

Abstract
BACKGROUND:
Hepatitis C virus (HCV) infection may induce insulin resistance (IR) irrespective of the severity of liver disease, and there is evidence of a central role for IR in failure to achieve sustained virological response (SVR) in HCV patients.
OBJECTIVE:
To assess IR as a predictor of the severity of hepatic fibrosis in Egyptian HCV patients, and its effect on early viral kinetics and virological response to HCV therapy.
METHODS:
A total of 140 chronic HCV patients were divided into two groups according to the homeostasis model assessment-IR (HOMA-IR). Group 1 consisted of 48 chronic HCV patients with HOMA-IR ≥2, and group 2 consisted of 92 chronic HVC patients without IR (HOMA IR <2). All patients were treated with combination therapy (pegylated interferon-alpha 2a plus ribavirin) for 48 weeks and studied for viral kinetics throughout the period of therapy.
RESULTS:
The study revealed that older age, higher body mass index and HOMA-IR≥2 were significantly associated with advanced fibrosis. Rapid virological response, complete early virological response and SVR were significantly lower in the IR-HCV group compared with the non-IR-HCV group. Univariate and multivariate analyses revealed that older age, fibrosis (F≥3), high viral load (>600,000 IU/mL) and HOMA-IR ≥2 were significantly associated with a lack of viral kinetics as well as SVR. However, HOMA-IR ≥2 was the main independent variable associated with lack of SVR. On the other hand, body mass index, plasma insulin level and HOMA-IR decreased significantly compared with starting levels in patients who achieved SVR. This suggests a cause and effect relationship between HCV infection and IR.
CONCLUSION:
IR in chronic HCV patients is associated with progressive fibrosis and slow viral kinetics, and could be a predictor for lack of rapid and early virological response. Therefore, HOMA-IR levels should be measured and improved before starting antiviral treatment.
Keywords: HCV, HCV therapy response, HOMA-IR, Insulin resistance, Liver fibrosis
Abstract
HISTORIQUE :
L’infection par le virus de l’hépatite C (VHC) peut être induite par l’insulinorésistance (IR), quelle que soit la gravité de la maladie hépatique, et des données probantes laissent supposer le rôle central de l’IR dans l’incapacité à obtenir une réponse virologique soutenue (RVS) chez les patients atteints du VHC.
OBJECTIF :
Évaluer l’IR comme prédicteur de la gravité de la fibrose hépatique chez des patients égyptiens atteints du VHC, ainsi que son effet sur la cinétique virale précoce et la réponse virologique à la thérapie du VHC.
MÉTHODOLOGIE :
Au total, 140 patients atteints du VHC chronique ont été répartis en deux groupes d’après l’évaluation du modèle d’homéostase-IR (ÉMHO-IR). Le groupe 1 se composait de 48 patients atteints du VHC chronique dont l’ÉMHO-IR était supérieure à 2, et le groupe 2, de 92 patients atteints du VHC chronique sans IR (ÉMHO-IR inférieure à 2). Tous les patients ont été traités par polythérapie (interféron-gamma 2a pégylé associé à de la ribavirine) pendant 48 semaines et ont fait l’objet d’une étude de cinétique virale tout au long de la période de thérapie.
RÉSULTATS :
L’étude a révélé qu’un âge plus avancé, un indice de masse corporelle plus élevé et une ÉMHO-IR supérieure à 2 s’associaient considérablement à une fibrose avancée. La réponse virologique rapide, la réponse virologique précoce complète et la RVS étaient considérablement plus faibles dans le groupe d’IR-VHC que dans celui sans IR. Les analyses univariée et multivariée ont révélé qu’un âge plus avancé, une fibrose F≥3, une charge virale élevée (supérieure à 600 000 UI/mL) et une ÉMHO-IR supérieure à 2 s’associaient considérablement à un manque de cinétique virale et à une RVS. Cependant, l’ÉMHO-IR supérieure à 2 était la principale variable indépendante associée à un manque de RVS. Par contre, l’indice de masse corporelle, le taux d’insuline plasmatique et l’ÉMHO-IR diminuaient considérablement par rapport aux taux de départ chez les patients qui parvenaient à une RVS. Ce phénomène laisse croire à une relation de cause à effet entre l’infection par le VHC et l’IR.
CONCLUSION :
L’IR chez les patients atteints du VHC chronique s’associe à une fibrose évolutive et à une cinétique virale lente. Elle pourrait prédire une absence de réponse virologique rapide et précoce. Par conséquent, il faudrait mesurer les taux de l’ÉMHO-IR et les améliorer avant d’amorcer l’antivirothérapie.
Hepatitis C virus (HCV) infection is a common problem worldwide and affects millions of people. Most infected patients develop chronic hepatitis, and as many as one in five will develop cirrhosis and its complications (1) in addition to a wide spectrum of extrahepatic manifestations (2).
