

Abstract
The authors describe two siblings, each with a different, rare genetic condition that affects liver function. The index case, the 18-year-old asymptomatic brother of a young man recently diagnosed with Wilson disease, presented for Wilson disease screening and was also found to have abnormal liver function suggestive of cholestasis. However, ceruloplasmin level, 24 h urine copper concentration and liver synthetic function were normal. Further hepatic investigations and genetic mutation analysis were performed, ultimately leading to a diagnosis of Alagille syndrome. He was treated with ursodiol, which resulted in normalization of his liver function tests. Subsequently, he was found to be a carrier for a mutation in the Wilson disease gene, ATP7B. In the present report, the potential implications of being a heterozygote for Wilson disease in the context of Alagille syndrome are discussed. Also stressed is that care must be exercised by the clinician when diagnosing family members who may present with two different disorders closely mimicking one another.
Keywords: ATP7B, Cholestasis, Hereditary liver disease, JAG1
Abstract
Les auteurs décrivent le cas de deux frères, dont chacun présentait une maladie génétique rare différente qui touche la fonction hépatique. Le cas de référence, le frère asymptomatique de 18 ans d’un jeune homme ayant récemment reçu un diagnostic de maladie de Wilson, a subi un test de dépistage de la maladie de Wilson, et on a découvert qu’il avait également une fonction hépatique anormale, évocatrice d’une cholestase. Cependant, son taux de céruloplasmine, sa concentration de cuivre dans les urines sur 24 heures et sa fonction synthétique du foie étaient normales. Les médecins ont procédé à de nouvelles explorations hépatiques et à une analyse de mutation génétique et fini par diagnostiquer un syndrome d’Alagille. Ils ont traité le patient à l’ursodiol, ce qui a normalisé les épreuves de fonction hépatique. Par la suite, ils ont découvert qu’il était porteur d’une mutation du gène de la maladie de Wilson, le ATP7B. Dans le présent rapport, les auteurs exposent les conséquences potentielles d’être un hétérozygote de la maladie de Wilson dans le contexte d’un syndrome d’Alagille. Ils soulignent également que le clinicien doit être prudent lorsqu’il diagnostique des membres d’une même famille qui peuvent présenter deux troubles différents étroitement similaires.
There are many different hereditary causes of hepatic impairment, and the clinician needs to be alert to these when investigating otherwise unexplained abnormalities in liver function. Occasionally, rare genetic diseases can coexist in the same family, which may provide difficult diagnostic challenges and lead to situations in which outcome and treatment response is more difficult to predict. In the present article, we describe two brothers who both presented with liver disease in early adulthood and were diagnosed with two different genetic conditions affecting liver function.
RESULTS
The index case, an asymptomatic 18-year-old man of mixed Italian and Hungarian descent (Figure 1, individual IV: 2), was screened for Wilson disease following the same diagnosis in his brother. Routine blood tests including liver synthetic function, ceruloplasmin, alpha-1 antitrypsin, ferritin, antimitochondrial antibodies, antismooth muscle antibody, antihepatitis C and antihepatitis B virus core antibodies were normal. However, his liver enzyme levels were abnormal (alanine aminotransferase 82 IU/mL [two times the upper limit of nornmal (ULN)], alkaline phosphatase 241 IU/mL [two times ULN] and gammaglutamyl transferase 227 IU/mL [five times ULN]), suggesting a mainly cholestatic process. Twenty-four hour urine copper and ceruloplasmin levels were both found to be within the normal range (urine copper 0.5 μmol/day, ceruloplasmin 294 mg/L), making a diagnosis of Wilson disease unlikely. Additional investigations were arranged: an abdominal ultrasound showed moderate hepatosplenomegaly; magnetic resonance cholangiopancreatography showed no evidence of primary sclerosing cholangitis; and an upper gastrointestinal endoscopy was normal. A liver biopsy was performed and revealed nonpathognomonic changes including marginating ductules in the portal tracts and a positive rhodanine copper stain (Figure 2). The latter change can be seen in Wilson disease, but is also present in various types of cholangiopathy and in chronic cholestasis. Notably, there was a lack of bile duct paucity.
Figure 1).
