

Abstract
Background
The apolipoprotein E (APOE) ε2 allele has been suggested as having a protective effect and delaying the age at onset of Alzheimer disease.
Objective
To describe a dissociation between findings neuropathologic with normal cognition in a woman with severe Alzheimer disease with the APOE ε2/ε2 genotype.
Design
Case report from a community based prospective study of persons 90 years or older (The 90+ Study).
Participant
A 92-year-old woman without dementia with the APOE ε2/ε2 genotype who lived independently without significant cognitive or functional loss and was a participant in The 90+ Study. She died in December 2004, and postmortem examination of her brain was performed.
Intervention
Neurologic examination and a battery of neuropsychological tests were performed 6 months and 1 month before death. Neuropathologic examination included Braak and Braak staging for senile plaques and neurofibrillary tangles.
Results
Neuropathologic examination of the brain revealed advanced senile plaque and neurofibrillary tangle disease consistent with a high likelihood of Alzheimer disease. At clinical evaluation, the participant demonstrated no dementia and only mild cognitive deficits.
Conclusions
The APOE genotype may have contributed to maintenance of cognition despite advanced neuropathologic findings of Alzheimer disease. This case suggests that the APOE ε2 isoform may have a protective effect against cognitive decline in Alzheimer disease that may be independent from senile plaques and neurofibrillary tangles.
In the Last Decade, Many Studies (for a review, see Laws et al1) have examined the role of the apolipoprotein E (APOE) genotype in the development of Alzheimer disease (AD) and neuropathologic findings in AD. There are 3 APOE alleles: ε2, ε3, and ε4. The ε3 allele is the most common, representing approximately 78% of all alleles; the ε2 and ε4 alleles are much less common, representing 7% and 15%, respectively, of all alleles.2 Of the 6 possible genotypes, ε2/ε2 is the rarest, occurring in less than 1.4% of most populations3 and severely limiting research opportunities.
The ε2 allele (APOE2) has been suggested as having a protective effect and delaying the age of onset of AD.4 In addition, APOE2 appears with reduced frequency in sporadic cases of AD5 and patients with AD with APOE2 have less (β-amyloid (Aβ) density in the frontal and parietal cortices compared with patients with AD with the ε3/ε3 genotype.6
We report the case of a woman with the rare ε2/ε2 genotype, who died at age 92 years. At brain autopsy, substantial AD-like neuropathologic features were noted, but the participant had not demonstrated any cognitive deficits indicative of AD. Many studies have found evidence of a weak association between neuropathologic features of AD and cognition in the elderly,7,8 a finding that is more common in studies that included oldest old subjects.9-11 However, these studies rarely reported dissociation of the magnitude observed in our participant, and most do not examine the contribution of the APOE genotype. It is suggested that the unusual APOE status in our participant may have had a protective effect, delaying or possibly preventing the onset of cognitive symptoms but not AD-like neuropathologic features.
Report of a case
The subject was a 92-year-old right-handed woman with hypertension but no diabetes mellitus who was a participant in The 90+ Study, a prospective population based study of cognition and longevity in the oldest old. She reported mild shortterm memory loss in 2004 but was still living independently despite blindness in the left eye as a result of shingles.
As part of the Leisure World Cohort Study,12 the participant had completed a health questionnaire in 1984 and followup surveys in 1992 and 1998. These surveys indicated a history of vitamins A and E and ascorbic acid usage for 25 years and estrogen supplementation (1.25 mg) for more than 15 years. The participant indicated that she exercised, on average, 7 hours per week.
Her medical history included hypertension and hypercholesterolemia diagnosed in 1980, a transient ischemic attack in 1990, and atrial fibrillation diagnosed in 1999. Her medications were atenolol, levothyroxine sodium, hydrochlorothiazide, acyclovir, calcium, and potassium chloride. There was a family history of stroke (her brother had a stroke at an unknown age), but no family history of dementia was reported. The participant completed high school and some college. She smoked onefourth pack of cigarettes a day for 17 years, but stopped at age 37 years. No alcohol consumption was reported.