HCV may induce insulin resistance (IR) regardless of the severity of liver disease, and IR may be associated with severe hepatic fibrosis and contribute to fibrotic progression in chronic HCV infection (3,4). The association between type 2 diabetes mellitus (DM) and HCV infection has been reported (5), and is associated with a greater risk for the development of hepatocellular carcinoma (HCC), providing additional evidence that DM plays an important role in the progression of liver disease and development of complications in this population (6).
Glucose abnormalities and alteration of beta-cell function have an important role in the development of IR (7), although the specific mechanisms involved in the pathogenesis of IR associated with HCV infection remain to be elucidated (8). High levels of proinflammatory cytokines (9) and impairment of insulin receptor substrate-1 (IRS-1) and IRS-2 expression have been observed in patients with chronic hepatitis C. Moreover, HCV mediates dysfunction in the insulin signalling pathways by upregulating the expression of suppressors of cytokine signalling 3 expression (10) and increased tumour necrosis factor-alpha secretion (8). In addition to interleukin (IL)-6 and free fatty acids, cellular stress can induce IR by activating serine phosphorylation of IRS-1, thereby inhibiting its function (11).
Combination therapy with peginterferon-alpha (PEG-IFN-α 2a)/ribavirin has been recommended as standard therapy for patients with HCV infection and has shown favourable efficacy (12,13). Although it is known that some pretreatment variables, such as age, viral load, level of fibrosis and IR, affect sustained virological response (SVR) in HCV-infected patients, little information is available concerning which factors interfere with the early phase of viral dynamics in these patients (14). The aim of the present study was to assess IR as a predictor of the severity of hepatic fibrosis in Egyptian HCV patients, as well as its effect on early viral kinetics and early virological response (EVR) to HCV therapy.
METHODS
Patients
Ethics approval from the Tanta University Ethics Committee and the Faculty of Medicine (Tanta, Egypt) was obtained before commencement of the present study and written informed consent was obtained from the study participants.
Patients included in the present study were nonobese (body mass index [BMI] <30 kg/m2) chronic hepatitis C patients 18 to 60 years of age with no evidence of renal function impairment, anemia or significant reduction of their neutrophil or platelet blood counts (>2×109/L to 10.8×109/L, and 100×109/L, respectively), and fasting blood glucose level >6.7 mmol/L. Chronic hepatitis C was diagnosed by positivity for anti-HCV (third-generation enzyme immunoassay), HCV-RNA levels >1000 IU/mL, a liver biopsy showing chronic hepatitis and/or increased serum alanine aminotransferase (ALT) levels.
Patients were excluded if they had decompensated liver disease, hepatitis B virus infection, HIV infection, autoimmune disorders, clinically significant cardiac or cardiovascular abnormalities, clinically significant bleeding disorders, evidence of malignant neoplastic diseases, concomitant immunosuppressive medication, significant retinal disease or active seizures, a fasting blood glucose level >120 mg/L or antidiabetic treatment, or any alcohol intake or drug abuse within six months before study entry.
Clinical and laboratory examination
Age, sex and BMI (kg/m2) were recorded for all patients at baseline. Measurements of serum levels of ALT, aspartate aminotransferase, serum bilirubin and albumin, alpha-fetoprotein, thyroid stimulating hormone, antinuclear antibody, serum creatinine and complete blood count were performed. Serum hepatitis B surface antigen and third-generation HCV antibody (anti-HCV) were detected using a commercially available, third-generation ELISA kit (AxSYM 3.0, Abbott Laboratories, USA).
An overnight (12 h) fasting blood sample was drawn for measurement of fasting plasma glucose and radioimmunoassay determination of fasting serum insulin levels (Diagnostic Products Co, USA). Insulin sensitivity was determined by the homeostasis model assessment (HOMA) method (15) using the following equation:
Detection/quantification of serum HCV RNA and viral kinetics
Serum HCV-RNA levels were quantified in all patients at the beginning of treatment and then at weeks 4, 12, 24, 48 and 72 using a quantitative reverse-transcription polymerase chain reaction (RT-PCR) assay. Detection of serum HCV RNA was performed using a standardized automated qualitative RT-PCR assay (COBAS AMPLICOR Hepatitis C Virus Test, version 2.0; Roche, USA). The detection limit was 50 IU/mL (17). Pretreatment HCV RNA levels were determined using a branched DNA assay (Quantiplex HCV RNA 3.0, Bayer, USA) in strict accordance with the manufacturer’s instructions. Because the majority of HCV patients in Egypt are infected with HCV genotype 4, genotyping was not performed in the present study (18).