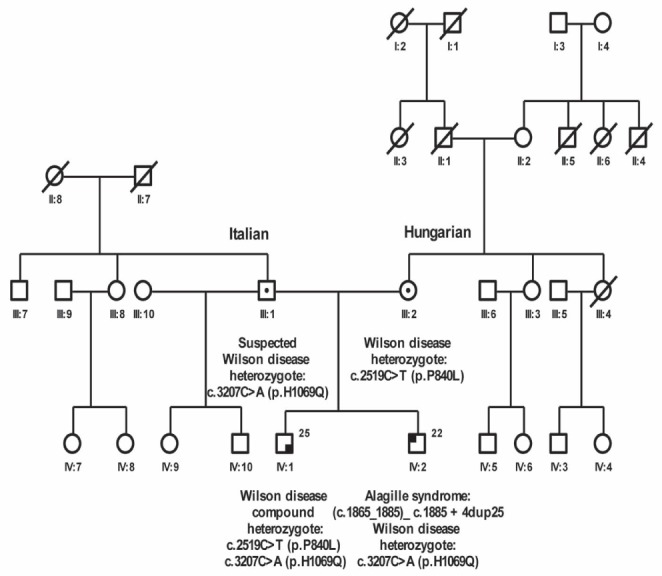
Family pedigree showing the index case (IV:2) and his brother (IV:1)
Figure 2).
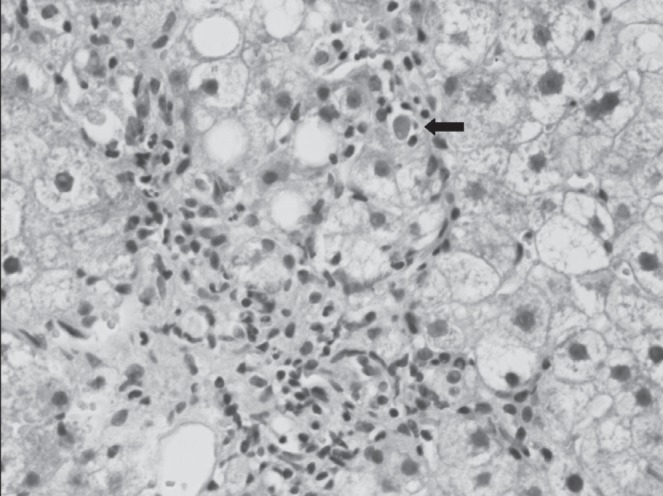
Liver biopsy showing interface hepatitis, steatosis, hydropic change and an apoptotic body (arrow). Hematoxylin and eosin stain, original magnification ×40
A medical record review revealed that the index case had been diagnosed with peripheral pulmonary artery stenosis and bilateral posterior embryotoxin when he was a child. Moreover, a childhood thoracolumbar x-ray demonstrated butterfly vertebrae, raising the possibility of Alagille syndrome. To investigate this further, genetic mutation analysis of JAG1 was performed. This revealed a heterozygous duplication of 25 nucleotides, identified in exon 14 of the JAG1 gene: (c.1865_1885)_c.1885 + 4dup25. His brother and both parents tested negative for this mutation, indicating that it originated de novo in the index case. In the meantime, the molecular defect causing Wilson disease in the family had been identified, and an ATP7B mutation analysis in the index case revealed that he was a carrier of the ATP7B p.H1069Q mutation. The mother of these two brothers was found to be a heterozygous carrier of the ATP7B p.P840L mutation previously identified in individuals of Hungarian descent (1); therefore, the index case had inherited the other mutation from his father.
DISCUSSION
The present article is the first to report a case of Alagille syndrome and Wilson disease in the same family. Alagille syndrome is an autosomal dominant disorder in which three of the following major clinical criteria must be present (along with liver bile duct paucity) to make a clinical diagnosis: cholestasis, cardiac defects, skeletal anomalies, ophthalmological abnormalities and characteristic facial features, including a prominent forehead, deep set eyes with moderate hypertelorism, a pointed chin and a straight nose (2). The reported prevalence of Alagille syndrome is one in 40,000 to one in 100,000 (3). The most common features are bile duct paucity on liver biopsy (present in 90% of affected individuals), peripheral pulmonary stenosis (4) and the ophthalmological finding of posterior embyrotoxon, which is found in up to 89% of affected individuals, compared with approximately 15% of the general population (5).The most common skeletal abnormality is butterfly vertebrae, which is reported in 70% of cases. Alagille syndrome shows reduced penetrance and variable expressivity, and approximately 45% of patients are found to have a new mutation while the remainder have an inherited mutation that may or may not affect the parent. The majority of symptomatic patients present in early childhood with hepatic disease manifesting as cholestasis and pruritis or, in more severe cases, progressive liver failure. Growth retardation is frequently associated with Alagille syndrome-related hepatic disease. Less commonly, patients present in infancy with hemodynamically significant congenital heart disease (2). In 1997, the disease-causing gene, JAG1, was identified (6) and most identified mutations were found to cause a frameshift resulting in a truncated protein product (7). The JAG1 protein, Jagged-1, is a ligand for the transmembrane receptor Notch1, and Notch signalling has been found to play an essential role in cell fate determination during embryogenesis (8).
