

Abstract
Study Design
The inflammatory responses of primary human intervertebral disc (IVD) cells to tumor necrosis factor (TNFα) and an antagonist were evaluated in vitro.
Objective
To investigate an ability for soluble TNF receptor type II (sTNFRII) to antagonize tumor necrosis factor alpha (TNFα) induced inflammatory events in primary human IVD cells in vitro.
Summary of Background Data
TNFα is a known mediator of inflammation and pain associated with radiculopathy, and IVD degeneration. Soluble TNF receptors (sTNFR) and their analogues are of interest for the clinical treatment of these IVD pathologies although information on the effects of sTNFR on human IVD cells remains unknown.
Methods
IVD cells were isolated from surgical tissues procured from 15 patients and cultured with or without 1.4 nM TNFα (25ng/ml). Treatment groups were co-incubated with varying doses of sTNFRII (12.5–100nM). Nitric oxide (NO), prostaglandin E2 (PGE2), and interleukin-6 (IL6) levels in media were quantified to characterize the inflammatory phenotype of the IVD cells.
Results
Across all patients, TNFα induced large, statistically significant increases in NO, PGE2, and IL6 secretion from IVD cells compared to controls (60, 112, and 4-fold increases, respectively; p < 0.0001). Coincubation of TNFα with nanomolar doses of sTNFRII significantly attenuated the secretion of NO and PGE2 in a dose-dependent manner, while IL6 levels were unchanged. Mean IC50 values for NO and PGE2 were found to be 35.1 nM and 20.5 nM, respectively.
Conclusions
Nanomolar concentrations of sTNFRII were able to significantly attenuate the effects of TNFα on primary human IVD cells in vitro. These results suggest this soluble TNF receptor to be a potent TNF antagonist with potential to attenuate inflammation in IVD pathology.
INTRODUCTION
Tumor necrosis factor (TNF) is one of several pro-inflammatory cytokines believed to contribute to the painful symptoms and progressive pathology of intervertebral disc (IVD) disorders, including disc degeneration, disc herniation and lumbar or cervical radiculopathy[1–4]. TNFα is spontaneously produced by IVD tissues ex vivo[5,6], is expressed at higher levels in herniated IVD tissues from symptomatic patients compared to asymptomatic or autoptic controls, and its expression levels increase with increasing radiographic grade of degeneration in the IVD[1,7,8]. Macrophages are key producers of TNFα and their increased presence in herniated and degenerated IVD tissues has been suggested as one of several sources in the IVD[9–12].
Cells of the IVD respond to proinflammatory stimuli (TNFα, lipopolysaccharide (LPS), and interleukin-1 beta (IL-1β)) in vitro with a net catabolic response characterized by decreased collagen and aggrecan synthesis, increased gene or protein expression for degradative collagenases, gelatinases and aggrecanases, as well as increased release of inflammatory mediators, such as nitric oxide (NO), prostaglandin E2 (PGE2), and interleukin-6 (IL6)[2,5,11,13–16]. TNFα exposure can also promote nerve growth factor (NGF) and NGF receptor expression in primary IVD cells in vitro[17]. Exogenous TNFα placed on dorsal root ganglia can induce decreased nerve conduction velocities[6,18] and increased mechanical allodynia and thermal hyperalgesia characteristic of IVD herniation in rat models[18,19], which suggests a potential role for TNFα in regulating pain-related IVD pathology. Together, these studies have motivated interest in developing and characterizing therapies able to antagonize the inflammatory and catabolic events associated with TNFα exposure in the IVD[20–24].
While TNFα binds multiple receptors, two cell surface receptors, TNF receptors (TNFR) type I and II, are the most abundant and potent receptors for stimulating multiple apoptotic and pro-inflammatory pathways[25,26]. The molecular basis of TNFα signaling is complex and depends on cell lineage, but is known to involve the activation of NF-κB, AP-1, and MAPK transduction factors in IVD cells[15,16]. Both p38 MAPK[14] and NF-κB inhibitors[27] have been studied as therapeutic targets for antagonizing intracellular effects of TNF in IVD cells, and two anti-proliferative antibiotics, minocycline[28,29] and pentoxifylline[6], have been evaluated for treating neuropathic pain models in rats. TNF-neutralizing antibodies have also been examined for mediating inflammation in IVD tissues in vitro[30], evaluated in animal models[3,6,31,32], and clinically for IVD herniation-associated radiculopathy[21].