SVR was defined as undetectable serum HCV RNA levels at week 72 (ie, 24 weeks after the end of treatment, which was also the end of follow-up). The definition of on-treatment response was as follows: rapid virological response (RVR) was defined as undetectable HCV RNA at week 4; complete EVR (cEVR) was defined as undetectable HCV RNA at week 12; ‘non-RVR’-cEVR was defined as positive HCV RNA at week 4 but undetectable at week 12; and ‘partial’ EVR (pEVR) was defined as positive for HCV RNA at weeks 4 and 12 but with ≥2 log10 drop in viral load at week 12 compared with baseline. Patients whose viral load declined more slowly (<2 log10 drop at week 12 compared with baseline), those whose viral load dropped >2 log10 at week 12 compared with baseline and who were still positive for HCV RNA at week 24, those who became HCV-RNA positive after negativization before the end of treatment (breakthrough response), as well as those who became HCV-RNA positive after negativization at the end of treatment were all considered to be non-SVR subjects (19). Abdominal ultrasonography examination was performed for all study participants.
Liver biopsy
All patients underwent ultrasound-guided liver biopsy before treatment. The biopsies were examined after paraffin-embedded sections of specimens were stained with hematoxylin and eosin. Liver biopsy specimens not <15 mm in length or the with presence of at least 10 complete portal tracts were required. Necroinflammatory activity was graded and fibrosis was staged according to the histological activity index (20).
Study design
The current observational case control study was started with 180 non-diabetic patients with chronic HCV who had not previously been treated with interferon alpha and/or ribavirin from Kafr El-Sheikh Liver Institute, Domiate Fever Hospital & Tanta Fever Hospital during the period between August 2008 and June 2010. Only 140 patients continued in the study, while the other 40 patients dropped out of the study in addition to those who showed a <2 log10 drop in viral load at week 12 compared with baseline or those whose HCV RNA level was positive at week 24 discontinued treatment on the basis of international guidelines. According to the HOMA-IR, 140 chronic HCV patients were divided into two groups: group 1 consisted of 48 chronic HCV patients with IR (HOMA-IR ≥2), group 2 consisted of 92 chronic HCV patients without IR (HOMA-IR <2). All patients were treated with combination PEG-IFN-2α 180 μg/week (Pegasys, Roche, Switzerland) plus oral ribavirin 1000 mg/day combination therapy for those weighing ≤75 kg and 1200 mg/day for those weighing >75 kg for 48 weeks. Patients were evaluated at monthly intervals to monitor compliance and side effects (ie, blood cell count, serum creatinine and liver function tests).
All 140 patients who experienced a ≥2 log10 decline in viral load at week 12 compared with baseline and whose HCV-RNA level was negative at week 24 continued the treatment schedule for a total of 48 weeks.
Statistical analysis
Database management and statistical analyses were performed using SPSS version 12 (IBM Corporation, USA). The t test, χ2 and comparison proportion tests were used to compare the two groups. To predict the presence or absence of a dependent variable based on a set of predictor variables (independent variables), linear multiple logistic regression analyses were performed. Statistical significance was set at P<0.05 for interpretation of results and tests of significance.
RESULTS
Baseline characteristics of the patients are summarized in Table 1. There was no significant difference in age, levels of serum aminotransferase levels and fasting plasma glucose between IR HCV pateints (group 1) and non-IR HCV patients (group 2). However, female sex, high BMI, increased fasting insulin level, higher viral load, steatosis >5% and advanced fibrosis (F≥3) were statistically significant in group 1 compared with group 2.
TABLE 1.
Demographic, virological and histopathological features of chronic hepatitis C (HCV) patients with or without insulin resistance (IR)
| IR chronic HCV Group 1 (n=48) | Non-IR HCV Group 2 (n=92) | P | |
|---|---|---|---|
| Age, years | 42±3.1 | 32.333±6.658 | 0.084 |
| Male sex, n/n (%) | 21/48 (43.75) | 54/92 (58.69) | 0.039* |
| BMI, kg/m2 | 27.889±1.571 | 21.01±2.646 | 0.001* |
| ALT, IU/L | 167±106.864 | 99.80±64.794 | 0.264 |
| AST, IU/L | 146.800±75.191 | 130.000±97.747 | 0.768 |
| Fasting glucose, mmol/L | 5.20±0.39 | 5.22±0.29 | 0.866 |
| Fasting insulin, pmol/L | 105.6±1.15 | 43.2±11.4 | 0.001* |
| Viral load, log10 copies/mL | 6.34±0.54 | 5.50±0.90 | 0.022 * |
| Grade of inflammation ≥3, n/n (%) | 16/48 (33.33) | 23/92 (25.1) | 0.415 |
| Fibrosis F≥3, n/n (%) | 23/48 (47.91) | 22/92 (23.91) | 0.007* |
| Steatosis >5%, n/n (%) | 35/48 (72.9) | 27/92 (29.34) | 0.0001* |
Data presented as mean ± SD unless otherwise indicated.
