

Abstract
Carbonic anhydrases (CAs) are ubiquitous enzymes that catalyze the reversible hydration/dehydration of carbon dioxide/bicarbonate. As such, there is enormous industrial interest in using CA as a bio-catalyst for carbon sequestration and biofuel production. However, to ensure cost-effective use of the enzyme under harsh industrial conditions, studies were initiated to produce variants with enhanced thermostability while retaining high solubility and catalytic activity. Kinetic and structural studies were conducted to determine the structural and functional effects of these mutations. X-ray crystallography revealed that a gain in surface hydrogen bonding contributes to stability while retaining proper active site geometry and electrostatics to sustain catalytic efficiency. The kinetic profiles determined under a variety of conditions show that the surface mutations did not negatively impact the carbon dioxide hydration or proton transfer activity of the enzyme. Together these results show that it is possible to enhance the thermal stability of human carbonic anhydrase II by specific replacements of surface hydrophobic residues of the enzyme. In addition, combining these stabilizing mutations with strategic active site changes have resulted in thermostable mutants with desirable kinetic properties.
Keywords: carbonic anhydrase, proton transfer, thermostability
Introduction
Carbonic anhydrases (CA, EC 4.2.1.1) are ubiquitous enzymes that catalyze the reversible hydration/dehydration of carbon dioxide/bicarbonate. As such, there is an increasing industrial interest in using CA as bio-catalyst for carbon sequestration out of flue gas from coal-fired power plants (Saville and Lalonde, 2011). In addition, there is also interest in exploiting CA in algae as a way to capture CO2 and convert it into biofuels or other valuable products (Fulke et al., 2010; Ramanan et al., 2010). Human carbonic anhydrase II (HCA II) is a suitable candidate for these applications: It is easy and cost-effective to express and purify, from overexpression in Escherichia coli; it has fast kinetic parameters, with a turnover rate of 106 s−1; it is very soluble, to concentrations of >100 mg/ml; and it has an intermediate melting temperature, TM of ∼58°C (Avvaru et al., 2009). Also, from a rational design and bio-engineering perspective much is known about the structure and catalytic mechanism of this enzyme. However, for industrial applications, small ‘improvements’ in stability, without detriment to yield, activity or solubility can add greatly in the development of HCA II as a better bio-catalyst, as the environment of action may be at an extreme pH and/or elevated temperature. Use of the free enzyme in solution has also many serious drawbacks, such as low stability that limits re-usability, recovery and cost in an industrial setting (Kanbar and Ozdemir, 2010). Hence, several groups have immobilized HCA II on a variety of surfaces as a way to overcome the stability and recovery problems (Vinoba et al., 2011). The enzyme remains very active in its immobilized form, but the enzyme shows significant loss in activity >55°C (Kanbar and Ozdemir, 2010). Having a stable HCA II variant with wild-type kinetic features will be essential for industrial applications—immobilized or in solution—in carbon sequestration and/or biofuel production as it will help limit costs.
The HCA II isozyme is the best-characterized CA to date. It is a monomeric Zn containing metalloenzyme with a molecular weight of ∼29 kDa (Fig. 1a). It is classified as an ultra-fast enzyme with a kcat/KM of 1.5×108 M−1 s−1 and a kcat of 1.4×106 s−1 and among the fastest CA isozyme characterized so far. The catalysis can be described as two half reactions and follows a ping-pong mechanism. In the hydration direction, the first stage starts with the diffusion of CO2 into the active site where it replaces the deep water (DW), next is the nucleophilic attack on CO2 by the zinc-bound OH− to form HCO3−. The HCO3− is then released from the active site leaving an H2O molecule bound to the zinc, termed Zn-water (ZW) (reaction (1)). The second stage is the transfer of a proton from the ZW to a regenerate Zn-OH− for the next round of catalysis, as shown in reaction (2) below, where B is an endogenous H+ acceptor of bulk solvent (Silverman and Lindskog, 1988):
| (1) |
| (2) |
Fig. 1.

Ribbon diagram of HCA II with the active site and mutated surface residues indicated in ball-and-stick. (a) HCA II in blue with the mutated Leu residues in red, the zinc is shown as a magenta sphere with the hydrophilic residues involved with catalysis shown in yellow ball and stick. For panels (b)–(d) wild type and TS2 was superimposed to show the differences, wild type is in blue and TS2 is in yellow ball-and-stick; (b) Leu100His variant and inferred H-bonds (based on distance and angles) are shown as black dashed lines, (c) Leu224Ser, (d) Leu240Pro and waters and H-bonds. This figure was generated and rendered in PyMOL (DeLano, 2002).
The catalytic hydration of CO2 is most affected by changes to Thr199 and Glu106 that are involved in H-bonding to Zn-bound solvent (either H2O or OH−) and mutations altering the hydrophobic CO2 binding pocket (Krebs and Fierke, 1993; Krebs et al., 1993; Nair and Christianson, 1993; Huang et al., 2002; Domsic et al., 2008). The proton-transfer step is however also sensitive to electrostatic changes, especially for the hydrophilic residues that line the active site: Tyr7, Asn62, His64, Asn67 and Thr200 (Fig. 1; Fisher et al., 2007a; Mikulski et al., 2011).
The production of thermostabilized enzymes is still a significant challenge and there are many approaches to this, each with varying success. One popular strategy is to create large libraries of mutants through random mutagenesis and directed evolution while selecting for a specific criteria (Jochens et al., 2010). Other studies have focused on a more rational approach with a small set of targeted changes, like introducing Arg residues as stabilizing elements (Mrabet et al., 1992). Unlike improving substrate-binding or enzyme kinetic properties, protein stability is a function of many variables, from protein folding, core packing, surface electrostatics, to overall rigidity and it appears that these determinants have varying importance in different proteins (Filikov et al., 2002; Permyakov et al., 2005; Strickler et al., 2006). To address these uncertainties computational tools have been developed that can assist with rational thermostability design, and while some of these methods are informative they do not suggest a generalizable strategy that will work for all proteins (Filikov et al., 2002; Potapov et al., 2009). Another successful approach is known as the B-factor iterative test principle method where areas in a protein with high thermal fluctuations are identified from the X-ray crystal structures. These areas are then subjected to iterative rounds of mutagenesis while selecting for thermostability (Reetz et al., 2006). The development of rational design approaches based on specific crystallographic data that inform on surface electrostatics, hydrophobic interactions as well as hydration and H-bonding are appealing because this can lead to the development of guidelines for thermostabilization of all proteins. Owing to the complex nature of protein folding, kinetics and stability, the most effective strategy will likely be a combination of many techniques, both computational and experimental.
