease and length of repeat expansion in human polyglutamine diseases (Morley et al, 2002).
Table 2.
Summary of genetic screens for modifiers of protein-misfolding toxicity and inclusion formation in small model organisms
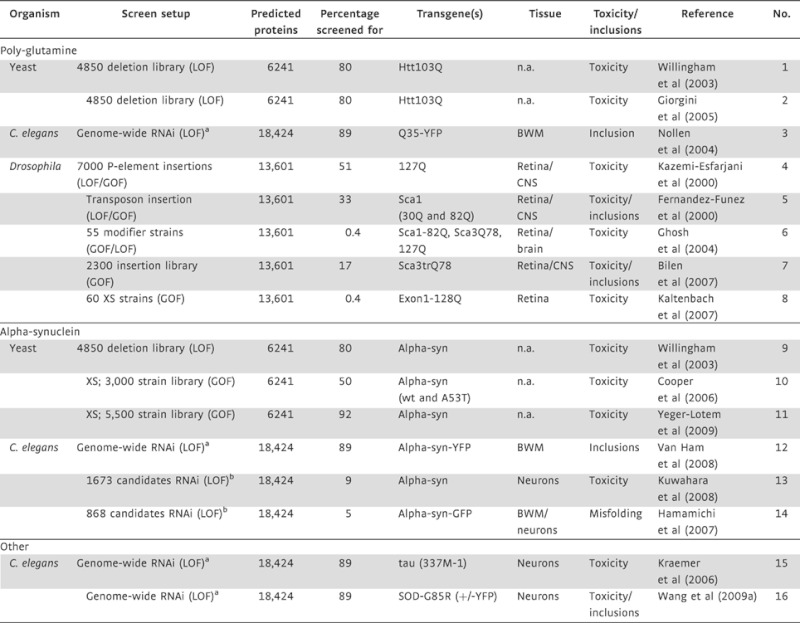 |
Screen setup indicates whether screen was performed using mutagenesis (deletion libraries, transposon based insertion), transgenic overexpression (XS) or knockdown (RNAi, RNA interference). Estimations for the number of encoded proteins in the genome are based on Ruben et al (2000). The ‘percentage screened for’ indicates the number of genes screened for/manipulated (GOF and LOF) as a percentage of the predicted number of proteins. Toxicity and inclusions indicate whether the screen included the formation of inclusions or the exertion of toxicity as a read for the screen, this does not indicate all genes screened for where tested accordingly. BWM, body wall muscle; LOF, loss-of-function; GOF, gain-of-function; n.a., not applicable.
aGenome-wide RNAi screens correspond to the assaying of RNAi knockdown ∼17,000 genes using the Ahringer RNAi library (ref).
bCandidates based on previous findings. SOD, superoxide dismutase; mutation of which is involved in amyloid lateral sclerosis (ALS), Sca1/3 Spinocerebellar ataxia genes 1/3.
Figure 2. Screening phenotypes in small model organisms related to polyglutamine diseases.

A major feature of all of these protein-misfolding diseases is that they manifest late in life, usually after middle age. With respect to this age dependency, an important practical advantage of using small animal models is that, while they do show a well-defined pathobiology of aging that resembles that of humans, they do have a much shorter lifespan. C. elegans, for example, lives for only about 20 days and shows progressive cellular aging, including a deterioration of muscles that resembles aging-related sarcopenia in humans (Herndon et al, 2002). The short lifespan and defined aging properties of these model organisms enable the progressive stages of age-related disease pathology to be monitored during all developmental stages up to old age.
A link between the molecular mechanisms of aging and protein- misfolding pathology has been found in these models; molecular pathways that regulate the lifespan of these organisms also influence progression of misfolding disease phenotypes (Cohen et al, 2006; Morley et al, 2002; Outeiro et al, 2007; Van Ham et al, 2008). In sum, small model organisms are available that capture paramount genetic features of protein-misfolding diseases, including the age-dependent aggregation and toxicity of disease-related proteins. These features can be modified by independent processes, as illustrated by mutations in genes that regulate lifespan and alter misfolded protein pathology. The ease of culturing and genetic screening in yeast, worm and fly models of protein-misfolding diseases means that these organisms are uniquely suited for the unbiased identification of novel modifiers and cellular processes involved in these aging-associated disease phenotypes.