Soluble cleavage products of the Type I and Type II TNF receptors (sTNFRI and sTNFRII respectively) retain a high affinity for TNFα[33] and function as decoy binding sites for this cytokine[34,35]. TNFα activity can hence be antagonized by sequestering soluble TNFα away from target receptors or by interacting with membrane-associated TNFα that will promote cytokine internalization and degradation. Clinical interest in the use of soluble receptors as TNF antagonists has led to the development of sTNFRI and sTNFRII analogues formed by conjugation or fusion to a macromolecular carrier, such as polyethylene glycol (PEG) or immunoglobulin G (IgG) constant domain (pegsunercept[36]; lenercept[37] and etanercept[38]). Clinical trials of systemic administration of etanercept, a sTNFRII:IgG fusion protein[38], have been conducted with the hope of reducing symptoms associated with lumbar radiculopathy[22,23]. While these and prior studies have shown interest in use of soluble TNF receptors or TNF-blocking antibodies for mediating nerve root-associated radiculopathy, data on the direct biological effects of sTNFR on IVD cells challenged with TNFα is not available. The objective of this study was to characterize the inflammatory response of human primary IVD cells following TNFα stimulation and test for a dose-dependent attenuation of these effects with sTNFRII.
METHODS
Isolation of human IVD cells
Pathologic intervertebral disc tissues were obtained according to IRB approved protocols from 15 different patients undergoing surgery for treatment of degeneration or adult scoliosis (n=15). Tissues were procured from levels between L1 and L5 in patients ranging from 14 to 74 years of age. Regions corresponding to anulus fibrosus and nucleus pulposus were mechanically separated where possible, and only anulus fibrosus cells were isolated via enzymatic digestion as described previously[39] (0.3% pronase (Boehringer-Mannheim, Gaithersburg, MD, USA) and 0.3% collagenase II (Worthington Biochemical Corp., Lakewood, NJ, USA) per gram tissue). All reagents were obtained from Gibco (Invitrogen), unless otherwise noted. Cells were plated onto 25 cm2 tissue culture plastic flasks, overlaid with culture medium (F-12 medium with 10% FBS (HyClone, Thermo Scientific, Waltham, MA, USA), 100 U/ml penicillin and 100 μg/ml streptomycin, 10 mM HEPES, 1 μg/ml Fungizone) and allowed to grow to confluence with a change of culture medium every 2–3 days. Cells for all experiments were used within 2 passages of culture after isolation.
Cell stimulation with TNFα and sTNFRII
Cells were seeded in gelatin-coated wells of 48-well plates (50,000 cells/well; 3 replicates per treatment group per patient sample), overlaid with 300μl of culture medium, and allowed to attach for 24hrs. Fresh culture medium (300μl) supplemented with 1.4 nM rhTNFα (Abcam, Cambridge, MA, USA; 25 ng/ml; 17.4 kDa) was added to attached cells; control cells received an exchange of culture medium without TNFα. Cells in one of four treatment groups were co-treated with rh-sTNFRII (Abcam; 18.9kDa) at 12.5, 25, 50, or 100 nM, which was added at the same time as TNFα. All cells were cultured at 37°C and 5% CO2 for an additional 72 hrs and supernatants were collected at the termination of all experiments and stored at −20°C until quantitation.
Quantitation of inflammatory mediators
All supernatants for each patient were assayed for release of NO (Griess reaction)[40], PGE2 (ELISA, R&D Systems, Minneapolis, MN, USA), and IL6 (ELISA, R&D Systems). Standard curves for ELISAs were generated as prescribed by the manufacturer, and absorbance values of standards for NO quantitation were adjusted for the absence of culture medium.