Statistically significant (P<0.05). ALT Alanine aminotransferase; AST Asparatate aminotransferase; BMI Body mass index
The association between IR and progressive fibrosis was evaluated by using univariate logistic regression analysis to detect factors associated with progressive fibrosis before starting treatment. Older age at the time of biopsy (P=0.007), higher BMI (P>0.001) and HOMA-IR ≥2 (P<0.001) were factors that were significantly associated with advanced fibrosis (Table 2). On multivariate logistic regression analysis, HOMA-IR ≥2 (P=0.040) remained independently associated with progressive fibrosis (Table 2). The influence of HOMA-IR ≥2 on early viral dynamics was assessed by detecting RVR, non-RVR-cEVR, cEVR, pEVR and SVR in both groups. In the present study, group 1 patients showed significantly lower RVR (18.36%), cEVR (32.65%) and SVR (26.53%) compared with group 2 patients who had RVR (31.52%) cEVR (54.34%) and SVR (63.04%) (P=0.046, 0.002 and 0.001, respectively), while, non RVR-cEVR and pEVR were insignificantly lower in group 1 compared with group 2 (Table 3). Viral kinetics throughout the treatment period are shown in Figure 1.
TABLE 2.
Pretreatment variables associated with progressive fibrosis among chronic hepatitis C patients
|
Univariate analysis
|
Multivariate analysis
|
||||
|---|---|---|---|---|---|
| Fibrosis F≤1 (n=68) | Fibrosis F≥3 (n=45) | P | OR (95% CI) | P | |
| Age, years, mean ± SD | 31.333±2.082 | 47.667±5.13 | 0.007* | 0.99 (0.74–1.33) | 0.258 |
| Male sex | 36 (52.9) | 21 (46.6) | 0.643 | – | – |
| Body mass index, kg/m2, mean ± SD | 21.80±03.701 | 28.200±1.789 | 0.001* | 0.82 (0.63–1.08) | 0.174 |
| High viral load† | 34 (50) | 19 (42.2) | 0.535 | – | – |
| HOMA-IR ≥2 | 14 (20.58) | 26 (57.78) | 0.001* | 1.92 (0.97–3.40) | 0.049* |
| Grade of inflammation ≥3 | 32 (47.05) | 21 (42.9) | 0.810 | – | – |
| Steatosis >5% | 22 (32.35) | 23 (51.1) | 0.077 | ||
Data presented as n (%) unless otherwise indicated.
Statistically significant;
>600,000 IU/mL. HOMA-IR Homeostasis model assessment-insulin resistance
TABLE 3.
Early viral kinetics among chronic hepatitis C patients with and without insulin resistance
| Group 1 (n=48) | Group 2 (n=92) | P | |
|---|---|---|---|
| RVR | 9 (18.36) | 29 (31.52) | 0.046* |
| Non-RVR-cEVR | 7 (14.28) | 21 (22.82) | 0.151 |
| cEVR | 16 (32.653) | 50 (54.43) | 0.002* |
| pEVR | 9 (18.36) | 26 (28.26) | 0.275 |
| SVR | 13 (26.53) | 58 (63.04) | 0.001* |
| Non-SVR | 35 (72.91) | 34 (36.95) | 0.001* |
Data presented as n (%) unless otherwise indicated.
Statististically significant (P<0.05). cEVR Complete early virological response; pEVR Partial early virological response; RVR Rapid virological response; SVR Sustained virological response
Figure 1).
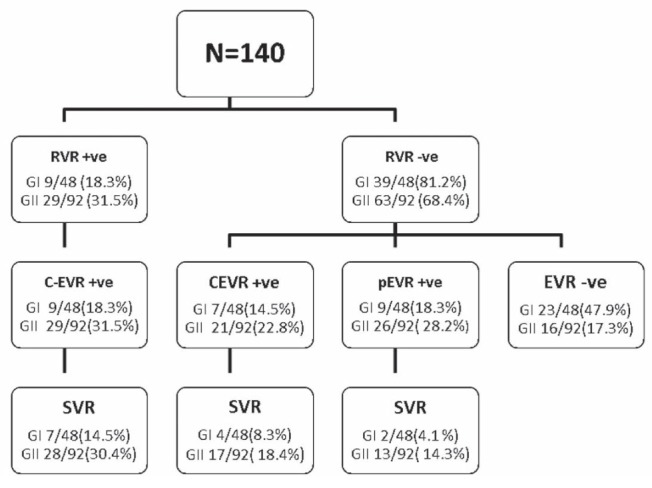
Viral kinetics throughout the treatment period. +ve Positive; −ve Negative; cEVR Complete early virological response; G Group; pEVR Partial early virologiacal response; RVR Rapid virological response; SVR Sustained virological response
To understand the pretreatment variables influencing SVR, both univariate and multivariate logistic regression analyses were performed. Univariate analysis showed that older age, fibrosis (F≥3), high viral load (>600,000 IU/mL) as well as HOMA-IR ≥2 were significantly associated with a lack of SVR. However, multivariate logistic regression analysis revealed that HOMA-IR ≥2 was the main independent pretreatment variable statistically associated with a lack of SVR, whereas fibrosis showed a weaker association, as shown in Table 4.