Genetic screens for modifiers of protein misfolding and aggregation
To systematically identify modifiers of aggregation and toxicity of misfolded disease proteins, yeast, worm and fly models have been used for a variety of high-throughput genetic screens, ranging from deletion libraries to genome-wide RNAi screens (Table 2 and Table 1 of Supporting Information). Some researchers doubt the relevance of small model organisms for identifying modifiers of human disease, but several observations demonstrate that the results can be extrapolated to human neuronal cells. For example, in the C. elegans genome-wide RNAi screen for polyglutamine aggregation, TCP-1 chaperonin orthologues were identified as modifiers (Nollen et al, 2004). Several studies later confirmed the role of this chaperonin in aggregation of mutant huntingtin in mammalian cells, proving the validity of such a screen to find bona fide candidate disease modifiers (Behrends et al, 2006; Kitamura et al, 2006; Tam et al, 2006). Along the same line, the ER-Golgi transport regulator, Rab1, picked up in a yeast screen as a suppressor of α-synuclein toxicity, showed rescue of different toxicity phenotypes related to α-synuclein in mammalian neurons as well, indicating that the underlying processes are evolutionarily conserved (Cooper et al, 2006). Finally, the yeast orthologue of a Parkinson's disease-linked gene, ATP13A2, has been found as a modifier of α-synuclein toxicity in yeast (Gitler et al, 2009). This finding has revealed a functional connection between α-synuclein and another Parkinson's disease susceptibility gene (Gitler et al, 2009). Together, these examples demonstrate the potential of genome-wide screens in simple organisms to provide insight into the molecular mechanisms of neurodegenerative diseases in humans.
Screens in flies
The first high-throughput genetic screens for modifiers of misfolded protein toxicity used transgenic Drosophila models for polyglutamine (Fernandez-Funez et al, 2000; Kaltenbach et al, 2007; Kazemi-Esfarjani and Benzer 2000). Expression of mutant ataxin-1 (SCA1), ataxin-3 (SCA3), which cause Spinocerebellar ataxias 1 and 3 in humans, or 127Q huntingtin in neuronal retina cells causes depigmentation and collapse of eye morphology (Fig 2) (Bilen and Bonini, 2007; Fernandez-Funez et al, 2000; Kazemi-Esfarjani and Benzer, 2000). This toxicity phenotype can be rescued by overexpression of heat shock protein 70 (ASP7), an evolutionarily highly conserved molecular chaperone involved in refolding of misfolded proteins (Warrick et al, 1999). Using libraries of genetically altered transposon (P-element) insertion strains, additional modifiers of toxicity of polyglutamine proteins have been screened for. These screens have identified Hsp70 co-chaperones, such as Hsp40 and DNAJ1, and genes involved in proteasomal degradation as modifiers of toxicity (Bilen and Bonini, 2007; Fernandez-Funez et al, 2000; Kazemi-Esfarjani and Benzer, 2000).
Screens in yeast
Comprehensive genetic screens in yeast have been performed with libraries comprised of thousands of strains that have a deletion in a single yeast gene or conversely overexpress one. Genes that enhance or suppress toxicity of misfolded α-synuclein or huntingtin proteins, as scored for by assessing their influence on colony forming ability, were considered modifiers of toxicity (Fig 2) (Cooper et al, 2006; Giorgini et al, 2005; Willingham et al, 2003). Classification of these genes revealed that modifiers of huntingtin toxicity mainly play a role in stress response pathways, protein folding and proteasomal degradation. Modifiers of α-synuclein toxicity, on the other hand, are overrepresented in vesicle-mediated transport and lipid metabolism, suggesting that, in contrast to expectations based on similarities between α-synuclein protein aggregation pathology to other diseases, these disease proteins cause toxicity via different mechanisms.