To address the industrial need to have more stable CAs that retains desirable kinetic properties, five mutants of HCA II were constructed using site-specific mutagenesis. The three residues changed to investigate stability were selected out of 10 possible mutations discovered using a random mutagenesis approach as described in US patent no. 7521217. The mutants were scored for thermal stability after incubating the mutants at elevated and defined temperatures for 2 h and then measuring residual activity. The results were then expressed as percent residual activity compared with wild-type HCA II. The authors of the patent showed that one mutation at a time had a very modest effect and that they could achieve much higher melting temperatures by combining the mutations. There was no specific order that mattered and that a minimum of three mutations yielded increased percent residual activity after incubation for 2 h at 10°C higher than wild type. After careful inspection of the crystal structure of wild-type HCA II we selected three Leu residues of the 10 mutations for the following reasons: (i) they are all on the surface of HCA II, (ii) they are all hydrophobic Leu residues, (iii) they cluster together in a patch on the surface of the enzyme (Fig. S1) and (iv) they are sufficiently far away from the active site that we were sure not to disturb the pKa of the proton donor/acceptor during catalysis. The other mutations that contributed to increased thermal stability identified in the patent are Ala65Thr, Phe93Leu, Gln136His/Tyr, Lys153Asn, Leu198Met and Ala247Thr. Our three Leu mutations served as the background to which strategic active site mutations were added to create active HCA II variants with improved stability and kinetics in some cases. Surprisingly, some of the mutants displayed improved proton transfer rates compared with wild type while CO2 hydration rates were unaffected. To better understand the biophysical effect of thermostabilizing mutations, X-ray structures of the mutants were solved and enzyme kinetics were determined under a variety of possible industrial environmental conditions. These data show that changing hydrophobic surface residues in HCA II to polar/charged ones can improve stability through an electrostatic mechanism. Simultaneously, it is possible to fine tune some of the enzyme kinetic parameters while creating variants with improved thermal stability.
Experimental
Crystallization and X-ray crystal structure determination
Site-specific mutations of HCA II were made by GenScript. The first mutant, thermostable 1 (TS1) was constructed based on results reported in US patent no. 7521217 (filed by CO2 solutions) and contained the following single amino acid substitutions: Leu100His as well as Leu224Ser and Leu240Pro. This triple mutant then served as the background for TS2–TS5. In addition to the starting triple mutations, TS2 also contained Tyr7Phe, TS3 had Tyr7Phe + Asn62Leu, TS4 had Tyr7Phe + Asn67Gln and TS5 had six mutations with Tyr7Phe + Asn62Leu + Asn67Gln added. The proteins were expressed and purified as described elsewhere (Fisher et al., 2009). After purification the proteins were concentrated and buffer exchanged into 50 mM Tris pH 7.8 using Amicon Ultra concentration devices with a 10 kDa molecular weight cut-off. The proteins were concentrated to 35–50 mg/ml prior to all subsequent experiments and characterizations.
The HCA II variants did not readily crystallize with the usual published conditions of ammonium sulfate or sodium citrate for HCA II (Fisher et al., 2007a,b). Therefore the Gryphon robotic drop-setter from Art Robbins Instruments was used to set up vapor diffusion sitting drops against different commercial screens. Diffraction quality crystals were obtained within a week from Hampton Screen 1, condition #6 (0.2 M magnesium chloride heptahydrate, 0.1 M Tris pH 8.5 and 30% Peg 4000) using a sample concentration of 50 mg/ml. The crystals were flash-cooled by rapidly placing them in cold gas stream with no added cryoprotectant. X-ray diffraction data at 100 K were collected on an RAXIS IV++ using an in-house rotating Cu anode HU-H3R. The frames were collected with 1° oscillation steps and 5 min per exposure. The crystal to detector distance was 80 mm. All crystals diffracted between 1.56 and 2.0 Å resolution. Data processing and reduction were done with either d*TREK or the HKL2000 suite of programs (Otwinowski and Minor, 1997; Pflugrath, 1999). The starting model was derived from protein data bank (PDB) accession number 2ili with all the waters and Zn removed, and with the mutated residues changed to Gly (Fisher et al., 2007b). All the structures were refined using PHENIX and manual inspection and model building was done using Coot (Emsley and Cowtan, 2004; Adams et al., 2010). The models for all of the variants were refined consistently in that there were no deleted or truncated residues and water molecules with a B-factor >40 Å2 were removed. All crystallographic figures were generated using PyMOL (DeLano, 2002). Table I shows a summary of the data set and refinement statistics. Experimental data and structural coordination have been deposited with the Protein Data Bank and have the following accession numbers: TS1 = 3V3F, TS2 = 3V3G, TS3 = 3V3H, TS4 = 3V3I and TS5 = 3V3J.
Table I.
