

Abstract
BACKGROUND:
Wilson disease (WD) is a rare disorder of copper metabolism.
OBJECTIVE:
To describe the authors’ clinical experience with a cohort of 48 adult patients followed in an ambulatory setting.
METHODS:
A retrospective chart review of patients with a diagnosis of WD was performed.
RESULTS:
Fifty-nine charts were identified and 11 were excluded on further review. At diagnosis, 14 patients were asymptomatic, with 13 hepatic, 15 neurological and six mixed hepatic/neurological presentations. Ceruloplasmin levels were low (<0.20 g/L) in 94%, and 24 h urinary copper levels high (>0.60 μmol/L) in 95% of cases. D-penicillamine was the most common initial therapy (48%), with zinc the most common at review (65%). Overall, biopsy and ultrasound reports documented cirrhosis in 53%. Portal hypertension, defined as splenomegaly (>12.0 cm), reversed portal venous flow on ultrasound or varices/gastropathy on endoscopy was seen in 63%. At last review, 39% had elevated aspartate aminotransferase (>34 U/L) and/or alanine aminotransferase levels (>40 U/L). One death and one transplant occurred, while three patients had encephalopathy, two became jaundiced, two developed ascites and one experienced variceal bleed. Of 21 neurological presenting patients, 14 improved compared with baseline, with four making almost complete recovery. Eleven patients experienced documented episodes of neurological decline, including four with non-neurological presentation. Diagnostic magnetic resonance imaging showed basal ganglia (64%), brainstem (64%) abnormalities and atrophy (36%); follow-up showed basal ganglia lesions (50%) and atrophy (55%).
CONCLUSION:
WD is a diverse chronic disease with generally favourable outcomes for patients who respond to initial therapy, which can be managed predominantly in an ambulatory setting.
Keywords: Ceruloplasmin, Hepatolenticular degeneration, Movement disorders, Wilson disease
Abstract
HISTORIQUE :
La maladie de Wilson (MW) est un trouble rare du métabolisme du cuivre.
OBJECTIF :
Décrire l’expérience clinique des auteurs auprès d’une cohorte de 48 patients adultes suivis en milieu ambulatoire.
MÉTHODOLOGIE :
Les auteurs ont procédé à une analyse rétrospective des dossiers des patients ayant une MW diagnostiquée.
RÉSULTATS :
Les auteurs ont trouvé 59 dossiers, dont 11 ont été exclus lors de l’analyse. Au diagnostic, 14 patients étaient asymptomatiques, tandis que 13 avaient des manifestations hépatiques, 15, des manifestations neurologiques et six, des manifestations hépatiques et neurologiques mixtes. Le taux de céruloplasmine de 94 % d’entre eux était faible (moins de 0,20 g/L), et dans 95 % des cas, le taux de cuivre dans les urines sur 24 heures étaient élevés (plus de 0,60 μmol/L). La d-pénicillamine était le traitement initial le plus courant (48 %), et le zinc était le plus utilisé lors de la révision (65 %). Dans l’ensemble, les rapports de biopsie et d’échographie ont indiqué une cirrhose dans 53 % des cas. Les chercheurs ont constaté une hypertension portale, définie comme une splénomégalie (plus de 12,0 cm), un flux veineux portal rétrograde à l’échographie ou des varices ou une gastropathie à l’endoscopie dans 63 % des cas. À la dernière analyse, 39 % avaient des taux élevés d’aspartate-aminotransférase (plus de 34 U/L) ou d’alanine-aminotransférase (plus de 40 U/L), ou les deux. On a signalé un décès et une greffe, tandis que trois patients ont souffert d’une encéphalopathie, deux d’une jaunisse, deux d’ascites et un d’une hémorragie variqueuse. L’état de 14 des 21 patients ayant des manifestations neurologiques s’est amélioré par rapport aux données de départ, et quatre se sont presque entièrement rétablis. Onze patients ont vécu des épisodes étayés de déclin neurologique, dont quatre n’avaient pas de manifestations neurologiques. L’imagerie par résonance magnétique diagnostique a révélé des noyaux gris centraux (64 %), des anomalies du tronc cérébral (64 %) et une atrophie (36 %). Le suivi a révélé des lésions des noyaux gris centraux (50 %) et une atrophie (55 %).
CONCLUSION :
La MW est une maladie chronique diversifiée dont l’issue est généralement favorable chez les patients qui répondent à la thérapie initiale, et peut majoritairement être prise en charge en milieu ambulatoire.
Wilson disease (WD) is an inherited genetic disorder of liver copper metabolism with predominant hepatic and neurological manifestations (1). WD is relatively rare, occurring in approximately 30 per one million individuals (1), with lower prevalence rates in North America compared with other parts of the world (2). Given this, coupled with Canada’s low population density, clinicians in Canada often have difficulty acquiring adequate exposure to this disease. The Toronto Western Hospital, located in the urban and multicultural setting of Toronto, Ontario, houses two specialized ambulatory adult clinics, the Liver Centre (LC) and the Movement Disorders Centre (MDC), for treating patients with liver disease and movement disorders, respectively. Both sites have accrued experience in the management of WD over time because of the referral nature of the practices.
Accordingly, in the present study, we sought to draw on our relatively large cohort of WD patients who attend our clinics and describe the WD experience in Toronto, comparing disease course, therapy and outcomes of patients based on mode of presentation.
METHODS
A retrospective chart review was performed after approval from the local institutional research ethics board. Electronic databases were searched at both the LC and MDC to identify all past and present patients with a possible diagnosis of WD. Identified charts were then reviewed individually. Type of presentation (see below) and presenting symptoms were recorded. Laboratory values at presentation were recorded if available and, if unavailable, the first set of values available was recorded. Initial treatment, treatment changes and treatment side effects were documented throughout the patient’s history. The presence or absence of Kayser-Fleicher (KF) rings were recorded where documentation existed. Results from special investigations, including brain magnetic resonance imaging (MRI), liver ultrasound (US), upper gastrointestinal tract endoscopy (EGD) and liver biopsy (LB), were recorded. Significant events such as neurological deterioration, variceal bleeding, ascites, hepatic encephalopathy or jaundice were noted. Neurological symptoms at the last neurological follow-up were also recorded. Neurological improvement was defined as having fewer neurological symptoms, and deterioration as having more symptoms compared with presentation. A change in therapy was defined as a switch from one medication to another, the addition or subtraction of a medication from the regimen or a discontinuation of therapy for >3 months.