Values for each inflammatory mediator in culture medium only were quantified and averaged across 3 replicates for each assay; this mean value was subtracted from corresponding data for each assay. A fold change from control (i.e., cells receiving no TNFα) was also calculated for each sample receiving TNFα to determine relative increases in inflammatory mediator secretion. Values of each inflammatory mediator for cells receiving sTNFRII were normalized by corresponding values for cells receiving TNFα only as a measure of TNFα attenuation. Mean fold changes and attenuation fractions for each patient were calculated by averaging the range of values obtained from three control and three experimental replicates.
Statistical analyses
Box plots of NO, PGE2, and IL6 data for all patients were generated to calculate a median, upper quartile (Q3), lower quartile (Q1), and interquartile range (IQR) for each data set. Medium-corrected data outliers—values greater or less than 1.5 IQR deviations from Q3 or Q1, respectively—were removed before calculating mean absolute responses to TNFα for all patients and fold-changes from control. A one-way ANOVA was used to detect differences in fold changes of NO, PGE2, and IL6 levels between control and TNFα-treated cells at a significance level of 0.05. A one-way ANOVA with a post-hoc Tukey’s HSD test was used to detect differences in attenuation of NO, PGE2, and IL6 amongst groups receiving sTNFRII. Dose-dependent attenuation of TNFα-induced effects was further quantified by calculating an IC50 value for each inflammatory mediator. The attenuation fractions of NO, PGE2, and IL6 for each patient for each dose of sTNFRII normalized to TNFα only treatment groups were fit by nonlinear regression to a logistic curve to derive half maximal inhibitory concentrations (IC50):
| (1) |
where X is the attenuation fraction of inflammatory mediator present relative to TNFα only controls, Xmin is the minimal observed fraction, and Xmax is the maximal observed fraction upon stimulation with TNFα. The Hill slope, k, and b are fit parameters. Mean values for the normalized concentration of each inflammatory mediator for each patient were used to determine patient-specific IC50, and average values across all patients were reported. Outlier values for each mediator were not used in determination of IC50. Goodness of fit was reported as the coefficient of determination, R2; and upper and lower bounds of the 95% confidence interval were graphically generated.
RESULTS
Human IVD cell response to TNFα
Across all patients, TNFα (1.4 nM; 25ng/ml) induced a very large and statistically significant increase in release of NO, PGE2, and IL6 (Table 1) from human primary IVD cells in vitro compared to controls cultured without TNFα, as expected. Control inflammatory mediator levels varied substantially amongst patient samples with individual sample responses to TNFα that ranged over 3 to 4 orders of magnitude for NO, PGE2, and IL6 (Figure 1 and Table 1). Patient-matched fold-change responses to TNFα were calculated to account for inter-patient variability (Table 1) and mean fold-changes across all patients were reported.
Table 1.
Increased release of NO, PGE2, and IL6 from IVD cells induced by TNFα
| NO (μM) | PGE2 (pg/ml) | IL6 (pg/ml) | ||||
|---|---|---|---|---|---|---|
| Control | TNF | Control | TNF | Control | TNF | |
| Median | 0.65 | 14.0 | 438 | 15,700 | 525 | 2210 |
| Mean ± SEM* | 0.58 ± 0.16 | 16.6 ± 2.36 | 476 ± 99.3 | 40500 ± 7520 | 556 ± 45.0 | 2290 ± 153 |
| Mean Fold Increase from Control | 60.0 | 112 | 4.12 | |||
A very large, statistically significant fold increase in NO, PGE2, and IL6 release occurred 72hr after addition of TNFα (25ng/ml; p < 10−4).
Calculated after removing outliers, as determined from box plot (Figure 1).
Figure 1.
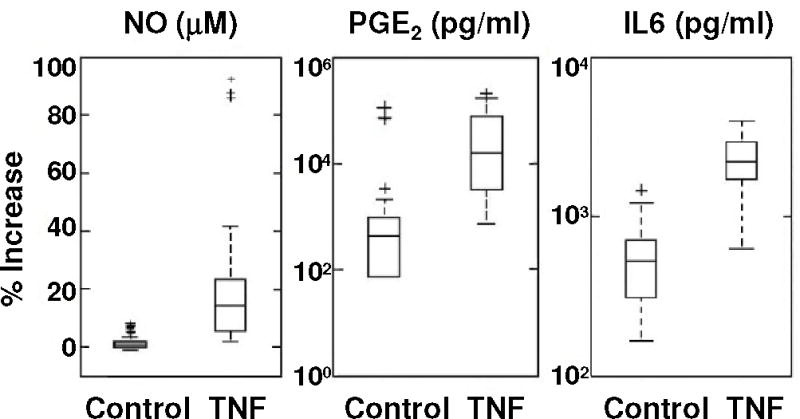
Box plots of NO, PGE2, and IL6 release for all patients, including outliers (+) (see methods). Patient responses to TNFα ranged over multiple log scales for all three mediators. See Table 1 for additional presentation.