X-ray crystallographic data set and refinement statistics
| TS1 | TS2 | TS3 | TS4* | TS5 | |
|---|---|---|---|---|---|
| PDB accession number | 3V3F | 3V3G | 3V3H | 3V3I | 3V3J |
| Resolution (Å) | 20–2.0 (2.07–2.0) | 20–1.56 (1.60–1.56) | 20–1.85 (1.95–1.85) | 20–1.74 (1.80–1.74) | 20–1.63 (1.69–1.63) |
| Total number of measured reflections | 96743 | 227265 | 101034 | 110298 | 178801 |
| Total number of unique reflections | 14737 | 32937 | 19063 | 20425 | 28381 |
| Rsyma (%) | 12.4 (36.7) | 7.8 (48.8) | 9.6 (47.7) | 9.6 (33.5) | 7.0 (48.9) |
| I/σI | 7.6 (3.5) | 19.9 (3.3) | 13.2 (3.1) | 7.2 (2.9) | 18.8 (3.3) |
| Completeness (%) | 92.9 (90.2) | 99.4 (97.9) | 95.5 (93.7) | 82.7 (99.4) | 99.3 (97.6) |
| Redundancy | 6.7 (6.7) | 6.9 (6.6) | 5.3 (5.3) | 3.0 (3.1) | 6.3 (5.6) |
| Rcrystb (%) | 18.8 | 17.0 | 16.5 | 19.5 | 18.3 |
| Rfreec (%) | 25.6 | 20.0 | 22.4 | 25.3 | 21.9 |
| Residue numbers | 257 | 258 | 258 | 257 | 258 |
| Number of protein atoms (including alternate conformations) | 4031 | 4112 | 3994 | 3942 | 4001 |
| Number of water molecules | 142 | 277 | 220 | 89 | 195 |
| R.m.s.d: bond lengths (Å), angles (°) | 0.012, 1.366 | 0.009, 1.245 | 0.010, 1.263 | 0.011, 1.308 | 0.009, 1.290 |
| Ramachandran statistics (%): most favored, additionally allowed and generously allowed regions | 96.1, 3.9, 0.0 | 97.0, 3.0, 0.0 | 96.9, 3.1, 0.0 | 97.3, 2.7, 0.0 | 96.5, 3.5, 0.0 |
| Average B factors (Å)2: all, main-, side-chain and solvent | 24.9, 22.5, 26.9, 25.5 | 21.0, 17.5, 22.5, 27.0 | 21.6, 19.3, 22.9, 24.6 | 24.2, 21.1, 26.9, 22.9 | 16.8, 14.2, 18.2, 21.7 |
| R.m.s.d. Cα (Å)d | 0.45 | 0.45 | 0.47 | 0.47 | 0.53 |
Values for the highest resolution shell are shown in parenthesis.
aRsym = (∑|I−<I>|/∑ <I>) × 100.
bRcryst = (∑|Fo−Fc|/∑|Fo|) × 100.
cRfree is calculated in the same way as Rcryst except it is for data omitted from refinement (5% of reflections for all data sets).
dr.m.s.d. of the Cα backbone compared with wild-type HCA II (PDB: 3KS3).
*Diffraction sets for TS4 contained ice rings limiting completeness in resolution shells 4.16–3.30, 2.29–2.18 and 1.93–1.87.
18O exchange kinetics
The 18O exchange method is based on mass spectrometric measurements using a membrane inlet of the depletion 18O from CO2 (Silverman, 1982). The isotopic content of CO2 in solution is measured when it passes across a membrane and into an Extrel EXM-200 mass spectrometer. The measured variable is the atom fraction of 18O in CO2. The first step of catalysis has a probability of reversibly labeling the Zn-bound OH− with 18O (reaction (3)). During the next step the 18OH− can be protonated and results in the release of H218O to the bulk solvent where it is essentially infinitely diluted by H216O (reaction (4)). In this process, His64 acts as a proton shuttle (Tu et al., 1989):
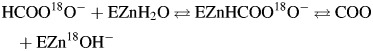 |
(3) |
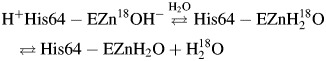 |
(4) |
The 18O-exchange method obtains two different rates at chemical equilibrium (Silverman, 1982): R1, which is the rate of exchange of CO2 and HCO3− (reaction (5)) and 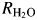 , which is the rate of release of H218O from the enzyme. In reaction (5), kcatex is the rate constant for maximal conversion between substrate and product while KeffS is the effective binding constant of the substrate ([S] is the concentration); [S] can be either CO2 or HCO3− depending on the direction of the reaction. The ratio expressed in reaction (5) of kcatex/KeffS is in principle the same as kcat/KM obtained under steady-state conditions:
, which is the rate of release of H218O from the enzyme. In reaction (5), kcatex is the rate constant for maximal conversion between substrate and product while KeffS is the effective binding constant of the substrate ([S] is the concentration); [S] can be either CO2 or HCO3− depending on the direction of the reaction. The ratio expressed in reaction (5) of kcatex/KeffS is in principle the same as kcat/KM obtained under steady-state conditions:
| (5) |
In the second part of catalysis the rate 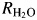 is the part of 18O exchange that is dependent on the rate of proton transfer from His64 to the labeled enzyme-bound OH− (i.e. in the dehydration direction) (Tu et al., 1989). Reaction (6) shows the relationship between kB, the rate constant for proton transfer to Zn-bound OH− and (Ka)donor and
is the part of 18O exchange that is dependent on the rate of proton transfer from His64 to the labeled enzyme-bound OH− (i.e. in the dehydration direction) (Tu et al., 1989). Reaction (6) shows the relationship between kB, the rate constant for proton transfer to Zn-bound OH− and (Ka)donor and 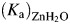 that are the ionization constants for the proton donor and Zn-bound water, respectively:
that are the ionization constants for the proton donor and Zn-bound water, respectively:
| (6) |
Except for the temperature dependence studies, all enzyme kinetic measurements were done at 25°C in the absence of buffer using a total substrate concentration (all species of CO2) of 25 mM. The temperature dependence studies used 10 mM total species of CO2. Kinetic constants and ionization constants shown in reactions (5) and (6) were determined through nonlinear least squares methods (Enzfitter, Biosoft).
Chemical and thermal stability
Enzyme activity was measured while increasing amounts of urea up to 8 M in 1 M increments. The solutions contained urea, 25 mM of all species of CO2 and 100 mM 4-(2-hydroxyehtyl)-1-piper-azineethane sulfonic acid (HEPES) at pH 7.6 and 25°C. An enzyme was added to the reaction vessel and catalytic 18O exchange activity was measured as R1/[E] (reaction (5)) over a period of up to 5min. Enzyme activity as R1/[E] was also measured at temperatures from 10 to 70°C in the similar solutions (100 mM HEPES and 10 mM total substrate) at pH 7.6. After the reaction solution was equilibrated to each different temperature, a small sample of enzyme (at 0.1–0.2% of reaction volume) was added. Measurements of 18O content of CO2 were made over the following 1–5min.