The Leipzig scoring system (3), which uses seven clinical parameters (Supplementary Table 1 [go to www.pulsus.com]), was applied to each chart once reviewed to determine the appropriateness of a WD diagnosis. Charts with scores <4 (indicating either probable or unlikely WD) were individually reviewed again by a second investigator and only included in the analysis if other evidence consistent with WD existed in the chart to support the diagnosis. Such evidence considered the presence of classic mitochondrial changes within hepatocytes or nuclear changes seen on electron microscopy (4).
In the final analyses, patients were divided into groups based on initial presentation. Asymptomatic patients included patients diagnosed through family screening and patients diagnosed due to incidental (but asymptomatic) abnormal laboratory or clinical findings. Symptomatic patients at presentation were divided into hepatic initial presentation, neurological initial presentation, and dual neurological and hepatic initial presentation (referred to as ‘mixed’ presentation). Leipzig scores (LS) and presenting laboratory values, which included 24 h urinary copper and serum copper concentrations, and ceruloplasmin, liver enzyme and total bilirubin levels, were compared among groups using the non-parametric ranking Kruskal-Wallis test. Statistical analysis was performed using R (5). Statistical significance was set at P<0.05.
RESULTS
Disease presentation
Fifty-nine charts were identified and reviewed (Figure 1), with 44 having an LS≥4. Of 15 patients with LS<4, five were included in the analyses due to further supporting evidence of WD not reflected by the LS. One patient with LS = 5 was removed from the analyses due to his symptoms being attributed to a pituitary germinoma. Of 48 patients included in the final analyses, 27 attended the LC, eight attended the MDC and 13 attended both clinics.
Figure 1).
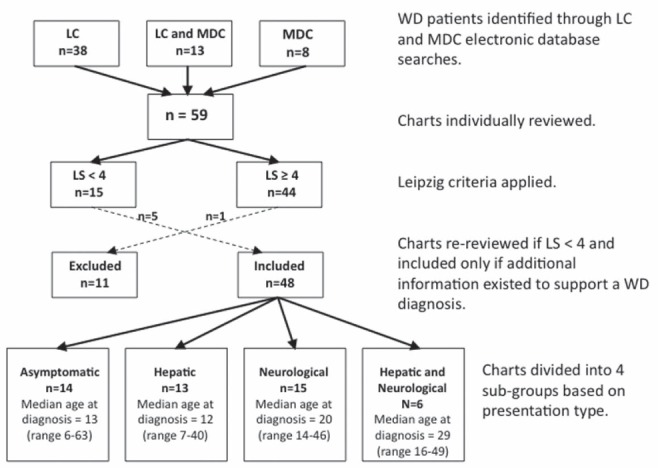
Flow chart depicting the methodology used to determine inclusion in the study based on Leipzig criteria and our division of charts into groups based on presentation type. LC Liver Centre; LS Leipzig score; MDC Movement Disorders Centre; WD Wilson disease
Median age at diagnosis was 17 years (range six to 63 years); six patients were diagnosed at age ≥40 years. At time of review, median time since diagnosis was 11.5 years (range zero to 44.6 years). Nine patients were diagnosed at the LC, six at the MDC and 22 at The Hospital for Sick Children (Toronto, Ontario), with the remainder having been diagnosed elsewhere. The median length of follow-up was 4.0 years (range zero to 26.8 years) at the LC and 1.85 years (range zero to 19.8 years) at the MDC. Ethnicity was available for 38 patients: 20 were Caucasian, 11 were eastern or southeastern Asian (China, Korea and Vietnam), six were south Asian and one was Middle-Eastern. No patients of African descent were identified. Thirteen patients had siblings with confirmed WD or a history strongly suggestive of WD (siblings who died at young age[s] due to fulminant liver failure of unknown cause); two patients had siblings with symptoms suggestive of WD. Beyond immediate siblings, two had confirmed family histories and three with suggestive family histories of WD in their extended families. Documented consanguinity existed between the parents of three patients; in all three cases, the parents were first cousins.
Clinical presenting features based on Leipzig criteria are summarized in Table 1. Neurological presentation was the most common (n=15), followed by asymptomatic (n=14), hepatic (n=13) and mixed presentation (n=6). There were no notable variations in distribution of presentation type among the identified ethnic groupings.
TABLE 1.
Clinical characteristics of patient cohort according to presentation type
| Presentation | K-F rings, n (%) |
Liver copper, μmol/g, median (range)
|
Hemolytic anemia, n (%) | Intitial ceruloplasmin, g/L, median (range) | Presenting urinary copper, μmol/24 h, median (range) | Leipzig score, median (range) | |
|---|---|---|---|---|---|---|---|
| Diagnostic | Follow-up | ||||||
| Asymptomatic (n=14) | 6 (43) | 4.11 (1.13–13.53) (n=3) | 10.84 (1.93–19.0) (n=3) | 0 (0) | 0.05 (UD–0.206) | 1.51 (0.472–4.97) | 6 (2–9) |
| Hepatic (n=13) | 8 (62) | 5.65 (n=1) | 4.58 (0.26–8.89) (n=2) | 3 (23) | 0.04 (UD–0.393) | 2.98 (1.52–3.98) | 5 (2–8) |
| Neurological (n=15) | 13 (87) | 3.46 (0.22–6.70) (n=2) | 2.25 (1.57– 20.43) (n=3) | 0 (0) | 0.06 (0.02–0.33) | 3.22 (1.91–15.6) | 8 (5–10) |
| Hepatic and neurological (n=6) | 4 (67) | – | 3.75 (n=1) | 1 (17) | 0.06 (0.02–0.169) | 3.40 (2.87–4.13) | 6.5 (6–10) |
| Total (n=48) | 31 (65) | 4.88 (0.22–13.53) | 3.75 (0.26–20.43) | 4 (8) | 0.05 (UD–0.393) | 3.22 (0.472–15.6) | 6 (2–10) |
| P | – | – | – | – | 0.502 | 0.421 | 0.002 |
Liver copper concentration is divided into diagnostic measurements before and up to one year postdiagnosis and follow-up measurements performed one year or greater postdiagnosis. Median time between diagnostic measurements and treatment initiation, and follow-up measurements and treatment initiation is −2.1 months (range −4.4 years to +2.0 months) and 8.1 years (range 1.5 to 23.2 years), respectively. Ceruloplasmin values are first available while urinary copper values are pretreatment only. P values are based on comparisons between presentation groups using the Kruskal-Wallis test (note: genetic data is not included due to the general lack of availability for our cohort; however, if available (n=6), it was used in the Leipzig score). K-F Kaiser-Fleicher; UD Undetectable.