Table 4.
Factors associated with lack of sustained virological response (SVR) determined by univariate and multivariate logistic regression analysis
|
Univariate analysis
|
Multivariate analysis
|
||||
|---|---|---|---|---|---|
| SVR (n=71) | Non-SVR (n=69) | P | OR (95% CI) | P | |
| Age, years, mean ± SD | 32.20±10.474 | 49.400±7.925 | 0.019* | 0.94 (0.97–1.16) | 0.605 |
| Male sex | 43 (61.97) | 31 (44.9) | 0.052 | ||
| Body mass index, kg/m2, mean ± SD | 24.34±3.782 | 26.100±3.36 | 0.007* | 1.93 (0.63–2.31) | 0.096 |
| High viral load† | 28 (39.4) | 40 (57.97) | 0.042* | 1.17 (0.82–1.65) | 0.379 |
| HOMA-IR ≥2 | 13 (18.30) | 35 (50.72) | <0.001* | 2.44 (1.11–5.47) | 0.003* |
| Grade of inflammation ≥3 | 27 (38.02) | 34 (49.27) | 0.241 | ||
| Advanced fibrosis (F≥3) | 14 (19.710) | 31 (44.93) | 0.003* | 2.01 (0.75–3.31) | 0.030* |
| Steatosis >5% | 28 (39.34) | 39 (56.52) | 0.241 | ||
Data presented as n (%) unless otherwise indicated.
Statistically significant;
>600,000 IU/mL. HOMA-IR Homeostasis model assessment-insulin resistance
BMI, fasting glucose and fasting insulin levels, and HOMA-IR were measured before starting the treatment and again at week 48. A significant decrease among the SVR patients was revealed in BMI, insulin level and HOMA-IR (Table 5).
TABLE 5.
Metabolic and biochemical evaluation during treatment in patients who achieved sustained virological response
| Baseline | Week 48 | P | |
|---|---|---|---|
| Body mass index, kg/m2 | 28.0±1.581 | 22.20±2.387 | 0.002* |
| Fasting glucose, mmol/L | 5.236±0.223 | 5.194±0.295 | 0.104 |
| Fasting insulin, pmol/L | 65.67±18.64 | 49.9±8.34 | 0.038* |
| HOMA-IR | 2.280±0.540 | 1.660±0.207 | 0.009* |
Data presented as mean ± SD.
Statistically significant. HOMA-IR Homeostasis model assessment - insulin resistance
DISCUSSION
Chronic HCV infection is associated with a greater risk for the development of IR, and greater IR is more prevalent among patients with HCV infection compared with those with other liver diseases and with the general population (21). On the other hand, HCV patients with IR experience faster progression of hepatic fibrosis (3), extrahepatic manifestations (22) and development of HCC (23), suggesting that IR plays a crucial role in HCV infection.
In the present study, HCV patients with IR were characterized by a preponderance of women, higher BMI, insulin levels, viral load, steatosis >5% and fibrosis (F≥3) compared with HCV patients without IR. A significant increase in steatosis and fibrosis confirms the association between fatty liver (nonalcoholic fatty liver disease/nonalcoholic steatohepatitis) and chronic HCV-infected patients with IR. Whether IR could be the cause or consequence of steatosis and fibrosis is unknown. Fartoux et al (24) found that IR in patients infected with HCV genotype 1 is the cause rather than the consequence of hepatic steatosis and fibrosis, and suggests that increased circulating insulin is a risk factor for fibrosis through IR-induced steatosis.
Our study found that older age, higher BMI and HOMA-IR ≥2 were associated factors with progressive fibrosis, and that HOMA-IR ≥2 remained independently associated with progressive fibrosis. These results are in agreement with Cua et al (25), who studied 346 nondiabetic patients with HCV and concluded that IR is a major independent determinant of fibrosis in chronic HCV infection, regardless of the genotype and the severity of liver damage. The results are also in agreement with those of Halfon et al (26), who studied the influence of IR on hepatic fibrosis and steatosis in HCV-infected patients and found that liver fibrosis is significantly associated with IR independent of liver steatosis only among HCV-monoinfected patients. In fact, it has been demonstrated that high levels of insulin and glucose could promote fibrogenesis by stimulating the release of connective tissue growth factor – a fibrogenic growth factor – from hepatic stellate cells (27).