Differential scanning calorimetry and circular dichroism
Differential scanning calorimetry (DSC) experiments were performed using a VP-DSC calorimeter (Microcal, Inc., North Hampton, MA, USA) with a cell volume of ∼0.5 ml. All wild type, Tyr7Phe and TS mutant HCA II samples were buffered in 50 mM Tris-HCl, pH 7.8, at protein concentrations of ∼30 μM. The samples were degassed while stirring for 1 h before data collection. The DSC scans were collected from 20 to 100°C with a scan rate of 60°C/h. The calorimetric enthalpies of unfolding were calculated by integrating the area under the peaks in the thermograms after adjusting the pre- and post-transition baselines. The thermograms were fit to a two-state reversible unfolding model to obtain van't Hoff enthalpies of unfolding. The melting temperature (TM) values of the HCA II samples were obtained from the midpoints on the DSC curves, indicating a two-state transition. All samples were measured in triplicate with a buffer baseline subtracted. The results are shown in Table II.
Table II.
Differential scanning calorimetry measurements of the major unfolding transition (TM in °C) of each TS mutant compared with wild-type HCA II and the Y7F mutant
| Enzyme | TM (°C) | Tinact (°C) |
|---|---|---|
| Wild-type HCA II | 58 (±1) | 60 |
| Y7F | 53 (±1) | ND |
| Y7F + N67Q | 56.9 (±0.3) | ND |
| TS1 (L100H, L224S, L240P) | 65 (±2) | 65 |
| TS2 (TS1 + Y7F) | 61.1 (±0.5) | 65 |
| TS3 (TS1 + Y7F + N62L) | 58.1 (±0.3) | 60 |
| TS4 (TS1 + Y7F + N67Q) | 62.7 (±0.5) | 65 |
| TS5 (TS1 + Y7F + N62L + N67Q) | 59.2 (±0.4) | 60 |
Scans for each HCA II variant were repeated at least three times and standard deviations are shown in parentheses. Tinact is an estimate of the thermal inactivation temperature of catalysis measured by 18O exchange methods.
ND, not determined.
Circular dichroism (CD) scans of all HCA II samples were obtained using an AVIV spectropolarimeter in the far UV range from 200 to 260 nm. All the samples were diluted in 50 mM Tris pH 7.8 to 0.1–0.2 mg/ml concentration and loaded into a 1-mm quartz cuvette. Ellipticity was measured while the temperature was increased from 28 to 70°C. Sample scans were performed in duplicate and averaged and baseline buffer scans were subtracted (data not shown).
Results and discussion
X-ray crystallography and structural analysis
All variants crystallized in the orthorhombic P212121 space group and were highly isomorphous with approximate unit cell dimensions a = 42, b = 72 and c = 75 Å and diffracted between 1.56 and 2.0 Å resolution. All models refined to Rcryst and Rfree between ∼16–19 and 20–25%, respectively. Table I shows a summary of all data set and refinement statistics.
Figure 1a shows the active site relative to the location of a hydrophobic cluster of surface Leucine residues that were mutated to confer stability. Leu100, Leu224 and Leu240 are all between 8 and 14 Å from each other (Cα–Cα distance) and form a small hydrophobic patch on the surface of HCA II (Fig. 1a). This patch is at least 20 Å away from the zinc active site. The mutated residues are located within the interface of two crystallographic symmetry-related chains that might contribute to different crystal packing compared with wild-type HCA II. Based on a comparison of the mutant and wild-type X-ray crystal structures it is clear that changing these hydrophobic residues to polar residues results in increased stability through differences in enthalpic contributions with the gain of H-bonds and favorable electrostatics (Fig. 1b–d). This decrease in surface hydrophobicity likely contributes to the increased solubility and different crystallization conditions required to crystallize the mutants.
Leu100His accommodates weak H-bonds (3.0–3.4 Å) with the backbone amide of Gly102 and the side chain of Gln103 (Fig. 1b). The average B-factors of all atoms in the loop consisting of residue 97–104 is ∼15 Å2 and are similar compared with wild-type structures also determined at 100 K. This supports the notion that a gain in H-bonding at position 100, and not thermal movement, dominates the observed increased stability. The presence of a small hydrophobic residue at this position is highly conserved in several homologous human CAs.
Leu224Ser is observed in a dual conformation making H-bond contacts to either backbone amides of residues Ser220 or Glu221, as well as to a water molecule that is also coordinated to the guanido group of Arg227 (Fig. 1c). A comparison of the average B-factors for all atoms from residue 223–225 is interesting: for wild type it is ∼12 Å2 and 19 and 26 Å2 for TS2 and TS4, respectively. This implies an increased thermal fluctuation in the mutant compared with wild type that is also reflected by the disorder of Ser224. Leu at this position is completely conserved in several other human CAs such as isoforms IV and XII, but interestingly is a Ser in the mitochondrial form, CA V.
Leu240Pro creates a solvent accessible, hydrophilic pocket that allows for the ordering of two water molecules to make H-bonds with the backbone carbonyls of residues Lys228 and Leu229. The introduction of a Pro at position 240, which sits at the end of a surface loop, may be expected to cause a reduction in the loop flexibility. However, similar to the mutations introduced at positions 100 and 224, changing Leu 240 to Pro appears to increase the average B-factor of the loop from ∼ 12 to 35 Å2 (Fig. 1d). Having a Leu at position 240 does not seem conserved among different human isoform, but there are Pro residues in three human CAs: I, III and XII. This is interesting as the phi/psi space that Pro can occupy is narrowly defined as between ∼ −60° and either ∼ 30° or +135°. These angles are virtually identical in the wild type and TS1–5 mutants.
Altered loop conformations at residues Val37–Ser50, Phe70–Lys80 and Lys225–Leu240 are observed in these mutants relative to the wild-type structure. The Cα backbone trace of the Phe70–Lys80 loop is displaced by up to 3.3 Å compared with wild type with the most dramatic effect arising from the movement of Lys76 that now makes an H-bond with Asp71 with Gln74 (Fig. S2). These alternative loop conformations have been observed with HCA II:inhibitor structures solved in the orthorhombic space group as well as for other HCA II structures with mutations in or near the opening of the active site (Ippolito et al., 1995; Lloyd et al., 2005). These displaced surface loops are most likely a direct consequence of the crystal packing forces.