The median LS in neurological presenting patients was 8 (range 5 to 10), which was significantly higher (P=0.002) compared with those with hepatic presentation (median 5; range 2 to 8) and asymptomatic presentation (median 6; range 2 to 9). Of 47 patients with available initial ceruloplasmin data, three (6.3%) were above the lower limit of normal (LLN, ≥0.20 g/L) while 31 (66%) had initial values less than one-half the LLN (<0.10 g/L). There was no significant difference in initial ceruloplasmin level among presentation groups (P=0.502). Of 22 patients with available pretherapy, 24 h urinary copper measurements, one patient was within the upper limit of normal (ULN, ≤0.60 μmol/24 h), with 19 (86%) having levels greater than two times the ULN (≥1.20 μmol/24 h). There was no significant difference among presentation groups (P=0.421). Of six patients with liver copper concentration measured within one year of treatment initiation, only one was within the ULN (<0.80 μmol/g); four of six had liver copper concentrations greater than five times the ULN (>4.0 μmol/g).
Median serum copper concentration at presentation was 7.1 μmol/L (range 4.0 μmol/L to 20.9 μmol/L), with only 19% of patients (four of 21) having values greater than the LLN (>11.3 μmol/L). Median pretreatment alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) levels were 37.5 U/L (range: 7 U/L to 236 U/L), 36 U/L (range 3 U/L to 200 U/L) and 100 U/L (range 15 U/L to 504 U/L), respectively. Forty per cent (eight of 20) had elevated ALT levels (>40 U/L), 50% (11 of 22) had elevated AST levels (>34 U/L) and 30% (seven of 23) had elevated ALP levels (>150 U/L). Elevations were generally mild to moderate; only four patients had an initial ALT or AST level >100 U/L. Median pretreatment total bilirubin level was 12 μmol/L (range 6 μmol/L to 76 μmol/L) with 19% (four of 21) having elevated levels (>22.0 μmol/L).
The only initial laboratory value to differ significantly among groups was pretreatment ALT level (P=0.013): it was significantly lower for neurological presenting patients (median 12 U/L; range 7 U/L to 39 U/L) compared with asymptomatic (median 79 U/L; range 14 U/L to 236 U/L) and mixed presenting patients (median 53 U/L; range 36 U/L to 162 U/L). Hepatic patients presented with intermediate ALT values (median 33 U/L; range 21 U/L to 125 U/L).
Asymptomatic presentation:
Seven of 14 asymptomatic patients were diagnosed through family screening. Seven were diagnosed though incidental findings, three through discovery of KF rings on routine eye examinations, two through abnormal aminotransferase levels or liver function tests and two through abnormal complete blood count tests during workup for other issues.
Hepatic presentation:
For hepatic presenting patients, jaundice was the most common presenting symptom (Figure 2A). Compared with neurological presenting patients and asymptomatic patients, hepatic patients had increased levels of cirrhosis and fibrosis on diagnostic LBs (Table 2), although this was not significant at a level of P<0.05. One patient in this group presented with concomitant psychiatric symptoms (but no associated movement disorders). Other presenting symptoms not shown in Figure 2A include coagulopathy (n=1), amenorrhea (n=1) and glomerulonephritis (n=1).
Figure 2).
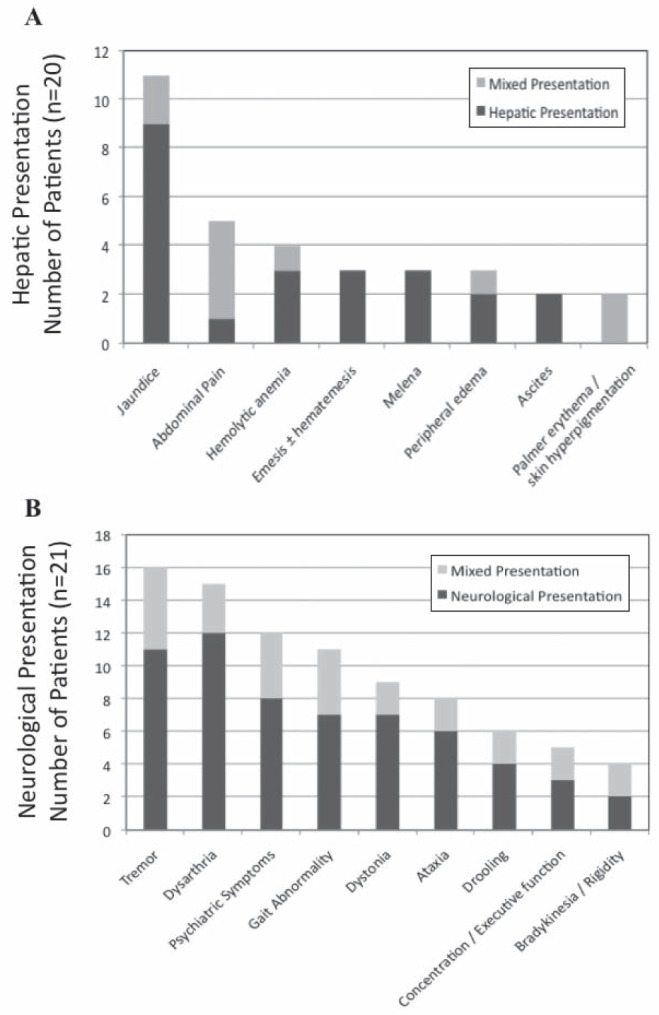
Presenting hepatic (A) and neurological symptoms (B) for patients who had hepatic and mixed (n=20), and neurological and mixed (n=21) presentations, respectively
TABLE 2.