Attenuation of TNFα-induced inflammatory events
Coincubation with sTNFRII significantly abated TNFα-induced release of NO and PGE2 from human IVD cells in a dose-dependent manner (Figure 2). In contrast, TNFα-induced release of IL6 was not attenuated by sTNFRII. Antagonist treatment groups were normalized to patient-matched TNFα only levels to account for variable absolute responses between patients. A directly proportional relationship between dose of sTNFRII and fractional attenuation of inflammatory mediator release was evident for NO and PGE2, with maximal attenuation fractions of 0.61 and 0.70, respectively, at the highest sTNFRII dose tested (100 nM).
Figure 2.
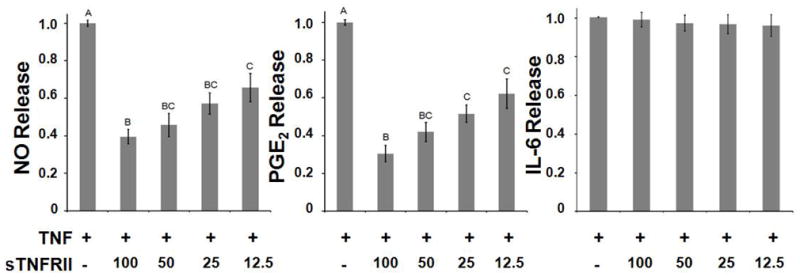
Dose-dependent attenuation of TNFα-induced release of NO (left; p < 10−10), PGE2 (middle; p < 10−3), and IL6 (right) after 72hr incubation with 25ng/ml TNFα averaged across all patients (n=15). Values are reported as mass fractions, normalized to TNFα only control values (mean ± SEM) (12.5–100 nM); groups without common letters are significantly different (one-way ANOVA with post-hoc Tukey’s HSD).
Using two-parameter logistic non-linear regression models, IC50 values for secreted NO and PGE2 were calculated for each patient (see Equation 1). Goodness-of-fit was generally excellent (R2 > 0.95). The mean IC50 doses of sTNFRII for NO and PGE2 release were found to be 35.1 ± 9.0 nM and 20.5 ± 9.0 nM, respectively (mean ± SEM). Representative data fits for patients with IC50 values near median values for NO and PGE2 are shown in Figure 3. These results indicate that relatively low molar excesses of sTNFRII substantially attenuated the proinflammatory effects of TNFα on human IVD cells in vitro.
Figure 3.
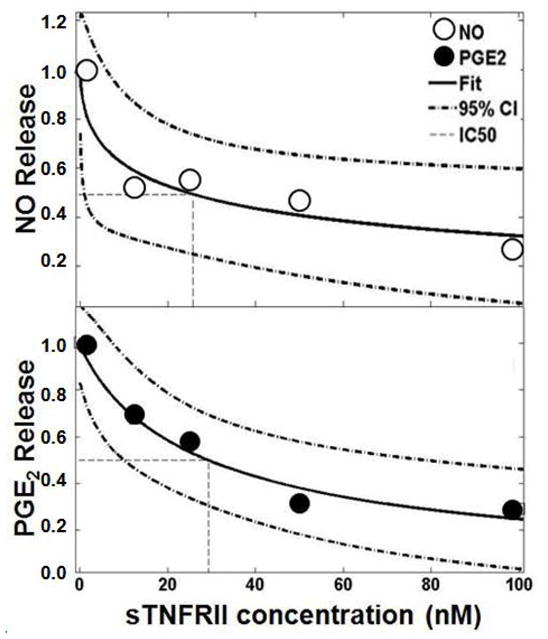
Logistic regression modeling of dose-dependent attenuation of NO and PGE2 release plotted against dose of sTNFRII. A representative data set is shown. IC50 values were calculated for each patient from fit parameters b and k, and a mean ± SEM was calculated. NO figure parameters (R2=0.95, IC50 = 24, b = 5.3, k = 0.52). PGE2 figure parameters (R2 = 0.97, IC50 = 29, b = 21, k = 0.90).