As a result of the crystallization conditions used for the TS mutants of HCA II, a single Cl− is seen along the surface within H-bonding distance of the amide groups of Gln158 and Lys225 for TS1–4. In the TS5 structure Lys225 is not in position to H-bond with Cl−, which allows Glu158 to be present at the Cl− binding site. As a result, a water molecule is seen in this position instead of the Cl− ion. It is not obvious why Lys225 occupies this unique conformation in TS5 compared with TS1–4. In addition, previous studies of HCA II in an orthorhombic space group report a Zn coordinated to His4 located near the opening of the active site cavity (Lloyd et al., 2005; PDB: 2X7S). Interestingly, in our TS2 structure (PDB: 3V3G), there is similar density observed in this region. However, the Nɛ group of residue His3 is coordinating to the observed density along with possible interactions from His64, the symmetry-related His36 residue and a water molecule. Attempts at placing Zn in this density resulted in increased B-factors (> 80 Å2) in addition to appearance of negative density in the Fo–Fc map. Owing to the uncertainty at this position, a water molecule was built and refined (B-factor < 20 Å2). Nevertheless, it is worth noting that the possible Zn coordination site at the N-terminus is very similar to the canonical Zn–His arrangement found in the active site.
The active site of wild type and each of the variants is shown in Fig. 2. Wild-type HCA II is shown for comparison and TS mutants are as labeled. As expected, there are no significant active site differences between wild type and TS1 (Fig. 2, top left and right). The waters and positions of the residues are essentially conserved, except that His64 in TS1 is observed in the ‘outward’ conformation compared with wild-type HCA II, where an ‘in’ and ‘out’ conformation is observed (Nair and Christianson, 1991; Fisher et al., 2005). In TS2 (Fig. 2, middle left) where Tyr7 is replaced with a Phe, the solvent W3a is displaced as compared with wild-type HCA II and can no longer participate in the H-bonding network in the active site. This is consistent with the results published previously (Fisher et al., 2007a,b). The rest of the waters and residues are the same as in wild type, except that W2 and H64 now appear to engage in an H-bond. In TS3 (Fig. 2, middle right) the solvent W3b has moved but is still H-bonded to Asn67 and W2. Similarly to TS2, the introduction of a hydrophobic residue causes displacement of active site water molecules. In TS4 (Fig. 2, bottom left) Gln67 mostly maintains a similar H-bond to W3b while in TS5 it is somewhat disordered and observed in two conformations. This displaces W3b completely and one conformer of Gln67 is observed to directly participate in a weak H-bond (O…O distance 3.4 Å) to W2. These results are expected based on previous structure–function studies of HCA II and indicate that the presence of the surface Leu mutations do not affect the active site structure in the TS mutants (Fisher et al., 2007a,b; Mikulski et al., 2011).
Fig. 2.

Ball-and-stick diagrams of wild type and stabilized variants of HCA II active sites. The Zn is shown as a magenta sphere and all the hydrophilic active site residues (and their counterparts in the mutants) are shown as sticks, waters are shown as red spheres. Residues and waters are as labeled and inferred H-bonds are shown as black dashed lines. Maps are omitted for clarity. Figures were generated and rendered with PyMOL (DeLano, 2002).
Catalytic activity
The pH profiles were determined for R1, the rate of catalyzed interconversion of CO2 and bicarbonate (reaction (5); Fig. S3) and 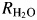 , the rate of dissociation of H218O from the active site (reaction (6); Fig. S4), using the 18O-exchange method. The background mutations (Leu100His, Leu224Ser and Leu240Pro) did not significantly affect the rate of CO2 hydration reflected through kcatex/KeffS (Table III) or R1/[E] (Figs S3 and S4, Supporting Information). Moreover, the replacements at positions 7, 62 and 67 also caused no significant changes in these measures of the first stage of catalysis (reaction (3)). This result was expected since the surface Leu mutations and the amino acid replacements in TS1–5 are sufficiently far from the catalytic Zn to avoid structural and electrostatic disruptions of the reaction of Zn-bound OH− with substrate.
, the rate of dissociation of H218O from the active site (reaction (6); Fig. S4), using the 18O-exchange method. The background mutations (Leu100His, Leu224Ser and Leu240Pro) did not significantly affect the rate of CO2 hydration reflected through kcatex/KeffS (Table III) or R1/[E] (Figs S3 and S4, Supporting Information). Moreover, the replacements at positions 7, 62 and 67 also caused no significant changes in these measures of the first stage of catalysis (reaction (3)). This result was expected since the surface Leu mutations and the amino acid replacements in TS1–5 are sufficiently far from the catalytic Zn to avoid structural and electrostatic disruptions of the reaction of Zn-bound OH− with substrate.
Table III.
Maximal, pH independent values of the kinetic constants for catalysis by variants of HCA II measured by 18O exchange
| Enzyme | kcatex/KeffSa (μM−1 s−1) | kBa (μs−1) |
|---|---|---|
| Wild type | 120 | 0.8 |
| Y7Fb | 120 | 3.9 |
| TS1 | 85 | 1.3 |
| TS2 | 110 | 5.6 |
| TS3 | 88 | ∼0.1 |
| TS4 | 94 | 4.9 |
| TS5 | 110 | ∼0.1 |
aThe standard errors in these rate constants determined by fitting reactions (5) and (6) to the data of Figs S2 and S3 are in the range of 10–20%.
bFrom Fisher et al. (2007a,b). Data at 10°C.