On November 14, 2003, the participant was examined using the Cognitive Abilities Screening Instrument via telephone and scored 33 of a possible 33, indicating intact global cognitive functioning. In 2004, she was examined twice. During these visits, she completed a neuropsychology battery including the MiniMental Status Examination (MMSE); California Verbal Learning Test (9 items); Digit Span (forward and backward) subtest of the Wechsler Adult Intelligence Scale, Third Edition; Boston Naming Test (15 items); animal and letter /F/ fluency; Constructional Praxis subtest of the Consortium to Establish a Registry for Alzheimer Disease Psychological Battery; and Trail Making Test, parts A through C. The results are given in Table. Her performance at the first visit was within normal limits for her age group13 on all measured domains including attention, language, visuoconstruction, verbal memory, and executive function. Between visits, in September 2004, the participant had a stroke that resulted in dysarthria, dysphagia, and rightsided weakness for 10 to 14 days. Her performance on the neuropsychological tests administered after the stroke remained within normal limits across domains, with 2 exceptions. Phonemic fluency declined, and she demonstrated mild executive dysfunction as reflected by inability to complete the Trail Making Test, part B. These deficits are best characterized as mild and, on the basis of family reports, did not result in functional loss. The participant died after a second stroke in December 2004.
Table. Neuropsychological Test Scores 6 Months and 1 Month Before Death.
| May 27, 2004 | November 4, 2004 | |||
|---|---|---|---|---|
|
|
|
|||
| Test | Raw Score | Percentilea | Raw Score | Percentilea |
| MMSE | 28/30 | 28/30 | ||
| Digit Span, forward | ND | 15 | 99 | |
| Digit Span, backward | ND | 4 | 20 | |
| Boston Naming Test, 15 items | 12 | 52 | 12 | 52 |
| Fluency tests, No. of words in 1 min | ||||
| Animal | 22 | 99 | 21 | 97 |
| Letter F | 13 | 52 | 7 | 15 |
| CVLT, 9 items, No. of words | ||||
| Trial 1 | 3 | 25 | 5 | 71 |
| Trial 4 | 6 | 38 | 6 | 38 |
| 10-Minute delay | 3 | 29 | 6 | 71 |
| CERAD copy total, 11 possible points | ND | 9 | 55 | |
| Clocks (11:10) total, 8 possible points | 7 | 79 | 6 | 60 |
| Trail Making Test, total s | ||||
| Part A | ND | 80 | 30 | |
| Part B | ND | 300b | 10 | |
| Part C | ND | 47 | 25 | |
Abbreviations: CERAD, Consortium to Establish a Registry for Alzheimer Disease; CVLT, California Verbal Learning Test; MMSE, MiniMental State Examination ND, not done.
Calculated using normative data from The 90+ Study.9
The participant was given maximum time, but the test was discontinued because she could not follow instructions
At autopsy, the brain weighed 1140 g. Senile plaque formation, which was moderately intense, was predominantly diffuse, although neuritic plaques were readily observed. Neurofibrillary tangle formation, in addition to being present within the hippocampus, subiculum, entorhinal-transentorhinal region, and amygdala, was also seen in moderately severe to severe degree within the frontal, temporal, parietal, and occipital layers of the neocortex (Figure 1). Overall, the pathological changes were judged to be of stage VI-C severity by Braak and Braak staging criteria (the stage that represents the highest frequency of senile plaques and neurofibrillary tangles). These neuropathologic changes are considered highly likely to be associated with dementia according to criteria of the National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer Disease,14 although our participant did not experience significant cognitive dysfunction. Additional immunostaining (Figure 2) for Aβ and tau proteins was performed to compare findings in our participant with those in a subject without dementia with a similar MMSE score but with few pathologic features of AD (age, 99 years; female sex; MMSE score 28; and Braak and Braak classification III-0) and a subject with dementia with similar neuropathologic features of AD but a much lower MMSE score (age, 94 years; female sex; MMSE score 0; and Braak and Braak classification VI-C). The results showed that senile plaques in our participant's brain were both diffuse and neuritic, compared with only neuritic as observed in the comparison subject with dementia.
Figure 1.
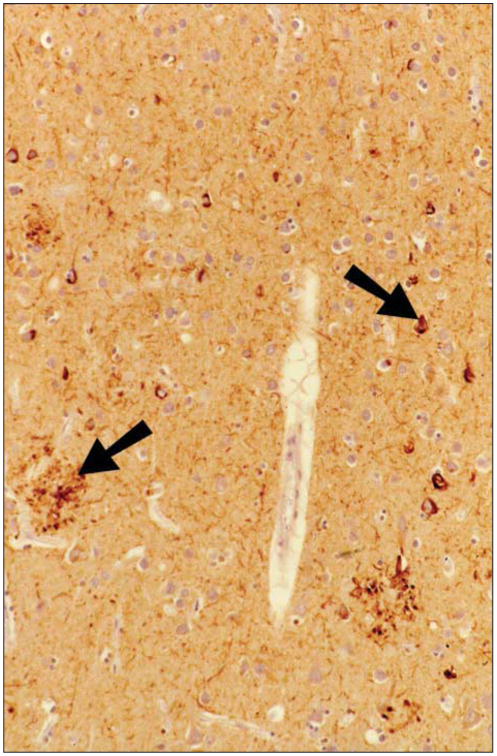
The case subject demonstrated neocortical neuropathologic features of Alzheimer disease. At histopathologic analysis, a section of inferior parietal cortex immunostained with polyclonal rabbit antihuman tau (DAKO, Carpenteria, California) showed neuritic plaque formation and neurofibrillary degeneration (arrows).