Screens in C. elegans
Finally, screens for modifiers in C. elegans have used genome-wide RNAi libraries composed of about 17,000 bacterial strains expressing double-stranded RNA against almost every C. elegans gene. The worms can be fed these strains to knock down each individual gene one by one. When applied after embryonic development, such screens have the advantage that, compared to yeast screens, they can also screen for essential genes. Screens for modifiers of aggregation initially took advantage of the transparency of C. elegans at all ages and the amenability of the body wall muscle cells for RNAi. These screens focused on identifying modifiers that caused a premature appearance of microscopically visible aggregates of fluorescently labelled misfolded proteins (Fig 2) (Nollen et al, 2004; Van Ham et al, 2008). A genome-wide RNAi screen for modifiers of polyglutamine aggregation identified close to 200 modifiers. The majority of these fell into five functional classes: translation, protein turnover, -folding and -transport and RNA metabolism, which suggested that even small changes in a wide variety of processes, including those not obviously related to known protein quality control pathways, can lead to pathological features of neurodegenerative diseases (Nollen et al, 2004).
In a similar C. elegans screen for α-synuclein aggregation, genome-wide RNAi screening identified modifier genes involved in protein degradation, vesicle trafficking, lipid metabolism and RNA metabolism. While very few individual genes were exact orthologues of previously identified modifiers for α-synuclein toxicity in yeast, they fell into gene classes that showed a clear overlap. Similar to the observations in yeast, the genes and the gene classes identified in the polyglutamine and α-synuclein screens in C. elegans showed little overlap, again suggesting that different processes underlie different protein-misfolding diseases (Van Ham et al, 2008).
As they were performed in non-neuronal cells, genome-wide screens in yeast and in C. elegans muscles have uncovered basal cellular processes that play a role in misfolded protein pathology. A serious concern about these results is that genes with a specific role in neurons, the actual site where the misfolded proteins in question cause disease, may have been missed. In an attempt to identify these factors in C. elegans, mutant strains have been used in which neurons, which are normally resistant to RNAi, are made more sensitive. Such strains have been used to find modifiers of toxicity and aggregation of misfolding disease-related proteins, including α-synuclein, in neurons (Kuwahara et al, 2008; Li et al, 2009). Unlike previous genome-wide screens, these screens used a pool of genes that were preselected based on their established role in neuronal function or for their predicted role in neurodegeneration. These preselected screens for α-synuclein neuronal toxicity identified modifiers in similar functional classes as the genes found in the genome-wide screen for α-synuclein inclusions formation in non-neuronal cells. In particular, genes with a role in vesicle transport were identified in both types of screens (Hamamichi et al, 2008; Kuwahara et al, 2008). Thus, this has revealed that the formation of inclusions and the toxicity of α-synuclein are influenced by related processes. Moreover, these findings indicate that the processes involved in pathology include basal cellular functions that are not exclusive to neurons, although neurons may be more sensitive to alterations in these processes.
One major question is whether the cellular mechanisms involved in protein-misfolding diseases are common to several such diseases or disease-specific. With the number of related screens performed thus far, it is now possible to compare these screens to start answering this question. One limitation to the comparison is that all high-throughput genetic screens for modifiers were performed in different model organisms and used very different screen set-ups, including different modes of gene targeting and scoring for different phenotypes (Table 2). It is therefore not surprising that, at first sight, the individual genes identified hardly overlap between the different screens, but taking a closer look at the results of these screens leads to a different conclusion. The molecular pathways and functional classes to which individual modifiers belong appear strikingly similar for each individual disease-related protein (Table 3 and Table 1 of Supporting Information).
Table 3.