However, there are interesting differences in the rate constants 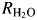 /[E] (Fig. S4) and the rate constant for proton transfer in catalysis kB (Table III). The rate constant kB measures in large part the proton transfer along an ordered water structure between His64 and the Zn-bound OH− in the dehydration direction and is determined from the bell-shaped pH profiles such as observed with wild-type HCA II (Fig. S4, Silverman, 1982; Tu et al., 1989; Silverman and McKenna, 2007). These results show that the background replacements of surface Leu residues in TS1 do not negatively affect R1 or kB compared with wild type (Table III), consistent with their location far from the active site and proton transfer pathway (Silverman and McKenna, 2007). However, combining the background mutations in TS1 with specific active site changes at positions 7 and 67 caused an unexpected, albeit modest, boost in proton transfer activity (bold numbers, Table III). Overall however, the proton transfer efficiency of the remaining variants can be understood in terms of the results for the corresponding variants with single amino acid replacements. The value of kB for Tyr7Phe HCA II is increased ∼5-fold compared with wild type (Table III, Fisher et al., 2007a,b); accordingly, it was expected that the variant TS2 has a higher value of kB compared with wild type. Based on our detailed analysis in a previous study on similar active site mutants, we concluded that the displacement of W3a and the loss of the hydroxyl group at position 7 led to changes in pKa for proton donor/acceptor groups. In addition to the electrostatic and related pKa changes that occurred also produced a shorter, unbranched chain of hydrogen-bonded waters that connect ZW to the proton shuttling residues His64. These electrostatic changes and unbranched water network boost the proton transfer activity of HCA II Tyr7Phe mutants (Fisher et al., 2007b). As reported in Table III, the values of kB for TS2 and TS4 are even better than for Tyr7Phe alone at 5.6 and 4.9, respectively. The value of kB for Asn67Gln HCA II is increased ∼2-fold compared with wild type (Mikulski et al., unpublished), and for Asn62Leu HCA II the value of kB is decreased 8-fold. These factors then influence the values of kB shown in Table III; for example, the two variants (TS3 and TS5) containing Asn62Leu have low values of
/[E] (Fig. S4) and the rate constant for proton transfer in catalysis kB (Table III). The rate constant kB measures in large part the proton transfer along an ordered water structure between His64 and the Zn-bound OH− in the dehydration direction and is determined from the bell-shaped pH profiles such as observed with wild-type HCA II (Fig. S4, Silverman, 1982; Tu et al., 1989; Silverman and McKenna, 2007). These results show that the background replacements of surface Leu residues in TS1 do not negatively affect R1 or kB compared with wild type (Table III), consistent with their location far from the active site and proton transfer pathway (Silverman and McKenna, 2007). However, combining the background mutations in TS1 with specific active site changes at positions 7 and 67 caused an unexpected, albeit modest, boost in proton transfer activity (bold numbers, Table III). Overall however, the proton transfer efficiency of the remaining variants can be understood in terms of the results for the corresponding variants with single amino acid replacements. The value of kB for Tyr7Phe HCA II is increased ∼5-fold compared with wild type (Table III, Fisher et al., 2007a,b); accordingly, it was expected that the variant TS2 has a higher value of kB compared with wild type. Based on our detailed analysis in a previous study on similar active site mutants, we concluded that the displacement of W3a and the loss of the hydroxyl group at position 7 led to changes in pKa for proton donor/acceptor groups. In addition to the electrostatic and related pKa changes that occurred also produced a shorter, unbranched chain of hydrogen-bonded waters that connect ZW to the proton shuttling residues His64. These electrostatic changes and unbranched water network boost the proton transfer activity of HCA II Tyr7Phe mutants (Fisher et al., 2007b). As reported in Table III, the values of kB for TS2 and TS4 are even better than for Tyr7Phe alone at 5.6 and 4.9, respectively. The value of kB for Asn67Gln HCA II is increased ∼2-fold compared with wild type (Mikulski et al., unpublished), and for Asn62Leu HCA II the value of kB is decreased 8-fold. These factors then influence the values of kB shown in Table III; for example, the two variants (TS3 and TS5) containing Asn62Leu have low values of 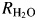 /[E]. It is important to point out that from an industrial and applications point of view, it is the value of kcatex/KeffS that is significant for low concentrations of CO2, say <10 mM. The values of kB measuring proton transfer come into significance when HCA II is under maximal velocity conditions for concentrations of CO2 >10 mM.
/[E]. It is important to point out that from an industrial and applications point of view, it is the value of kcatex/KeffS that is significant for low concentrations of CO2, say <10 mM. The values of kB measuring proton transfer come into significance when HCA II is under maximal velocity conditions for concentrations of CO2 >10 mM.
Stability
To test the stability of the variants against denaturation through thermal and chemical means, a series of experiments was carried out by DSC and by measuring the rate of catalyzed CO2/HCO3− interconversion. First, differential scanning calorimetry scans were measured for wild type, Tyr7Phe mutant and each of the TS HCA II variants. The melting temperatures or major unfolding transitions (TM) for each of the variants occurred at distinct peaks in the thermograms. The average peak values (with standard deviations shown in parentheses) from at least three runs are given in Table II. The CD data were consistent with the DSC data for each variant but showed higher temperatures of unfolding (∼2°C), probably reflecting the retention of secondary structure elements after initial melting (data not shown). Wild-type HCA II had a TM of ∼58°C under these experimental conditions and introducing the three background mutations present in TS1 (Leu100His, Leu224Ser and Leu240Pro) increased the TM to 65°C (Table II). Introducing the Tyr7Phe mutation to increase proton transfer activity had a destabilizing effect, reducing the TM to 53°C (Fisher et al., 2007a,b; Mikulski et al., 2011) while adding the background mutations ‘rescues’ the stability by increasing it to ∼61°C for TS2. Addition of Asn67Glu to an active site containing Tyr7Phe stabilizes HCA II almost back to wild-type levels while also displaying high catalytic efficiency (Tables II and III). The remaining variants of Table II contain different active site mutations and have varying effects on stability. Among all of the variants of Table II, TS1, TS2 and TS4 have values of TM that are significantly higher than wild-type HCA II. This is very encouraging as these variants have not only different stabilities but also somewhat different kinetic profiles (Fig. S4) compared with wild type that make them interesting from an industrial point of view (Fisher et al., 2007a,b; Zheng et al., 2008; Mikulski et al., 2011).