Wilson disease is an autosomal recessive copper storage disorder that can present with hepatic, neurological or psychiatric disturbances (9), and has a prevalence of one in 30,000 to one in 40,000 in the United States (10). While early liver disease manifests only as mildly abnormal liver enzyme levels, progressive liver disease can result, leading to recurrent jaundice, simple acute self-limited hepatitis-like illness, autoimmune-type hepatitis, fulminant hepatic failure or chronic liver disease. Ultimately, liver biopsy is essential for diagnosis of both symptomatic and asymptomatic patients (10). Wilson disease is caused by mutations in the ATP7B gene, which is located on chromosome 13q14.3, with missense mutations accounting for more than one-half of the 480 mutations listed in the Wilson Disease Mutation Database (www.wilsondisease.med.ualberta.ca/database.asp). The ATP7B gene product, a copper-transporting, P-type ATPase, is expressed in hepatocytes and localizes to the trans-Golgi compartment. There, it transports copper into the secretory pathway for incorporation into the plasma protein ceruloplasmin and excretion into bile. Defective ATP7B function results in hepatic copper accumulation, hepatocyte death, copper release into the plasma and decreased serum ceruloplasmin levels (11).
The present article describes brothers presenting with liver disease due to different genetic conditions. There are two aspects that warrant further discussion. First, the index case was diagnosed with Alagille syndrome at a relatively advanced age, raising the issue of whether this individual would have been diagnosed with Alagille syndrome had his brother not first been diagnosed with Wilson disease. Second, did his ATP7B heterozygote status have a modifying effect on his liver function? At presentation, he had abnormal liver function tests, suggesting cholestasis, but importantly, had no bile duct paucity on liver biopsy. In light of his ATP7B heterozygote status, we propose the following three scenarios and accompanying explanations:
First, the ATP7B heterozygous state has no effect on Alagille syndrome-related liver findings. The ATP7B heterozygous state did not contribute to the liver findings in the index case, and the presentation can be explained by variable expressivity in Alagille syndrome leading to milder, late-presenting disease. In general, there are no genotype-phenotype correlations for this syndrome, with one notable exception being the G274D mutation, which leads to a cardiac-specific phenotype (12). The phenotypic variability of Alagille syndrome within families has been proposed to be due, in part, to other modifying factors including variation in the proteins involved in Notch cleavage, nuclear translocation or Notch-mediated transcription (13). A recent mouse model study showed that the Fringe genes, which encode glycosyltransferases that act on Notch receptors altering their affinity for Jagged-1 ligand, can cause an increase in bile duct proliferation when mutated (14). These, and other as yet unknown factors, may have been protective to this patient’s liver, and could explain the lack of bile duct paucity on biopsy.
Second, the ATP7B heterozygous state has a deleterious effect on Alagille syndrome-related liver findings. It is possible that IV:2 may have had no liver findings at the time of his Wilson disease screening had he not also been an ATP7B heterozygote. However, it is more likely that his cholestasis was related to Alagille syndrome because this is a more common feature of Alagille syndrome than of Wilson disease. The increased alkaline phosphatase level found in this patient was indicative of cholestasis, but whether his increased alanine aminotransferease level was further increased by his ATP7B heterozygote status is debatable because the liver function tests in ATP7B heterozygotes without liver comorbidity have not, to our knowledge, been reported to be useful in their identification. The fact that this patient had less severe liver findings than his brother with Wilson disease could indicate that he had only recently developed significant liver disease. To date, there have been no published cases of ATP7B heterozygotes having clinically significant hepatic copper storage or hepatic disease related to the carrier state, but patients have been observed to exhibit a slower than average rate of incorporation of copper into ceruloplasmin, with the rate being between that of normal controls and ATP7B homozygotes (15). While a slightly decreased rate of copper excretion in heterozygotes has not been found to be associated with hepatic manifestations, there have been reports of mild movement disorders in heterozygotes (16). To date, these case reports have not been replicated in larger studies, and the most recent study investigating ATP7B heterozygotes found no correlation between the carrier state and either hepatic or neurological manifestations (17). More case-control studies examining large numbers of ATP7B heterozygotes are necessary to determine whether the carrier state truly has no deleterious impact on the liver findings of those with a pre-existing liver comorbidity.