Common classes of modifier genes identified in diverse screens
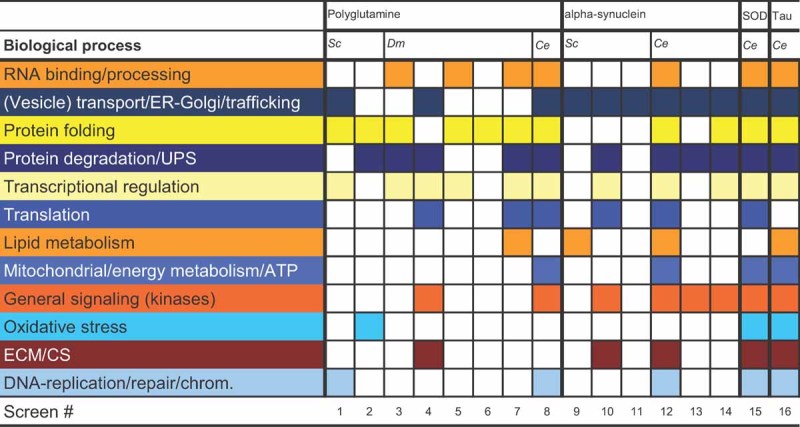 |
Screen numbers correspond to numbers and references in Table 2. Colour coding indicates classes (biological processes) described as given in the original publications (Table 1 of Supporting Information). White indicates that no genes in this class were found or not described as such in the original publication. Classes only found as one gene in one or two screens, without functional overrepresentation analysis or statistical analysis are not indicated here.
Disease-specific pathways
α-Synuclein diseases and vesicle transport defects
For instance, the vesicle transport defects identified as underlying the formation of α-synuclein inclusions and toxicity seem very specific to α-synuclein pathology. Modifiers in this functional class are hardly found in polyglutamine modifier screens (Table 3). This specificity could be explained by the lipid-binding properties of α-synuclein and its role in vesicle homeostasis, and suggests that this property is important for modulating an early toxic event (Chandra et al, 2005; Sharon et al, 2001). This early toxic event appears to also require aggregation of α-synuclein, because β-synuclein, a non-aggregating counterpart of α-synuclein, as well as mutant α-synuclein lacking the domain required for aggregation do not cause toxicity in yeast but do share the lipid-binding property with α-synuclein (Soper et al, 2008).
Neuronal function relies largely on the synthesis, maintenance, docking and fusion of synaptic vesicles. These processes depend on subtle changes in the stability, fluidity and curvature of the vesicle membranes, which are determined by the lipid composition of the membranes involved. The lipid composition of membranes changes as mammals age (Maguire & Druse, 1989; Yehuda et al, 2002). Due to the function and properties of in particular α-synuclein, its involvement in vesicle homeostasis, and its interactions with lipid membranes, altered lipid homeostasis due to old age may, therefore, specifically be important for the development of diseases related to α-synuclein misfolding (Welch & Yuan, 2003).
RNA metabolism in polyglutamine expansion diseases
Another example of a functional gene class that is specifically found for one type of neurodegenerative disease-related proteins are the many RNA-processing components as modifiers of polyglutamine toxicity and aggregation. It is noteworthy that this class of genes was consistently found in multiple screens for toxicity and aggregation of polyglutamine in Drosophila and C. elegans (Table 3). There could be several explanations for a role of general RNA metabolism in these diseases. One is that polyglutamine proteins interact with key regulators of transcription that often contain polyglutamine stretches themselves (Nucifora et al, 2001; Zhai et al, 2005). Another aspect of RNA metabolism that was recently shown to modify polyglutamine pathology is microRNA (miRNA) metabolism (Bilen et al, 2006a, b). Polyglutamine aggregation and toxicity are modified by several genes with a general role in miRNA processing, like C. elegans argonoute-like gene alg-1, and Drosophila gene R3D1 (Bilen et al, 2006a, b; Nollen et al, 2004). Thus, RNA processing as a polyglutamine disease-modifying pathway could involve the processing of non-coding RNAs. In mice, the expression of miRNAs is altered during aging, starting from middle-age onwards (Li et al, 2009). If such a change occurs in humans during aging, this would be consistent with miRNAs modifying the onset of polyglutamine disease, which typically takes place after middle-age. This potential mechanistic relation needs to be investigated more closely. Of particular note is the discovery of the fly orthologue of human SCA2, an RNA-binding protein, as a modifier of SCA3 and SCA1 toxicity, showing that SCA genes interact with each other (Al-Ramahi et al, 2007; Lessing & Bonini, 2008). Furthermore, the observation of muscleblind (mbl), another RNA-binding protein, as a modifier of toxicity has led to the discovery that the SCA3 transcript itself is toxic (Li et al, 2008). Altogether, these findings have provided explanations for the specificity of RNA metabolism genes as modifiers of polyglutamine toxicity.