Figure 2.
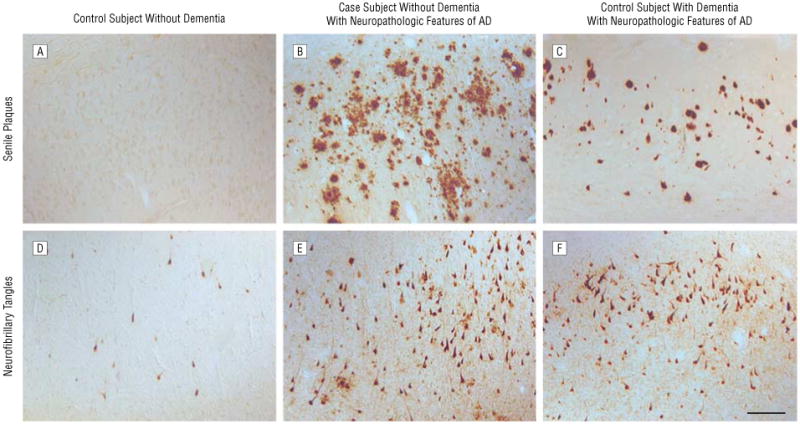
Neuropathologic features in the case subject compared with matched control subjects. Note pathologic features in area CA1/subiculum of the hippocampus in the case subject. β-Amyloid deposition is absent in area CA1/subiculum in a 99-year-old woman without dementia (A), primarily diffuse in the case subject (B), and associated with extracellular neurofibrillary tangles and compact neuritic plaques in a 94-year-old woman with dementia (C). Neurofibrillary tangle accumulation was sparse in the control subject without dementia (D) but was comparable in the case subject (E) and the control subject with dementia (F) AD indicates Alzheimer disease; bar, 100 μm.
Comment
This case shows a prominent dissociation between severe neuropathologic features of AD and preserved cognitive function. Although many potential mechanisms may be responsible, the rare APOE genotype (ε2/ε2) in our participant may have contributed to this dissociation. Current research suggests that APOE2 may delay the onset of clinical AD by increasing Aβ clearance15 and protecting against the formation of neurofibrillary tangles.16 Also, APOE2 is typically associated with reduced neuropathologic features of AD,17 and APOE2 injected in PDAPP mouse brains, a model of AD, reduces Aβ and facilitates degradation and turnover.18 Despite the extensive plaque deposition observed in our participant, many plaques appeared as diffuse (β-amyloid deposits, suggesting that the presence of APOE2 may mediate a more rapid turnover of Aβ deposits and lead to less neuronal injury.
Although our participant demonstrated diffuse plaque formation, extensive neuritic plaque and neurofibrillary tangles were also observed in the presence of intact cognition. Therefore, APOE2 may contribute to this dissociation independently of the formation of senile plaques and neurofibrillary tangles, exerting its protective effect through alternative mechanisms such as the reduction of oxidative stress damage19,20 and the preservation of synaptic viability.21
This case provides insight into a possible mechanism for preserved cognition with severe neuropathologic features of AD. In addition to providing evidence of a possible protective mechanism of APOE2, this case report also underscores the importance of clinical confirmation to support a neuropathologic diagnosis of AD, especially in the oldest old.
Acknowledgments
Funding/Support: This study was supported by grant RO1-AG 21055 from the National Institute on Aging (Dr Kawas).
Footnotes
Author Contributions:Study concept and design: Berlau. Acquisition of data: Kahle-Wrobleski, Head, Goodus, Kim, and Kawas. Analysis and interpretation of data: Berlau, Kahle-Wrobleski, and Head. Drafting of the manuscript: Berlau, Kahle-Wrobleski, and Head. Critical revision of the manuscript for important intellectual content: Berlau, Kahle-Wrobleski, Goodus, Kim, and Kawas. Statistical analysis: Kahle-Wrobleski. Obtained funding: Kawas. Administrative, technical, and material support: Berlau, Head, and Goodus. Study supervision: Berlau and Kim.