Liver histology results according to presentation type for 31 patients who underwent liver biopsies
| Presentation | Liver biopsy | Cirrhosis | Fibrosis | Steatosis | Inflammatory activity |
|---|---|---|---|---|---|
|
Diagnostic liver biopsies
| |||||
| Asymptomatic, (n=14) | 5 | 2 | 3 | 4 | 5 |
| Hepatic (n=13) | 6 | 4 | 6 | 5 | 5 |
| Neurological (n=15) | 5 | 2 | 4 | 1 | 3 |
| Hepatic and neurological (n=6) | 4 | 4 | 4 | 3 | 1 |
| Total (n=48) | 20 | 12 | 17 | 13 | 14 |
|
Follow-up liver biopsies
| |||||
| Asymptomatic, (n=14) | 4 | 1 | 4 | 3 | 1 |
| Hepatic (n=13) | 4 | 3 | 4 | 1 | 3 |
| Neurological (n=15) | 4 | 3 | 4 | 3 | 2 |
| Hepatic and neurological (n=6) | 2 | 2 | 2 | 1 | 1 |
| Total (n=48) | 14 | 9 | 14 | 8 | 7 |
Data presented as n. Results are divided into patients with diagnostic biopsies before and up to one year postdiagnosis, and those with follow-up biopsies, performed one year or greater postdiagnosis (some patients had both diagnostic and follow-up biopsies performed). Median time between diagnostic biopsies and treatment initiation, and follow-up biopsies and treatment initiation is −41 days (range −25.2 to +8.1 months) and 8.2 years (range 1.5 to 33.0 years), respectively. Due to lack of standardization in the pathology reporting, fibrosis was counted if it was documented in the report and may or may not have been graded. Similarly, cirrhosis and steatosis were counted if they were documented in the report and range from mild to severe. Inflammatory activity was counted if the report documented hepatitis or presence of inflammatory cell infiltrate
Neurological presentation:
Patients with pure neurological presentation presented with a median of four neurological symptoms (Figure 2B). The presence of KF rings was documented in 13 of 15 patients. Eight patients (53%) also presented with concomitant psychiatric symptoms, the most common being personality and behavioural changes but also included depression, anxiety and one case of psychosis with suicidal ideation. Other presenting symptoms not shown in Figure 2B include memory loss (n=3), hypophonia (n=3), dysphagia (n=1) and hyper-reflexia (n=1).
Mixed hepatic and neurological presentation:
Six patients presented with a mixed presentation. These patients presented with a median of four neurological symptoms (Figure 2B). Four patients had documented KF rings. Abdominal pain was the most common hepatic complaint and much more prevalent compared with those with purely hepatic presentations (Figure 2A). Four of six patients presented with concomitant psychiatric symptoms. Other presenting symptoms not included in Figure 2 include epistaxis (n=1), temporal wasting (n=1), asterixis (n=1), dysphagia (n=1) and hyper-reflexia (n=1).
Treatment
The most common initial therapy was D-penicillamine (D-P) monotherapy followed by zinc, trientine and D-P/zinc dual therapy (Table 3). Four neurological patients were enrolled in clinical trials elsewhere at presentation and participated in a double-blind study comparing tetra-thiomolybdate with trientine (6). Type of presentation did not appear to influence starting drug. Changes in treatment regimens were common. At the time of review, 75% of patients had changed from their initial regimen; 35% changed treatment once; 21% changed treatment twice; and 19% changed treatment three or more times. At the time of review, zinc monotherapy was the most common choice, followed by D-P and trientine (Table 3). This pattern appeared to be uninfluenced by mode of presentation.
TABLE 3.
Initial treatment regimen and treatment regimen at time of review according to presentation type
| Presentation, n | D-P | T | Z | Dual therapy (D-P + Z) | Tetrathiomolybdate or double-blinded study |
|---|---|---|---|---|---|
|
Initial treatment
| |||||
| Asymptomatic (n=14) | 8 (57) | 1 (7) | 5 (36) | 0 (0) | 0 (0) |
| Hepatic (n=13) | 4 (31) | 5 (38) | 2 (15) | 2 (15) | 0 (0) |
| Neurological (n=15) | 8 (53) | 1 (7) | 2 (13) | 1 (7) | 3 (20) |
| Hepatic and neurological (n=6) | 3 (50) | 0 (0) | 2 (33) | 0 (0) | 1 (17) |
| Total (n=48) | 23 (48) | 7 (15) | 11 (23) | 3 (6) | 4 (8) |
|
Treatment at review
| |||||
| Asymptomatic (n=14) | 1 (7) | 0 (0) | 11 (79) | 2 (14) | 0 (0) |
| Hepatic (n=13) | 3 (23) | 3 (23) | 6 (46) | 1 (8) | 0 (0) |
| Neurological (n=15) | 3 (20) | 1 (7) | 11 (73) | 0 (0) | 0 (0) |
| Hepatic and neurological (n=6) | 1 (17) | 2 (33) | 3 (50) | 0 (0) | 0 (0) |
| Total (n=48) | 8 (17) | 6 (12) | 31 (65) | 3 (6) | 0 (0) |
Data presented as n (%). D-P D-penicillamine; T Trientine; Z Zinc
Of the patients on D-P at some point during their disease course, 41% (12 of 29) experienced documented side effects. Most common were rash (n=8) and nephrotic range proteinuria (n=4). Patients also experienced hematuria (n=2), renal failure (n=2) and Stevens-Johnson syndrome (n=1). Median time on D-P before switching medication was 2.6 years (range two months to 30 years). Nineteen per cent (eight of 43) of patients experienced documented side effects from zinc. In all but one case, side effects were limited to nausea and abdominal discomfort. In one case, however, a patient reported fatigue, headaches, muscle aches, allodynia and emotional lability with zinc treatment. The median time on zinc before switching therapy was 1.5 years (range 17 days to 3.8 years). Two patients with zinc-related side effects remained on zinc. Trientine had the fewest side effects (two of 21 [9.5%]), with one case of minor skin rashes and another of arthralgia and proteinuria. One patient discontinued trientine and both had experienced side effects with other options.