Although there is much overlap in the modifiers found in the different polyglutamine models, a recent study comparing modifiers in two different Drosophila models shows that some modifiers are specific and some are common to SCA1 and huntingtin toxicity. Modifiers found in the mutant SCA1 (82Q) flies have been tested in an Htt-128Q fly model, revealing common and specific modifiers of toxicity (Branco et al, 2008). The modifiers fall into three classes: (i) modifiers of toxicity in only one model, (ii) modifiers that enhance or suppress toxicity in both models and (iii) modifiers that function as both enhancers and suppressors, depending on the background.
The first group contains two chaperones and two transcription factors. The second group of modifiers include chaperones (DNJA1), several components of the ubiquitin–proteasome system, including Carboxyl Terminus of HSP70-interacting protein (CHIP), and genes involved in signal transduction, RNA metabolism and apoptosis. The last group includes one gene involved in RNA metabolism and genes involved in signal transduction, including Akt, which is known to phosphorylate ataxin-1 (Branco et al, 2008). Such difference in modifier genes for different polyglutamine proteins is in part explained by findings that other domains of Atx1 besides the polyglutamine domain play a crucial role in toxicity (Lam et al, 2006; Lim et al, 2008; Tsuda et al, 2005). Thus, different expanded polyglutamine proteins have overlapping but also protein specific mechanisms of toxicity, which involve other, non-polyglutamine, segments of the disease proteins as well.
Common pathways in neurodegenerative diseases
The most obvious common class of genes that act on the pathology of all disease proteins is that involved in protein degradation (Table 3). The finding of this gene class is not surprising, as components of the ubiquitin proteasome system have been found to co-localize with aggregated proteins in the brain of patients; in fact, together with the co-localization of molecular chaperones, this was a first hint that protein misfolding might underlie neurodegeneration (Cummings et al, 1998; Rubinsztein, 2006). Still, finding these protein-degradation components powerfully confirms the validity of using model organisms to find modifiers of misfolding disease. In this common class of genes we still need to learn whether they all act on all misfolded proteins or whether some of them are specialized in mediating degradation of particular misfolded disease proteins. E3 ubiquitin ligases, for example, of which there are hundreds expressed in humans, each mark specific proteins for degradation (Robinson & Ardley, 2004). It is possible that there are E3 ligases that target all misfolded disease proteins or that there are ligases that act on specific disease proteins, but these remain to be identified.
Gene networks
In humans it is likely that a combination of gene mutations results in a sporadic neurodegenerative disease phenotype, given the small contribution to disease susceptibility for each gene found in genome wide association studies. The genetic screens in model organisms mentioned in this review all assess the role of a single gene at a time. Functional gene networks can be identified by large-scale synthetic and complementation screens and by systematic analyses of genetic dependencies (Kornmann et al, 2009; Jonikas et al, 2009; Van Haaften et al, 2004). It will be worthwhile to perform such studies for modifiers of protein misfolding in order to identify gene networks that modify disease pathology.
Systematic data integration
The systematic integration of the results obtained in a wide variety of high-throughput genetic screens in different model organisms has only just begun. The challenges are exemplified in a small-scale analysis our laboratory performed for genome-wide RNAi screens for α-synuclein and Q35 inclusion formation in C. elegans (Van Ham et al, 2008). Initially, a computational analysis of gene ontology classes of modifiers found in one such C. elegans screen revealed no significant overrepresentation of any functional class. However, analysis of the subcellular localization of the same set of modifiers, when compared to randomized sets of genes, discovered a clear overrepresentation of modifiers with a role in the endomembrane system (Van Ham et al, 2008). A parallel analysis of modifiers of polyglutamine aggregation revealed that these modifiers functioned mainly in the cytosol and in the nucleus, indicating that there are very different ways in which cells deal with misfolded polyglutamine proteins or misfolded α-synuclein (Van Ham et al, 2008).