The relationship between IR and fibrosis has been clarified by Fartoux et al (24), who found a significant link between circulating insulin levels and fibrosis stage using univariate analysis, while multivariate analysis revealed that steatosis, but not insulin, was independently associated with fibrosis, suggesting an indirect effect of the circulating insulin level on fibrosis stage through a steatosis-related pathway. This relationship between steatosis and fibrosis could be explained by several mechanisms, such as lipid peroxidation (28–32), and the ‘two hits hypothesis’, in which steatosis could increase the sensitivity of hepatocytes to oxidative stress, the second hit being HCV infection itself in patients with chronic HCV (28).
The present study evaluated the impact of pretreatment IR on SVR. We found that older age, fibrosis (F≥3), high viral load (>600,000 IU/mL) as well as HOMA-IR ≥2 were significantly associated with a lack of SVR. HOMA-IR ≥2 was found to be the main independent variable associated with lack of SVR. Our study reveals an insignificant association between steatosis and lack of SVR indicating that fatty liver (nonalcoholic fatty liver disease/nonalcoholic steatohepatitis) can lead to a lack of SVR indirectly through progressive fibrosis or IR.
These results are supported by the results reported by Romero-Gomez et al (13), who in a prospective study of Spanish patients with chronic hepatitis C found an SVR rate of only 32.8% in HCV genotype 1 patients with IR compared with an SVR rate of 60.5% in those without IR, and concluded that IR impairs the response to therapy in patients infected with chronic HCV. Furthermore, Poustchi et al (28) found that patients with HOMA-IR <2 were 6.5 times more likely to achieve SVR than those with HOMA-IR ≥2.
Our results differ from those of Merchante et al (33), who studied the impact of IR on SVR of HIV/HCV coinfection; they found that IR is not a relevant predictor of SVR to PEG-IFN plus ribavirin in HIV/HCV-coinfected patients. This can be explained by the different scenarios of hepatic fibrosis and steatosis in HCV monoinfection and HIV/HCV coinfection, which is supported by Halfon et al (26), who found that while IR is associated with significant liver fibrosis in HCV monoinfected patients this does not happen with HIV/HCV coinfected patients.
To understand whether the impact of IR on SVR occurs as a result of its effect on early viral kinetics, our study assessed the influence of IR on early viral kinetics by detecting RVR, non RVR-cEVR, cEVR, pEVR and SVR in IR and non-IR HCV patients. We found that RVR and cEVR were significantly lower in insulin-resistant HCV patients compared with non-insulin-resistant HCV patients, while non-RVR-cEVR and pEVR were lower in IR HCV compared with non-IR HCV patients, but did not reach statistical significance. The significant lack of SVR among the IR group compared with non-IR HCV patients was consequently attributed to slow viral kinetics with lack of RVR and EVR. These results are supported by those of Napoli et al (34), who found that RVR at week 4 of treatment was strongly associated with the likelihood of achieving SVR in genotype 1 HCV patients, regardless of the therapeutic regimen, and patients who did not clear the virus within the first 12 weeks of treatment had little chance of achieving SVR. In addition, Yu et al (35) studied the predictive value of RVR and EVR on SVR in HCV genotype 1 patients and found that genotype, RVR and EVR were independent factors for predicting the effect of antiviral therapy. Therefore, the pretreatment HOMA-IR could not only predict treatment outcome but could also predict RVR and EVR. These results go hand in hand with those of Grasso et al (36), who concluded that IR could predict RVR in non-diabetic, noncirrhotic genotype 1 HCV patients (36). According to these results, this study suggests that the improvement in IR either by weight loss, lifestyle change or insulin sensitizer will lead to rapid viral kinetics, improving the RVR and EVR, which will, therefore, improve SVR in chronic HCV patients. This is supported by Khattab et al (37), who found that pioglitazone improves virological response to PEG-IFN-2α/ribavirin combination therapy in hepatitis C genotype 4 patients with IR.
On the other hand, our study demonstrates that HOMA-IR, BMI and insulin levels showed significant decreases at 48 weeks in SVR patients compared with their starting levels. This improvement happened without exercise, dietary or lifestyle changes, or insulin sensitizer intake. This suggests a cause-effect relationship between HCV and IR, which warrants further study.
CONCLUSION.
IR in chronic HCV patients is associated with advanced fibrosis, slow viral kinetics and could be a predictor for lack of RVR and EVR. Therefore, HOMA-IR levels should be assessed and improved before initiating antiviral treatment in chronic HCV patients.
Acknowledgments
The authors thank Dr Abd El Moneim El Hadydy from Domiate Fever hospital for his great effort in supporting the work during this study.
Footnotes
DISCLOSURES: The authors have no financial disclosures or conflicts of interest to declare.