Other WD-related treatments included 16 patients on pyridoxine (doses ranged between 10 mg/day to 50 mg/day), and nine patients on Vitamin E (doses between 200 IU/day to 800 IU/day). Eleven patients were on beta-blockers (six on propranolol, four on nadolol and two on metoprolol). Of the neurological patients, four were receiving botox injections and four were on anticholinergic treatment (two on trihexyphenidyl and atropine, one on trihexyphenidyl and propantheline, and one on trihexyphenidyl exclusively). Sixteen patients were on psychiatric medications, including nine on a selective serotonin reuptake inhibitor or serotonin norepinephrine reuptake inhibitor, seven on a benzodiazepine, five on atypical antipsychotics, one on a typical antipsychotic and three on tricyclic antidepressants.
Latest laboratory values
At the time of the most recently available laboratory values (Supplementary Table 2 [go to www.pulsus.com]), median time on treatment was 10.1 years (range zero to 43.7 years). Of the 40 patients with access to follow-up 24 h urinary copper measurements, 25 were exclusively on zinc at the time of the last available measurement. Median urinary copper measurement for this group was 1.07 μmol/24 h (range 0.24 μmol/24h to 3.90 μmol/24 h). Eleven (44%) patients were above the American Association for the Study of Liver Diseases (AASLD) recommended level of 1.20 μmol/24 h (1), and two (8%) were greater than two times this level (>2.40 μmol/24 h). Of these two, one had been on zinc for only 34 days at the time of measurement and the second was grossly noncompliant with his medications, failing to even fill prescriptions. Of the nine with measurements between 1.20 μmol/24h to 2.40 μmol/24h, two had documented issues with noncompliance in their charts. It is unclear why the other seven had elevated urinary copper levels, other than raising the spectre that zinc monotherapy is not always sufficient therapy. Five had corresponding 24 h urinary zinc measurements, which ranged between 24.3 μmol/24 h and 66.8 μmol/24 h, indicating adequate compliance (1).
Of the 15 patients on chelator therapy at the time of the last 24 h urinary copper measurement (including three on a chelator plus zinc), the median level was 5.71 μmol/24 h (range 0.33 μmol/24 h to 18.8 μmol/24 h). Six patients (40%) were within the AASLD-recommended targets (3 μmol/24 h to 8 μmol/24 h) for patients on chelator therapy (1). Four patients (27%) were below the recommended targets, two of which had documented issues with non-compliance in their charts. Of three patients with corresponding ceruloplasmin and serum copper measurements available to calculate non-ceruloplasmin bound serum copper levels, one was above the ULN (>2.36 μmol/L), indicating possible subtherapeutic dosages of chelator therapy (1); none, however, had levels <0.79 μmol/L which could indicate potential overtreatment (1). Five patients (33%) were above the recommended urinary copper targets, which can indicate variable compliance if in the context of established chronic use (eg, increased chelator use just before urine collection) (1). Two of these patients did have documented issues in their charts with noncompliance, including one patient who went 26 years without treatment. It was unclear why the other three had urinary copper levels above the recommended targets. No patients were below the LLN for 24 h urinary copper (<0.06 μmol/24 h).
Ceruloplasmin levels were consistently low: one of 33 patients had a value above the LLN (>0.20 g/L) and 61% (20 of 33) were less than one-half the LLN. Eighty-one per cent (26 of 32) of patients had serum copper values below the LLN (<11.3 μmol/L). AST and ALT levels were within normal limits for 72% (31 of 43) and 67% (29 of 43) of patients, respectively. Aminotransferase level elevations were generally mild: only three patients (7%) had an ALT and/or AST level >100 U/L. Nine per cent (four of 46) of patients had total bilirubin levels above the ULN (>22.0 μmol/L).
Outcomes
Liver disease:
At the time of review, one patient in the cohort had progressed to liver transplant due to cirrhosis and progressive neurological and psychiatric decline. The outcome was generally favourable, with excellent graft function and stabilized neurological functioning. There was one death – the patient presented to the LC 15 months before his death with a mixed presentation that included psychiatric manifestations; he also had other comorbidities including chronic hepatitis B. The occurrence of major hepatic events while on treatment was rare. During follow-up, one patient developed ascites, experienced three episodes of jaundice and was also our only patient to experience a variceal bleed. This patient had documentation of excessive alcohol consumption, which was believed to be a contributing factor. Among the other patients, there was one other case of ascites after a prolonged period of treatment noncompliance, one patient with recurrent jaundice and three patients who developed hepatic encephalopathy (including the patient who died). Despite rare major hepatic events, both diagnostic and follow-up LBs universally showed some degree of liver injury or processes leading to injury. While three patients had no documented fibrosis on diagnostic biopsies (Table 2), they still demonstrated inflammatory activity, steatosis or both. Cirrhosis or coarse echotexture were also evidenced on US imaging across presentation types (Supplementary Table 3 [go to www.pulsus.com]). Of the patients with an LB, an US or both, 54% (25 of 46) had cirrhosis or suspected cirrhosis documented on at least one of these procedures.