The rate constant R1/[E] was determined while increasing temperature from 10 to 70̊C (Fig. 3). These are rough estimates of the thermal inactivation temperature with measurements made in intervals of 5°C at the higher temperatures the purpose of which was to determine whether there was inactivation of catalysis at a temperature less than the major unfolding determined by DSC. What is important here is that there appears no inactivation of catalysis at temperatures less than the TM determined by DSC (Fig. 3), and that the more accurate major unfolding transition determined as TM by DSC most likely also measures the thermal stability of catalysis.
Fig. 3.
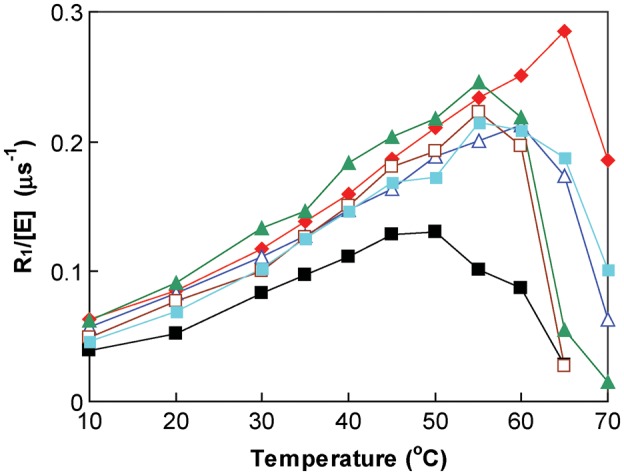
The rate constant R1/[E] (s−1) as a function of temperature for the interconversion of CO2 and bicarbonate catalyzed by variants of HCA II. Wild type = black filled square; TS1 = red filled rhombus; TS2 = dark blue triangle; TS3 = brown square; TS4 = light blue filled square and TS5 = green filled triangle. Data have been separated on the ordinate to avoid superposition of points. Solutions contained 10 mM of all species of CO2 and 100 mM HEPES at pH 7.6.
The chemical stabilities of the mutants were compared with wild-type HCA II increasing urea concentration up to 8 M in 1 M increments while measuring the kinetic constant R1/[E] (Fig. S5). The Leu surface mutations had no significant effect on enzyme ability to withstand denaturation by urea under these conditions. The addition of 4 M urea led to <10% relative activity compared to no urea for all variants, including wild type. This observation can be partly explained by urea denaturation thought to occur through unfolding of the hydrophobic core of the protein. It has also been pointed out that urea can interact with the protein through electrostatic, van der Waals interactions, and indirectly through the disruption of water structure (Samiotakis et al., 2010; Wang et al., 2011). Since the mutations reported here were all on the surface and active site of the protein, that is not in the core, the denaturation resulted in similar effects in the wild-type and mutant enzymes. Previous studies aimed at the relationship between chemical and thermal denaturation have also revealed discrepancies similar to the ones we have observed here for apoazurin, cytochrome c and apoflavodoxin (Wang et al., 2011). Our metric for chemical denaturation involved activity assays only and not DSC. These techniques measure different properties of the protein and it is possible that HCA II is still fairly well folded up to 4 M urea but that the assay conditions have been compromised. CO2 hydration and subsequent proton transfer is strongly dependent on the water structure in the active site and this can easily be disrupted by excess urea (Silverman and McKenna, 2007). The relationship between thermal and chemical denaturation is complex and appears to be unique to different proteins, it is prudent to determine these values empirically for each system under study.
From a structural perspective the rationale for how these mutations confer stability is not intuitive. In contrast to the B FIT approach, an increase in the thermal fluctuation of residues at positions 224 and 240 is observed with a concomitant increase in thermal stability (Reetz et al., 2006). This probably reflects the dominant effect of the gain in H-bonding and hydrophilicity over flexibility on the surface of HCA II. The underlying principle of thermal stability as a change in surface electrostatics reported here for HCA II is consistent with several other successful studies on diverse enzymes such as ubiquitin, acylphosphatase and α-lactalbumin (Permyakov et al., 2005; Strickler et al., 2006; Jochens et al., 2010).
Conclusions
We have shown that it is possible to enhance the thermal stability of HCA II by strategic replacement of amino acids on the surface of the enzyme. Moreover, these replacements had no significant effect on the active site structure and no effect on the catalytic rate of CO2 hydration and HCO3− dehydration. Single amino acid replacements that were previously found to enhance catalysis were also effective in enhancing catalysis in variants with these surface changes. The net result was a variant of HCA II (TS4 of Table II) with thermal stability enhanced by ∼6°C and with maximal proton transfer enhanced ∼6-fold compared with wild-type HCA II. Further analysis of the surface of HCA II shows that there are other areas to target using this approach. Phe20 and Leu57 are located on the surface also and could be modeled to engage in H-bonds with surrounding residues. Leu204 and Val135 are very close together on the surface and could be mutated so that changes at these positions make a salt bridge or H-bond to each other. The initial results reported here shed light on the underlying biophysical principle, that is removing surface hydrophobic residues and replacing them with polar or hydrophilic residues leads to a gain in H-bonding interaction and this results in increased thermal stability.
Supplementary data
Funding
This work was supported in part by grants from Los Alamos National Laboratory LDRD Early Career #20110535ER (to ZF) and National Institutes of Health GM25154 (to DNS and RM).
Supplementary Material
Acknowledgements
The authors would like to acknowledge Mary Jo Waltman and David Fox for assistance and useful discussions.