REFERENCES
- 1.Flamm SL. Chronic hepatitis C virus infection. JAMA. 2003;289:2413–7. doi: 10.1001/jama.289.18.2413. [DOI] [PubMed] [Google Scholar]
- 2.Mazzaro C, Panarello G, Tesio F, et al. Hepatitis C virus risk: A hepatitis C virus-related syndrome. J Intern Med. 2000;247:535–45. doi: 10.1046/j.1365-2796.2000.00627.x. [DOI] [PubMed] [Google Scholar]
- 3.Hui JM, Sud A, Farrell GC, et al. Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression. Gastroenterology. 2003;125:1695–704. doi: 10.1053/j.gastro.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 4.Svegliati-Baroni G, Bugianesi E, et al. Post-load insulin resistance is an independent predictor of hepatic fibrosis in virus C chronic hepatitis and in nonalcoholic fatty liver disease. Gut. 2007;56:1296–301. doi: 10.1136/gut.2006.107946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai CY, Chuang WL, Ho CK, et al. Associations between hepatitis C viremia and low serum triglyceride and cholesterol levels: A community-based study. J Hepatol. 2008;49:9–16. doi: 10.1016/j.jhep.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Taura N, Ichikawa T, Hamasaki K, et al. Association between liver fibrosis and insulin sensitivity in chronic hepatitis C patients. Am J Gastroenterol. 2006;101:2752–9. doi: 10.1111/j.1572-0241.2006.00835.x. [DOI] [PubMed] [Google Scholar]
- 7.Petit JM, Bour JB, Galland-Jos C, et al. Risk factors for diabetes mellitus and early insulin resistance in chronic hepatitis C. J Hepatol. 2001;35:279–83. doi: 10.1016/s0168-8278(01)00143-x. [DOI] [PubMed] [Google Scholar]
- 8.Shintani Y, Fujie H, Miyoshi H, et al. Hepatitis C virus infection and diabetes: Direct involvement of the virus in the development of insulin resistance. Gastroenterology. 2004;126:840–8. doi: 10.1053/j.gastro.2003.11.056. [DOI] [PubMed] [Google Scholar]
- 9.Masini M, Campani D, Boggi U, et al. Hepatitis C virus infection and human pancreatic β-cell dysfunction. Diabetes Care. 2005;28:940–1. doi: 10.2337/diacare.28.4.940. [DOI] [PubMed] [Google Scholar]
- 10.Kawaguchi T, Yoshida T, Harada M, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through upregulation of suppressor of cytokine signaling 3. Am J Pathol. 2004;165:1499–508. doi: 10.1016/S0002-9440(10)63408-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–7. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 12.Strader DB, Wright T, Thomas DL, Seeff LB. American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C. Hepatology. 2004;39:1147–71. doi: 10.1002/hep.20119. [DOI] [PubMed] [Google Scholar]
- 13.Romero-Gomez M, Del MV, Andrade RJ, et al. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology. 2005;128:636–41. doi: 10.1053/j.gastro.2004.12.049. [DOI] [PubMed] [Google Scholar]
- 14.Grasso A, Malfatti F, De Leo P, et al. Insulin resistance predicts rapid virological response in non-diabetic, non-cirrhotic genotype 1 HCV patients treated with peginterferon alpha-2b plus ribavirin. J Hepatol. 2009;51:984–90. doi: 10.1016/j.jhep.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 15.Narita R, Abe S, Kihara Y, Akiyama T, Tabarau A, Otsuki M. Insulin resistance and insulin secretion in chronic hepatitis C virus infection. J Hepatol. 2004;41:132–8. doi: 10.1016/j.jhep.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 16.Knobler H, Zhornicky T, Sandler A, Haran N, Ashur Y, Schattner A. Tumor necrosis factor-α-induced insulin resistance may mediate the hepatitis C virus-diabetes association. Am J Gastroenterol. 2003;98:2751–6. doi: 10.1111/j.1572-0241.2003.08728.x. [DOI] [PubMed] [Google Scholar]
- 17.Conjeevaram HS, Kleiner DE, Everhart JE, et al. Race, insulin resistance and hepatic steatosis in chronic hepatitis C. Hepatology. 2007;45:80–7. doi: 10.1002/hep.21455. [DOI] [PubMed] [Google Scholar]
- 18.Sievert W, Altraif I, Razavi HA, et al. A systematic review of hepatitis C virus epidemiology in Asia, Australia and Egypt. Liver Int. 2011;31(Suppl 2):61–80. doi: 10.1111/j.1478-3231.2011.02540.x. [DOI] [PubMed] [Google Scholar]
- 19.Hadziyannis SJ, Sette H, Jr, Morgan TR, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: A randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140:346–55. doi: 10.7326/0003-4819-140-5-200403020-00010. [DOI] [PubMed] [Google Scholar]