Portal hypertensive changes were also common across presentation groups. Compared with their symptomatic counterparts, asymptomatic patients had fewer signs of portal hypertension on US (as indicated by a spleen span >12.0 cm, splenic varices or reversal of portal venous flow [Supplementary Table 3]); however, they still had high prevalence of varices and portal hypertensive gastropathy seen on EGD (Supplementary Table 4 [go to www.pulsus.com]). Of the patients with an US, EGD or both, 63% (27 of 43) had evidence of portal hypertension on at least one of these images.
Neurology outcomes:
At the time of review, 14 of 21 patients with neurological or mixed presentation had improved neurologically; however, only four (19%) experienced a complete or almost complete recovery (defined as no symptoms or only one mild residual symptom). Two patients were unchanged from presentation, four had deteriorated and one was lost to follow-up. Four non-neurological patients developed neurological symptoms after diagnosis: two remained symptomatic, one, with the exception of his dysarthria, improved once compliant with therapy; the fourth patient recovered completely. Eleven patients, including the four without neurological presentation, had documented episodes of neurological deterioration. Nine improved relative to their episode of deterioration; however, at time of review, only five had improved relative to their baseline at presentation. Three episodes occurred within six months of treatment initiation (all with D-P) and two patients improved relative to their presentation baseline. Five episodes were due to documented or suspected non-compliance with WD medication, of which two patients recovered beyond their baseline at presentation. Two episodes were attributed to other medications (noncompliance with olanzapine and an adverse reaction with amiodarone); one patient improved beyond their baseline at presentation. One episode occurred postpartum and another following a suspected but undefined viral illness with cardiac manifestations. While the postpartum patient showed improvement, both were non-neurological presenting patients and had significant residual symptoms at the time of review.
Brain MRI at initial presentation:
Fourteen patients underwent MRI before or within one year of diagnosis (Table 4). Eleven were neurological or mixed presenting patients, and all had abnormal scans. One patient was hepatic, presenting with psychiatric symptoms but no associated movement disorders, while the other two had no neurological or psychiatric manifestations of their disease. All three of these patients had normal scans. Basal ganglia abnormalities (high signal on T2 imaging) were most common in the putamen (seven of 10), caudate nucleus (six of 10) and globus pallidus (five of 10), with brainstem abnormalities primarily in the midbrain (nine of 10) and pons (four of 10). Atrophy was documented in the brainstem (three of five), cerebellum (two of five) and cerebrum (two of five).
TABLE 4.
Findings in 26 patients who underwent brain magnetic resonance imaging (MRI)
| Presentation | MRI | Basal ganglia | Brain-stem | Atrophy | Thalamus | White matter | Normal |
|---|---|---|---|---|---|---|---|
|
Diagnostic MRI
| |||||||
| Asymptomatic (n=14) | 2 | 0 | 0 | 0 | 0 | 0 | 2 |
| Hepatic (n=13) | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| Neurological (n=15) | 8 | 7 | 7 | 4 | 3 | 4 | 0 |
| Hepatic and neurological (n=6) | 3 | 3 | 3 | 1 | 2 | 2 | 0 |
| Total (n=48) | 14 | 10 | 10 | 5 | 5 | 6 | 3 |
|
Follow-up MRI
| |||||||
| Asymptomatic (n=14) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hepatic (n=13) | 5 | 1 | 1 | 2 | 0 | 2 | 3 |
| Neurological (n=15) | 10 | 6 | 1 | 5 | 0 | 2 | 3 |
| Hepatic and neurological (n=6) | 5 | 3 | 0 | 3 | 0 | 1 | 2 |
| Total (n=48) | 20 | 10 | 2 | 10 | 0 | 5 | 8 |
Data presented as n. Results are divided into patients with diagnostic imaging, before and up to one year postdiagnosis, and those with follow-up imaging, performed one year or greater postdiagnosis (some patients had both diagnostic and follow-up imaging performed). Median time between diagnostic imaging and treatment initiation, and follow-up imaging and treatment initiation is +3 days (range −4.4 years to +1.0 year) and 10.4 years (range 1.1 years to 34.9 years), respectively. Structural abnormalities counted refer to high signal seen on T2 imaging for the brain structure indicated. Atrophy was counted if it was documented in the radiology report for any brain
Changes in brain MRI after treatment and over the follow-up period:
Twenty patients underwent follow-up MRIs one year or more postdiagnosis (Table 4). Basal ganglia abnormalities on follow-up MRIs were in the putamen (10 of 10) and globus pallidus (four of 10), with caudate and substantia nigra abnormalities seen in only one patient each. Of two patients with brainstem abnormalities, both had midbrain involvement and one also with pons involvement. Atrophy was more common on follow-up imaging compared with baseline imaging and most commonly seen in the basal ganglia (five of 10), cerebellum (five of 10), cerebrum (two of 10) and brainstem (one of 10). Three patients demonstrated the face of the giant panda diagnostic sign (7). Five patients with pure hepatic presentations underwent follow-up imaging (Table 4). Two had developed movement disorders during follow-up. Both had abnormal scans, while the other three had normal scans. Most recent follow-up imaging was also normal in five patients with neurological or mixed presentations.
Psychiatric outcomes:
In addition to patients presenting with psychiatric symptoms, four developed psychiatric manifestations postdiagnosis (three hepatic presenting and one neurological presenting patient), resulting in 35% of the cohort displaying psychiatric manifestations at some point. Similar to patients with psychiatric presentations, manifestations developed during follow-up included personality changes, depression, anxiety and two cases of psychosis.
DISCUSSION
Our study draws on the experience of 48 patients with diverse ethnic backgrounds demonstrating the variable presentation, treatment history and course of WD. The variability seen within our cohort reflects the variability in the WD experience reported in the literature worldwide. With six of our patients (12.5%) presenting ≥40 years of age, our study provides additional support that late-onset WD may not occur as infrequently as once believed (8–12). Our oldest patient, previously reported (11), came to medical attention at 58 years of age due to the incidental finding of KF rings. She was otherwise asymptomatic and observed for five years until she eventually developed signs of portal hypertension on US and a final diagnosis was made. Once on treatment, her disease course was very stable. Genetic testing revealed two different mutations, Pro992Leu and Ala861Thr.