On a larger scale, a comparison of all yeast genome-wide screens on a wide variety of phenotypes revealed that there was generally no overlap between expression profiling studies and genetic screens, regardless of the phenotype analysed (Yeger-Lotem et al, 2009). However, computational comparison of these screens using a newly developed algorithm, revealed a regulator–effector relationship between the datasets (Yeger-Lotem et al, 2009). These authors assembled an integrated network model of the yeast interactome containing protein–protein interactions, metabolic relations and protein–DNA interactions of various levels of reliability. In this network, they then identified high-probability paths of regulators that potentially connect modulators identified in the genetic screens to their differentially expressed target genes. By applying this algorithm to the yeast screens for α-synuclein toxicity and expression profiling, novel pathways were uncovered. For example, the algorithm predicted the involvement of the heat shock transcription factor Hsf-1 and Gip2 in α-synuclein disorders and this prediction was confirmed experimentally as overexpression of Gip2 was shown to induce an heat shock response and to suppress the toxicity of α-synuclein (Yeger-Lotem et al, 2009). Integrating RNAi screening data with orthogonal evidence, for instance protein–protein interaction data, can further help with refining the biological interpretation of the data (Wang et al, 2009b). Together, these meta-analyses demonstrate that a computational analysis of combined datasets from genome-wide screens in one model organism can reveal previously unidentified pathways with a role in protein aggregation pathogenesis in other species.
Pending issues
What is the initiating toxic event in each of the neurodegenerative diseases? Is the initiating toxic event common or disease-specific?
Which modifiers act on the initiating events of each disease?
Do the modifying processes found in small model organisms play a role in human disease? What additional modifiers are human-specific, due to the larger complexity of tissues or genes?
In the case of sporadic disease, why are specific misfolded proteins found in inclusions, and which modifying processes determine which proteins get misfolded?
What determines the tissue specificity of protein folding disease?
If misfolding of disease proteins occur all the time, what determines the onset of disease? Is age-of-onset dependent on an increased load of misfolded protein or on an altered function of modifiers?
What is the toxic conformation of misfolded disease proteins and is this form similar for different misfolding diseases?
As more data from a broader range of models are acquired, the need for a comprehensive data integration strategy is growing. The work done so far indicates several areas that need special consideration when an automated computational approach is applied: for instance, gene products need to be carefully matched to their orthologues in various organisms, and the number of genes screened for compared to the total number of genes in the genome needs to be taken into account when comparing the result statistics. The interpretation also needs to consider that the results of individual screens tend to provide an incomplete picture and to correct for how the genes that were screened for were selected in each case (see also Table 2). The most important remaining limitation, revealed by both our own analysis of α-synuclein and Q35 inclusion formation and the study of Yeger-Lotem et al, is the striking heterogeneity of modifier genes. Predefined functional classes or pathways will not always be overrepresented among them, as they have been delimited based on unrelated physiological processes that do not necessarily match the protein aggregation process. Here remains a fruitful area for the application of systems biology concepts (Kitano, 2002), which will allow mapping the results of genetic screens onto unbiased dynamic networks of cellular physiology to reveal the mechanistic connections between specific cellular pathways and specific misfolded protein pathologies.
Conclusion
The pathology of different aggregation disorders in small model organisms can be provoked by perturbations in a wide variety of basal cellular processes. Some of these processes affect the pathology of various disease proteins, while others appear to be specific for one type of disease protein. Although these findings remain to be validated in humans, they could provide an explanation for the differences in susceptibility and age-at-onset of aging-associated neurodegeneration, which may be due to natural genetic variation and differences in exposure to environmental factors that influence disease-specific modifying processes.