Third, the ATP7B heterozygous state has a protective effect on Alagille syndrome-related liver findings. This is the least likely possibility because these disorders are due to defects in different pathways, one involving copper metabolism in the liver (9), and the other involving cell fate determination and cell migration during the development of the liver, heart, skeleton, eye and kidney (8).
SUMMARY.
The present case illustrated the importance of a multidisciplinary approach to the diagnosis, management and treatment not only of index cases within a family, but also of their relatives. It is notable that liver damage in both Alagille syndrome and Wilson disease is generally treatable. It is important for the clinician to keep in mind that a similar phenotype (phenocopy) can be present in family members with two different disorders. It is unknown whether the ATP7B heterozygous state has any hepatic or other organ effects on the Alagille syndrome patient because such a situation in an individual is extremely rare.
Acknowledgments
The authors thank Diane Wilson Cox and Darren Bugbee for performing the ATP7B mutation analysis.
REFERENCES
- 1.Folhoffer A, Ferenci P, Csak T, et al. Novel mutations of the ATP7B gene among 109 Hungarian patients with Wilson’s disease. Eur J Gastroenterol Hepatol. 2007;19:105–11. doi: 10.1097/01.meg.0000223904.70492.0b. [DOI] [PubMed] [Google Scholar]
- 2.Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet. 1997;34:152–7. doi: 10.1136/jmg.34.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhadri VA, Stormon MO, Arbuckle S, Lam AH, Gaskin KJ, Shun A. Hepatocellular carcinoma in children with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2005;41:676–8. doi: 10.1097/01.mpg.0000179759.60048.c4. [DOI] [PubMed] [Google Scholar]
- 4.Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology. 1999;29:822–9. doi: 10.1002/hep.510290331. [DOI] [PubMed] [Google Scholar]
- 5.Pearce WG, Kerr CB. Inherited variation in Rieger’s malformation. Br J Ophthalmol. 1965;49:530–7. doi: 10.1136/bjo.49.10.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–51. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 7.Krantz ID, Colliton RP, Genin A, et al. Spectrum and frequency of Jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet. 1998;62:1361–9. doi: 10.1086/301875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–32. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 9.Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 10.Sternlieb I. Wilson’s disease. Clin Liver Dis. 2000;4:229–39. viii–ix. doi: 10.1016/s1089-3261(05)70105-7. [DOI] [PubMed] [Google Scholar]
- 11.Loudianos G, Gitlin JD. Wilson’s disease. Semin Liver Dis. 2000;20:353–64. doi: 10.1055/s-2000-9389. [DOI] [PubMed] [Google Scholar]
- 12.Eldadah ZA, Hamosh A, Biery NJ, et al. Familial Tetralogy of Fallot caused by mutation in the Jagged1 gene. Hum Mol Genet. 2001;10:163–9. doi: 10.1093/hmg/10.2.163. [DOI] [PubMed] [Google Scholar]
- 13.Weinmaster G. Notch signal transduction: A real rip and more. Curr Opin Genet Dev. 2000;10:363–9. doi: 10.1016/s0959-437x(00)00097-6. [DOI] [PubMed] [Google Scholar]
- 14.Ryan MJ, Bales C, Nelson A, et al. Bile duct proliferation in Jag1/fringe heterozygous mice identifies candidate modifiers of the Alagille syndrome hepatic phenotype. Hepatology. 2008;48:1989–97. doi: 10.1002/hep.22538. [DOI] [PubMed] [Google Scholar]
- 15.Cox DW, Fraser FC, Sass-Kortsak A. A genetic study of Wilson’s disease: Evidence for heterogeneity. Am J Hum Genet. 1972;24:646–66. [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhn J, Miyajima H, Takahashi Y, et al. Extrapyramidal and cerebellar movement disorder in association with heterozygous ceruloplasmin gene mutation. J Neurol. 2005;252:111–3. doi: 10.1007/s00415-005-0608-3. [DOI] [PubMed] [Google Scholar]
- 17.Torsdottir G, Gudmundsson G, Kristinsson J, Snaedal J, Johannesson T. Ceruloplasmin and superoxide dismutase (SOD1) in heterozygotes for Wilson disease: A case control study. Neuropsychiatr Dis Treat. 2009;5:55–9. doi: 10.2147/ndt.s4360. [DOI] [PMC free article] [PubMed] [Google Scholar]