- 20.Ishak KG. Chronic hepatitis: Morphology and nomenclature. Mod Pathol. 1994;7:690–713. [PubMed] [Google Scholar]
- 21.Mason AL, Lau JY, Hoang N, et al. Association of diabetes mellitus and chronic hepatitis C virus infection. Hepatology. 1999;29:328–33. doi: 10.1002/hep.510290235. [DOI] [PubMed] [Google Scholar]
- 22.Sumie S, Kawaguchi T, Komuta M, et al. Significance of glucose intolerance and SHIP2 expression in hepatocellular carcinoma patients with HCV infection. Oncol Rep. 2007;18:545–52. [PubMed] [Google Scholar]
- 23.El-Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology. 2004;126:460–8. doi: 10.1053/j.gastro.2003.10.065. [DOI] [PubMed] [Google Scholar]
- 24.Fartoux L, Poujol-Robert A, Guéchot J, Wendum D, Poupon R, Serfaty L. Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut. 2005;54:1003–8. doi: 10.1136/gut.2004.050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cua IH, Hui JM, Kench JG, George J. Genotype-specific interactions of insulin resistance, steatosis, and fibrosis in chronic hepatitis C. Hepatology. 2008;48:723–31. doi: 10.1002/hep.22392. [DOI] [PubMed] [Google Scholar]
- 26.Halfon P, Pénaranda G, Carrat F. Influence of insulin resistance on hepatic fibrosis and steatosis in hepatitis C virus (HCV) mono-infected compared with HIV-HCV co-infected patients. Aliment Pharmacol Ther. 2009;30:61–70. doi: 10.1111/j.1365-2036.2009.03995.x. Epub March 9, 2009. [DOI] [PubMed] [Google Scholar]
- 27.Paradis V, Perlemuter G, Bonvoust F, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34:738–44. doi: 10.1053/jhep.2001.28055. [DOI] [PubMed] [Google Scholar]
- 28.Poustchi H, Negro F, Hui J, et al. Insulin resistance and response to therapy in patients infected with chronic hepatitis C virus genotypes 2 and 3. J Hepatol. 2008;48:28–34. doi: 10.1016/j.jhep.2007.07.026. Epub October 1, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Fartoux L, Serfaty L, Lefevre G, et al. Steatosis, oxidative stress and fibrosis in chronic hepatitis C. Hepatology. 2003;38:363A. [Google Scholar]
- 30.Day CP, James OF. Steatohepatitis: A tale of two “hits”? Gastroenterology. 1998;114:842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 31.Lerat H, Honda M, Beard MR, et al. Steatosis and liver cancer in transgenic mice expressing the structural and non-structural proteins of hepatitis C virus. Gastroenterology. 2002;122:352–65. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- 32.Letteron P, Fromenty B, Terris B, et al. Acute and chronic hepatic steatosis lead to in vivo lipid peroxidation in mice. J Hepatol. 1996;24:200–8. doi: 10.1016/s0168-8278(96)80030-4. [DOI] [PubMed] [Google Scholar]
- 33.Merchante N, Macías J, Palacios RB, et al. Prevalence of non-significant liver fibrosis and rate of fibrosis progression in HIV/hepatitis C virus- co-infected patients: Still a role for liver biopsy? AIDS. 2004;18:1746–8. doi: 10.1097/01.aids.0000131389.83844.8a. [DOI] [PubMed] [Google Scholar]
- 34.Napoli N, Giannelli G, Parisi CV, Antonaci A, Maddalena G, Antonaci S. Predictive value of early virological response to treatment with different interferon-based regimens plus ribavirin in patients with chronic hepatitis C. New Microbiol. 2005;28:13–21. [PubMed] [Google Scholar]
- 35.Yu JW, Wang GQ, Sun LJ, Li XG, Li SC. Predictive value of rapid virological response and early virological response on sustained virological response in HCV patients treated with pegylated interferon alpha-2a and ribavirin. J Gastroenterol Hepatol. 2007;22:832–6. doi: 10.1111/j.1440-1746.2007.04904.x. [DOI] [PubMed] [Google Scholar]
- 36.Grasso A, Malfatti F, De Leo P, et al. Insulin resistance predicts rapid virological response in non-diabetic, non-cirrhotic genotype 1 HCV patients treated with peginterferon alpha-2b plus ribavirin. J Hepatol. 2009;51:984–90. doi: 10.1016/j.jhep.2009.07.008. Epub July 23, 2009. [DOI] [PubMed] [Google Scholar]
- 37.Khattab M, Emad M, Abdelaleem A. Pioglitazone improves virological response to peginterferon alpha-2b/ribavirin combination therapy in hepatitis C genotype 4 patients with insulin resistance. Liver Int. 2010;30:447–54. doi: 10.1111/j.1478-3231.2009.02171.x. Epub November 16, 2009. [DOI] [PubMed] [Google Scholar]