Pretreatment urinary copper concentration was the most sensitive test in identifying WD among our cohort. All symptomatic patients had 24 h urinary copper excretion >1.50 μmol/24 h (ULN = 0.6 μmol/24 h) at diagnosis. One patient, diagnosed through family screening at eight years of age, had a normal diagnostic urinary copper level; however, when she presented to the LC at 18 years of age, after 10 years of zinc monotherapy, her urinary copper level was 2.07 μmol/24 h. Similar to other studies (8,9), our patients presented with normal or only slightly elevated aminotransferase levels. While neurological patients had lower pretreatment ALT levels, they had comparable rates of inflammatory activity on LB, suggesting that liver inflammation still needs to be monitored in these patients.
Eighty-one per cent of our patients with neurological/mixed presentation had documented KF rings. While this is lower than numbers reported by others (8,13–16), our four neurological or mixed patients without documented KF rings were diagnosed at other clinics and no records pertaining to the testing for KF rings could be found. Among patients with hepatic or asymptomatic presentation, the presence of KF rings was substantially lower, reinforcing the fact that KF rings are not a sensitive test in diagnosing non-neurological WD.
The traditional use of D-P monotherapy as the mainstay of WD treatment no longer held true in our cohort, a reflection of additional therapeutic options. While before 1990, all but one patient initiated treatment with D-P monotherapy (n=13); since 1990, only 31% of patients were initiated on D-P monotherapy, and this dropped even more (to 20%) if considering only patients diagnosed after 2000. At review, only 25% of our patients were on the same medication they started with. This high level of treatment change occurred across presentation types. Reasons for change included adequate chelation prompting change to zinc maintenance therapy, side effects from therapy and cost inhibition, particularly for trientine, which is unlicensed for sale in Canada and, therefore, requires approval from Health Canada to purchase, and, therefore, must be paid for out of pocket by patients.
Despite good overall functional status, high levels of liver injury and portal hypertension persisted in our cohort. While asymptomatic patients exhibited less liver injury on LB and US, they exhibited comparable levels of inflammatory activity and steatosis, both of which precede fibrosis and indicate early stages of liver damage (17). All asymptomatic patients who underwent EGD imaging (five of 14) had evidence of abnormalities, and while the decision to image these patients (including two with prolonged documented episodes of non-compliance) was based on clinical indications of potential portal hypertension not evident in the other asymptomatic patients, these results show that patients who are asymptomatic at presention are also at high risk of portal hypertensive changes and require close monitoring. Liver transplantation was successful in stabilizing the neurological symptoms of our one transplant patient. While liver failure remains the most established indication for transplantation (1) there is limited – and debated – evidence that it is also an effective intervention for neurological WD (18–21). Consistent with previous studies (13,22–25) and despite adequate chelation therapy, our neurological patients experienced partial neurological improvement followed by stabilization of symptoms rather than full recovery.
Consistent with other MRI studies (7,22,26), T2 high-signal abnormalities at baseline were most commonly described in the putamen, caudate and brainstem, with persistent lesions on follow-up most common in the putamen and globus pallidus and sharply reduced incidence in the caudate, thalamus and brainstem. While others have reported signal abnormalities in hepatic patients (7,8,23,27), all of the patients with no neurological dysfunction in our cohort who were scanned (n=6) had normal imaging regardless of when the scan was completed. Of the five neurological patients with normal or essentially normal follow-up MRIs, two had only minimal symptoms remaining, two had improved but still had notable movement disorders and the fifth had actually developed new neurological symptoms despite normal imaging. While some studies have suggested strong associations between MRI signal improvements and improvements in neurological function (7,24,28), others, similar to our experience, have reported disconcordance between MRI changes and disease progression (22,29).
CONCLUSION.
Our retrospective case series presents a cohort of adult patients with WD who are managing their disease quite well in an ambulatory setting assuming adherence to therapy and lack of comorbidities or other insults to their liver. Even with good management, however, evidence of compensated liver injury and long-standing physical changes to the brain are common, demonstrating vulnerability within our cohort to further insult. The few cases of hepatic or neurological decline among this cohort underscore the importance of emphasizing life-long therapy adherence and vigilant monitoring of patients’ liver and neurological status regardless of presentation type.
Supplementary Materials
REFERENCES
- 1.Roberts E, Schilsky M. Diagnosis and treatment of Wilson’s disease: An update. Hepatology. 2008;47:2089–111. doi: 10.1002/hep.22261. [DOI] [PubMed] [Google Scholar]
- 2.Chu NS, Hung TP. Geographic variations in Wilson’s disease. J Neurologic Sci. 1993;117:1–7. doi: 10.1016/0022-510x(93)90145-o. [DOI] [PubMed] [Google Scholar]
- 3.Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver International. 2003;23:139–42. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 4.Roberts EA, Robinson BH, Yang S. Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Molecular Genet Metabol. 2008;93:54–65. doi: 10.1016/j.ymgme.2007.08.127. [DOI] [PubMed] [Google Scholar]
- 5.Institute of Statistics and Mathematics of the WU Wien The R Project for Statistical Computing. < http://www.r-project.org/> (Accessed April 2011).