References
- Adams P.D., Afonine P.V., Bunkoczi G., et al. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvaru B., Busby S.A., Chalmers M.J., Palmer P.R., Venkatakrishnan B., Agbandje-McKenna M., Silverman D.N., McKenna R. Biochemistry. 2009;48:7365–7372. doi: 10.1021/bi9007512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W.L. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. http://www.pymol.org . [Google Scholar]
- Domsic J.F., Avvaru B.S., Kim C.U., Gruner S.M., Agbandje-McKenna M., Silverman D.N., McKenna R. J. Biol. Chem. 2008;283:30766–30771. doi: 10.1074/jbc.M805353200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Filikov A.V., Hayes R.J., Luo P., Stark D.M., Chan C., Kundu A., Dahiyat B.I. Protein Sci. 2002;11:1452–1461. doi: 10.1110/ps.3500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S.Z., Hernandez-Prada J., Tu C.K., Duda D., Yoshioka C., An H., Govindasamy L., Silverman D.N., McKenna R. Biochemistry. 2005;44:1097–1105. doi: 10.1021/bi0480279. [DOI] [PubMed] [Google Scholar]
- Fisher S.Z., Kovalevky A.Y., Domsic J., Mustyakimov M., Silverman D.N., McKenna R., Langan P. Acta Crystallogr. 2009;F65:495–498. doi: 10.1107/S1744309109013086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S.Z., Tu C.K., Bhatt D., Govindasamy L., Agbandje-McKenna M., McKenna R., Silverman D.N. Biochemistry. 2007a;46:3803–3813. doi: 10.1021/bi602620k. [DOI] [PubMed] [Google Scholar]
- Fisher S.Z., Maupin C.M., Budayova-Spano M., Govindasamy L., Tu C., Agbandje-Mckenna M., Silverman D.N., Voth A., McKenna R. Biochemistry. 2007b;46:2930–2937. doi: 10.1021/bi062066y. [DOI] [PubMed] [Google Scholar]
- Fulke A.B., Mudliar S.N., Yadav R., et al. Bioresour. Technol. 2010;101:8473–8476. doi: 10.1016/j.biortech.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Huang S., Sjoblom B., Sauer-Eriksson E., Jonsson B.-H. Biochemistry. 2002;41:7628–7635. doi: 10.1021/bi020053o. [DOI] [PubMed] [Google Scholar]
- Ippolito J.A., Nair S.K., Alexander R.S., Kiefer L.L., Fierke C.A., Christianson D.W. Protein Eng. 1995;8:975–980. doi: 10.1093/protein/8.10.975. [DOI] [PubMed] [Google Scholar]
- Jochens H., Aerts D., Bornscheuer U.T. Protein Eng. Des. Sel. 2010;23:903–909. doi: 10.1093/protein/gzq071. [DOI] [PubMed] [Google Scholar]
- Kanbar B., Ozdemir E. Biotechnol. Prog. 2010;26:1474–1480. doi: 10.1002/btpr.452. [DOI] [PubMed] [Google Scholar]
- Krebs J.F., Fierke C.A. J. Biol. Chem. 1993;268:948–954. [PubMed] [Google Scholar]
- Krebs J.F., Ippolito J.A., Christianson D.W., Fierke C.A. J. Biol. Chem. 1993;268:27458–27466. [PubMed] [Google Scholar]
- Lloyd M.D., Pederick R.L., Natesh R., Woo L.W.L., Purohit A.P., Reed M.J., Acharya K.R., Potter B.V.L. J. Med. Chem. 2005;385:715–720. doi: 10.1042/BJ20041037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikulski R., Avvaru B.S., Tu C.K., Case N., McKenna R., Silverman D.N. Arch. Biochem. Biophys. 2011;506:181–187. doi: 10.1016/j.abb.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrabet N.T., Van den Broeck A., Van den brande I., et al. Biochemistry. 1992;31:2239–2253. doi: 10.1021/bi00123a005. [DOI] [PubMed] [Google Scholar]
- Nair S.K., Christianson D.W. J. Am. Chem. Soc. 1991;113:9455–9458. [Google Scholar]
- Nair S.K., Christianson D.W. Biochemistry. 1993;32:4506–4514. doi: 10.1021/bi00068a005. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Permyakov S.E., Makhatadze G.I., Owenius R., Uversky V.N., Brooks C.L., Permyakov E.A., Berliner L.J. Protein Eng. Des. Sel. 2005;18:425–433. doi: 10.1093/protein/gzi051. [DOI] [PubMed] [Google Scholar]
- Pflugrath J. Acta Crystallogr. 1999;D55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- Potapov V., Cohen M., Schreiber G. Protein Eng. Des. Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- Ramanan R., Kannan K., Deshkar A., Yadav R., Chakrabarti T. Bioresour. Technol. 2010;101:2616–2622. doi: 10.1016/j.biortech.2009.10.061. [DOI] [PubMed] [Google Scholar]
- Reetz M.T., Carballeira J.D., Vogel A. Angew. Chem. 2006;118:7909–7915. [Google Scholar]
- Samiotakis A., Homouz D., Cheung M.S. J. Chem. Phys. 2010;132:175101. doi: 10.1063/1.3404401. [DOI] [PubMed] [Google Scholar]
- Saville C., Lalonde J.J. Curr. Opin. Biotech. 2011;22:1–6. [Google Scholar]
- Strickler S.S., Gribenko A.V., Gribenko A.V., Keiffer T.R., Tomlinson J., Reihle T., Loladze V.V., Makhatadze G.I. Biochemistry. 2006;45:2761–2766. doi: 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]
- Silverman D.N. Methods Enzymol. 1982;87:732–752. doi: 10.1016/s0076-6879(82)87037-7. [DOI] [PubMed] [Google Scholar]
- Silverman D.N., Lindskog S. Acc. Chem. Res. 1988;21:30–36. [Google Scholar]
- Silverman D.N., McKenna R. Acc. Chem. Res. 2007;40:669–675. doi: 10.1021/ar7000588. [DOI] [PubMed] [Google Scholar]
- Tu C.K., Silverman D.N., Forsman C., Jonsson B.H., Lindskog S. Biochemistry. 1989;28:7913–7918. doi: 10.1021/bi00445a054. [DOI] [PubMed] [Google Scholar]
- Vinoba M., Lim K.S., Lee S.H., Jeong S.K., Alagar M. Langmuir. 2011;27:6227–6234. doi: 10.1021/la105029h. [DOI] [PubMed] [Google Scholar]
- Wang Q., Christianson A., Samiotakis A., Wittung-Stafshede P., Cheung M.S. J. Chem. Phys. 2011;135:175102. doi: 10.1063/1.3656692. [DOI] [PubMed] [Google Scholar]
- Zheng J., Avvaru B.S., Tu C.K., McKenna R., Silverman D.N. Biochemistry. 2008;47:12028–12036. doi: 10.1021/bi801473w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.