Financial Disclosure: None reported.
References
- 1.Laws SM, Hone E, Gandy S, Martins RN. Expanding the association between the APOE geneand the risk of Alzheimer's disease: possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J Neurochem. 2003;84(6):1215–1236. doi: 10.1046/j.1471-4159.2003.01615.x. [DOI] [PubMed] [Google Scholar]
- 2.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240(4852):622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 3.Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. 2002;155(6):487–495. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- 4.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7(2):180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 5.West HL, Rebeck GW, Hyman BT. Frequency of the apolipoprotein E epsilon 2 allele is diminished in sporadic Alzheimer disease. Neurosci Lett. 1994;175(1–2):46–48. doi: 10.1016/0304-3940(94)91074-x. [DOI] [PubMed] [Google Scholar]
- 6.Lippa CF, Smith TW, Saunders AM, Hulette C, PulaskiSalo D, Roses AD. Apoipoprotein E-epsilon 2 and Alzheimer's disease: genotype influences pathologic phenotype. Neurology. 1997;48(2):515–519. doi: 10.1212/wnl.48.2.515. [DOI] [PubMed] [Google Scholar]
- 7.Crystal H, Dickson D, Fuld P, et al. Clinicopathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease. Neurology. 1988;38(11):1682–1687. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- 8.Galvin JE, Powlishta KK, Wilkins K, et al. Predictors of preclinical Alzheimer's disease and dementia: a clinicopathologic study. Arch Neurol. 2005;62(5):758–765. doi: 10.1001/archneur.62.5.758. [DOI] [PubMed] [Google Scholar]
- 9.Katzman R, Terry R, DeTeresa R, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23(2):138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- 10.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 11.Prohovnik I, Perl DP, Davis KL, Libow L, Lesser G, Haroutunian V. Dissociation of neuropathology from severity of dementia in late-onset Alzheimer disease. Neurology. 2006;66(1):49–55. doi: 10.1212/01.wnl.0000191298.68045.50. [DOI] [PubMed] [Google Scholar]
- 12.Paganini-Hill A, Ross RK, Henderson BE. Prevalence of chronic disease and health practices in a retirement community. J Chronic Dis. 1986;39(9):699–707. doi: 10.1016/0021-9681(86)90153-0. [DOI] [PubMed] [Google Scholar]
- 13.Whittle C, Corrada MM, Dick M, et al. Neuropsychological data in nondemented oldest old: The 90+ Study. J Clin Exp Neuropsychol. 2007;29(3):290–299. doi: 10.1080/13803390600678038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Consensus recommendations forthe postmortem diagnosis of Alzheimer's disease. Neurobiol Aging. 1997;18(4)(suppl):S1–S2. [PubMed] [Google Scholar]
- 15.Yang DS, Smith JD, Zhou Z, Gandy SE, Martins RN. Characterization of the binding of amyloid-beta peptide to cell culture-derived native apolipoprotein E2, E3, and E4 isoforms and to isoforms from human plasma. J Neurochem. 1997;68(2):721–725. doi: 10.1046/j.1471-4159.1997.68020721.x. [DOI] [PubMed] [Google Scholar]
- 16.Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer's disease. Proc Natl Acad Sci U S A. 1995;92(11):4725–4727. doi: 10.1073/pnas.92.11.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tiraboschi P, Hansen LA, Masliah E, Alford M, Thal LJ, Corey-Bloom J. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62(11):1977–1983. doi: 10.1212/01.wnl.0000128091.92139.0f. [DOI] [PubMed] [Google Scholar]
- 18.Dodart JC, Marr RA, Koistinaho M, et al. Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102(4):1211–1216. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Y, Aono M, Laskowitz D, Warner DS, Pearlstein RD. Apolipoprotein E protects against oxidative stress in mixed neuronalglial cell cultures by reducing glutamate toxicity. Neurochem Int. 2004;44(2):107–118. doi: 10.1016/s0197-0186(03)00112-8. [DOI] [PubMed] [Google Scholar]
- 20.Miyata M, Smith JD. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and betaamyloid peptides. Nat Genet. 1996;14(1):55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- 21.Love S, Siew LK, Dawbarn D, Wilcock GK, Ben-Shlomo Y, Allen SJ. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging. 2006;27(6):797–803. doi: 10.1016/j.neurobiolaging.2005.04.008. [DOI] [PubMed] [Google Scholar]