Acknowledgments
We thank Mats Holmberg and Annemieke van der Goot for suggestions and J. L. Senior for editing the final manuscript. This work was supported by grants from ZonMw Research Institute of the Elderly, the Hersenstichting, the Prinses Beatrixfonds and the Vereniging van Huntington.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Al-Ramahi I, Perez AM, Lim J, Zhang M, Sorensen R, de HM, Branco J, Pulst SM, Zoghbi HY, Botas J. dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCAI. PLoS Genet. 2007;3:e234. doi: 10.1371/journal.pgen.0030234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Behrends C, Langer CA, Boteva R, Bottcher UM, Stemp MJ, Schaffar G, Rao BV, Giese A, Kretzschmar H, Siegers K, et al. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell. 2006;23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007;3:1950–1964. doi: 10.1371/journal.pgen.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J, Liu N, Bonini NM. A new role for microRNA pathways: modulation of degeneration induced by pathogenic human disease proteins. Cell Cycle. 2006a;5:2835–2838. doi: 10.4161/cc.5.24.3579. [DOI] [PubMed] [Google Scholar]
- Bilen J, Liu N, Burnett BG, Pittman RN, Bonini NM. MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol Cell. 2006b;24:157–163. doi: 10.1016/j.molcel.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Branco J, Al Ramahi I, Ukani L, Perez AM, Fernandez-Funez P, Rincon-Limas D, Botas J. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum Mol Genet. 2008;17:376–390. doi: 10.1093/hmg/ddm315. [DOI] [PubMed] [Google Scholar]
- Carrasquillo MM, Zou F, Pankratz VS, Wilcox SL, Ma L, Walker LP, Younkin SG, Younkin CS, Younkin LH, Bisceglio GD, et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer's disease. Nat Genet. 2009;41:192–198. doi: 10.1038/ng.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chen S, Ferrone FA, Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant cu/zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- Gaczynska M, Osmulski PA, Ward WF. Caretaker or undertaker? The role of the proteasome in aging. Mech Ageing Dev. 2001;122:235–254. doi: 10.1016/s0047-6374(00)00246-3. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Feany MB. Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum Mol Genet. 2004;13:2011–2018. doi: 10.1093/hmg/ddh214. [DOI] [PubMed] [Google Scholar]
- Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME. Huntington's disease: seeing the pathogenic process through a genetic lens. Trends Biochem Sci. 2006;31:533–540. doi: 10.1016/j.tibs.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Hamamichi S, Rivas RN, Knight AL, Cao S, Caldwell KA, Caldwell GA. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson's disease model. Proc Natl Acad Sci USA. 2008;105:728–733. doi: 10.1073/pnas.0711018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–1697. doi: 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- Kenyon CL, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kitamura A, Kubota H, Pack CG, Matsumoto G, Hirayama S, Takahashi Y, Kimura H, Kinjo M, Morimoto RI, Nagata K. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat Cell Biol. 2006;8:1163–1170. doi: 10.1038/ncb1478. [DOI] [PubMed] [Google Scholar]
- Kitano H. Computational systems biology. Nature. 2002;420:206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer BC, Burgess JK, Chen JH, Thomas JH, Schellenberg GD. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum Mol Genet. 2006;15:1483–1496. doi: 10.1093/hmg/ddl067. [DOI] [PubMed] [Google Scholar]
- Kuwahara T, Koyama A, Koyama S, Yoshina S, Ren CH, Kato T, Mitani S, Iwatsubo T. A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans. Hum Mol Genet. 2008;17:2997–3009. doi: 10.1093/hmg/ddn198. [DOI] [PubMed] [Google Scholar]
- Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–R59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- Lessing D, Bonini NM. Polyglutamine genes interact to modulate the severity and progression of neurodegeneration in Drosophila. PLoS Biol. 2008;6:e29. doi: 10.1371/journal.pbio.0060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LB, Yu Z, Teng X, Bonini NM. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453:1107–1111. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Bates DJ, An J, Terry DA, Wang E. Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.04.020. Epub May 30, doi: 10.1016/j.neurobiolaging.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, Orr HT, Zoghbi HY. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCAI. Nature. 2008;452:713–718. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire PA, Druse MJ. The influence of cholesterol on synaptic fluidity, dopamine D1 binding and dopamine-stimulated adenylate cyclase. Brain Res Bull. 1989;23:69–74. doi: 10.1016/0361-9230(89)90165-2. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Jorgensen E, Horvitz HR. Genes required for GABA function in Caenorhabditis elegans. Nature. 1993;364:334–337. doi: 10.1038/364334a0. [DOI] [PubMed] [Google Scholar]