- 6.Brewer GJ, Askari F, Dick R, et al. Treatment of Wilson’s disease with tetrathiomolybdate: V. Control of free copper by tetrathiomolybdate and a comparison with trientine. Translat Res. 2009;154:70–7. doi: 10.1016/j.trsl.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Sinha S, Taly A, Ravishankar S, et al. Wilson’s disease: Cranial MRI observations and clinical correlation. Neuroradiology. 2006;48:613–21. doi: 10.1007/s00234-006-0101-4. [DOI] [PubMed] [Google Scholar]
- 8.Taly A, Meenakshi-Sundaram S, Sinha S, Swamy H, Arunodaya G. Wilson disease: Description of 282 patients evaluated over 3 decades. Medicine. 2007;82:112–21. doi: 10.1097/MD.0b013e318045a00e. [DOI] [PubMed] [Google Scholar]
- 9.Gow P, Smallwood R, Angus P, Smith A, Wall A, Sewell R. Diagnosis of Wilson’s disease: An experience over three decades. Gut. 2000;46:415–9. doi: 10.1136/gut.46.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumago T, Horiike N, Michitaka K, et al. Recent clinical features of Wilson’s disease with hepatic presentation. J Gastroenterol. 2004;39:1165–9. doi: 10.1007/s00535-004-1466-y. [DOI] [PubMed] [Google Scholar]
- 11.Xuan A, Bookman I, Cox DW, Heathcote J. Three atypical cases of Wilson disease: Assessment of the Leipzig scoring system in making a diagnosis. J Hepatol. 2007;47:428–33. doi: 10.1016/j.jhep.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 12.Ferenci P, Czlonkowska A, Merle U, et al. Late-onset Wilson’s disease. Gastroenterology. 2007;132:1294–8. doi: 10.1053/j.gastro.2007.02.057. [DOI] [PubMed] [Google Scholar]
- 13.Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: A cohort study. Gut. 2007;56:115–20. doi: 10.1136/gut.2005.087262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steindl P, Ferenci P, Dienes HP, et al. Wilson’s disease in patients presenting with liver disease: A diagnostic challenge. Gastroenterology. 1997;113:212–8. doi: 10.1016/s0016-5085(97)70097-0. [DOI] [PubMed] [Google Scholar]
- 15.Brewer GJ, Yuzbasiyan-Gurkan V. Wilson disease. Medicine. 1992;71:139–64. doi: 10.1097/00005792-199205000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Rodrigo-Agudo J, Valdes-Mas M, Vargas-Acosta A, et al. Presentacionclinica, diagnostico y evolucion a largo plazo en 29 pacientes con enfermedad de Wilson. Revista Espanola Enfermedades Digestivas. 2008;100:456–461. doi: 10.4321/s1130-01082008000800002. [DOI] [PubMed] [Google Scholar]
- 17.Brewer GJ. Wilson’s disease: A clinician’s guide to recognition, diagnosis, and management. Norwell: Kluwer Academic Publishers; 2001. [Google Scholar]
- 18.Bax RT, Hassler A, Luck W, et al. Cerebral manifestation of Wilson’s disease successfully treated with liver transplantation. Neurology. 1998;51:863–5. doi: 10.1212/wnl.51.3.863. [DOI] [PubMed] [Google Scholar]
- 19.Duarte-Rojo A, Zepeda-Gomez S, Garcia-Leiva J, et al. Liver transplantation for neurologic Wilson’s disease: Reflections on two cases within a Mexican cohort. Rev Gastroenterol Mex. 2009;74:218–23. [PubMed] [Google Scholar]
- 20.Lui C, Chen C, Cheng Y, Lee T. Wilson’s disease after liver transplantation. Transplant Proc. 1998;30:3324–5. doi: 10.1016/s0041-1345(98)01048-3. [DOI] [PubMed] [Google Scholar]
- 21.Stracciari A, Tempestini A, Borghi A, Guarino M. Effect of liver transplantation on neurological manifestations in Wilson disease. Arch Neurol. 2000;57:384–6. doi: 10.1001/archneur.57.3.384. [DOI] [PubMed] [Google Scholar]
- 22.Leiros da Costa M, Spitz M, Bacheschi L, Leite C, Lucato L, Barbosa E. Wilson’s disease: Two treatment modalities. Correlations to pretreatment and post-treatment brain MRI. Neuroradiology. 2009;51:627–33. doi: 10.1007/s00234-009-0536-5. [DOI] [PubMed] [Google Scholar]
- 23.Tarnacka B, Szeszkowski W, Golebiowski M, Czlonkowska A. MR spectroscopy in monitoring the treatment of Wilson’s disease patients. Movement Disorders. 2008;23:1560–6. doi: 10.1002/mds.22163. [DOI] [PubMed] [Google Scholar]
- 24.Prashanth L, Taly A, Sinha S, et al. Prognostic factors in patients presenting with severe neurological forms of Wilson’s disease. Q J Med. 2005;98:557–63. doi: 10.1093/qjmed/hci095. [DOI] [PubMed] [Google Scholar]
- 25.Sinha S, Taly AB, Prashanth LK, Ravishankar S, Arunodaya GR, Vasudev MK. Sequential MRI changes in Wilson’s disease with de-coppering therapy: A study of 50 patients. Br J Radiol. 2007;80:744–9. doi: 10.1259/bjr/48911350. [DOI] [PubMed] [Google Scholar]
- 26.Starosta-Rubinstein S, Young A, Kluin K, et al. Clinical assessment of 31 patients with Wilson’s disease: Correlations with structural changes on magnetic resonance imaging. Arch Neurol. 1987;44:365–70. doi: 10.1001/archneur.1987.00520160007005. [DOI] [PubMed] [Google Scholar]
- 27.Kozic D, Svetel M, Petrovic B, Dragašević N, Semnic R, Kostić VS. MR imaging of the brain in patients with hepatic form of Wilson’s disease. Eur J Neurol. 2003;10:587–92. doi: 10.1046/j.1468-1331.2003.00661.x. [DOI] [PubMed] [Google Scholar]
- 28.Larnaout A, Ammar N, Mourad Z, Naji S, Hentati F. Wilson disease: Appreciable improvement of sub-cortical white matter abnormalities after copper chelating treatment: Five years follow-up. Neuropediatrics. 2008;39:176–8. doi: 10.1055/s-0028-1085464. [DOI] [PubMed] [Google Scholar]
- 29.Nazar H, Brismar, AI-Kawi MZ, Gunasekaran TS, Jorulf KH. Magnetic resonance imaging of the brain in Wilson’s disease. Neuroradiology. 1993;35:130–3. doi: 10.1007/BF00593969. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.