- Metzger S, Bauer P, Tomiuk J, Laccone F, Didonato S, Gellera C, Mariotti C, Lange HW, Weirich-Schwaiger H, Wenning GK, et al. Genetic analysis of candidate genes modifying the age-at-onset in Huntington's disease. Hum Genet. 2006;120:285–292. doi: 10.1007/s00439-006-0221-2. [DOI] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougeot JL, Richardson-Milazi S, Brooks BR. Whole-genome association studies of sporadic amyotrophic lateral sclerosis: are retroelements involved? Trends Mol Med. 2009;15:148–158. doi: 10.1016/j.molmed.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Nollen EA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, Plasterk RH. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci USA. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. O'Brien. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick AZ, Gershon D. The effect of age on the protein degradation system in the nematode Turbatrix aceti. Mech Ageing Dev. 1979;11:403–415. doi: 10.1016/0047-6374(79)90016-2. [DOI] [PubMed] [Google Scholar]
- Robinson PA, Ardley HC. Ubiquitin-protein ligases. J Cell Sci. 2004;117:5191–5194. doi: 10.1242/jcs.01539. [DOI] [PubMed] [Google Scholar]
- Rosenblatt A, Brinkman RR, Liang KY, Almqvist EW, Margolis RL, Huang CY, Sherr M, Franz ML, Abbott MH, Hayden MR, et al. Familial influence on age of onset among siblings with Huntington disease. Am J Med Genet. 2001;105:399–403. [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Sanchez I, Mahlke C, Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421:373–379. doi: 10.1038/nature01301. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with, the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci USA. 2001;98:9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtilerman MD, Ding TT, Lansbury PT., Jr Molecular crowding accelerates fibrillization of alpha-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson's disease? Biochemistry. 2002;41:3855–3860. doi: 10.1021/bi0120906. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Soper JH, Roy S, Stieber A, Lee E, Wilson RB, Trojanowski JQ, Burd CG, Lee VM. Alpha-synuclein-induced aggregation of cytoplasmic vesicles in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:1093–1103. doi: 10.1091/mbc.E07-08-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S, Geller R, Spiess C, Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda H, Jafar-Nejad H, Patel AJ, Sun Y, Chen HK, Rose MF, Venken KJ, Botas J, Orr HT, Bellen HJ, et al. The AXH domain of Ataxin-l mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122:633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Van Haaften G, Vastenhouw NL, Nollen EA, Plasterk RHA, Tijsterman M. Gene interactions in the DNA damage-response pathway identified by genome-wide RNA-interference analysis of synthetic lethality. Proc Natl Acad Sci USA. 2004;101:12992–12996. doi: 10.1073/pnas.0403131101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ham TJ, Thijssen KL, Breitling R, Hofstra RM, Plasterk RH, Nollen EA. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, Horwich AL. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009a;5:e1000350. doi: 10.1371/journal.pgen.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tu Z, Sun F. A network-based integrative approach to prioritize reliable hits from multiple genome-wide RNAi screens in Drosophila. BMC Genomics. 2009b;10:220. doi: 10.1186/1471-2164-10-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Welch K, Yuan J. Alpha-synuclein oligomerization: a role for lipids? Trends Neurosci. 2003;26:517–519. doi: 10.1016/j.tins.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- Yeger-Lotem E, Riva L, Su LJ, Gitler AD, Cashikar AG, King OD, Auluck PK, Geddie ML, Valastyan JS, Karger DR, et al. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat Genet. 2009;41:316–323. doi: 10.1038/ng.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda S, Rabinovitz S, Carasso RL, Mostofsky DI. The role of polyunsaturated fatty acids in restoring the aging neuronal membrane. Neurobiol Aging. 2002;23:843–853. doi: 10.1016/s0197-4580(02)00074-x. [DOI] [PubMed] [Google Scholar]
- Zhai W, Jeong H, Cui L, Krainc D, Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell. 2005;123:1241–1253. doi: 10.1016/j.cell.2005.10.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.