Attenuation of IL6 release followed an unexpected behavior for some patients. To investigate these results, sTNFRII (50 nM) without TNFα was also added to cells from 5 individual patients and cultured for the same period of time under the same conditions. Surprisingly, the presence of sTNFRII alone at a concentration of 50nM induced a significant release of IL6 (1.41 ± 0.04 fold-change; mean ± SEM) from human IVD cells (n=5), but did not affect NO or PGE2 release. This finding could explain the inability of sTNFRII to significantly moderate TNFα-induced release of IL6.
DISCUSSION
The primary objective of this study was to investigate an ability for sTNFRII to antagonize TNFα induced inflammatory events in primary human IVD cells in vitro. Across all patients, TNFα induced large, statistically significant increases in NO, PGE2, and IL6 secretion from IVD cells compared to controls. For the sample population tested, there was no apparent effect of patient age on release of NO, PGE2, or IL6. Coincubation of TNFα with nanomolar doses of sTNFRII significantly attenuated the secretion of NO and PGE2 in a dose-dependent manner. TNFα-induced secretion of IL6, however, was unchanged by the same concentrations of sTNFRII, a result that may be explained by a statistically significant induction of IL6 by sTNFRII without TNFα for a subset of patients. Attenuation data was fit to nonlinear regression models to calculate IC50 values for NO and PGE2 release for each patient. Mean IC50 values for NO and PGE2 were found to be 35.1 ± 9.0 nM and 20.5 ± 9.0 nM, respectively (mean ± SEM), indicating that nanomolar concentrations of sTNFRII were able to significantly attenuate the effects of TNFα on primary human IVD cells in vitro.
Pathologic IVD tissue is known to exhibit an inflammatory phenotype characterized by infiltrating innate and adaptive immune cells, along with their pro-inflammatory chemokine products. Herniated and degenerate IVD tissue contain significantly higher levels of TNFα[1,7], and TNFα is known to be a key mediator of pain and inflammation associated with radiculopathy following herniation[3,6,18,31,32]. Our observed trends of increased secretion of key inflammatory mediators, NO, PGE2, and IL6, from pathologic primary human IVD cells after exposure to TNFα corroborate previous reports of human nucleus pulposus (NP) cells cultured in alginate[14]. Studer et al.[14] challenged human NP cells in vitro with TNFα, at a lower dose of 5 ng/ml, and also saw significant increases in NO, PGE2, and IL6 secretion after 72 hours in culture. While induced secretion trends are similar to those reported here, differences in cell type, culture method, and dose of TNFα likely contribute to differences in absolute cell responses. The higher dose of TNFα, which was within the range of previous studies on IVD cell response to TNFα[2,14], was chosen to elicit a large enough inflammatory response such that attenuation with sTNFRII could be easily detected over a range of drug doses.
Quantitation of attenuation with sTNFRII revealed that low concentrations of sTNFRII were able to significantly attenuate TNFα effects, which further supports claims that targeting TNFα in IVD pathologies can reduce inflammation. The observed IC50 values for attenuating NO and PGE2 secretion with sTNFRII were roughly 20- to 30-fold higher than the supplemented concentration of TNFα, consistent with the IC50 range reported for inhibition of cell death for the L929 murine fibrosarcoma cell line (IC50 =6.6 nM sTNFRII, TNFα = 14 pM, or 470-fold molar excess) (Abcam)[41]. While anti-TNF antibodies have lower reported IC50 values than sTNFRI and sTNFRII in the same assay (Abcam), the smaller size of the soluble receptor-based drugs lends toward more facile synthesis, coupling to drug carrier vehicles, and more rapid clearance to minimize morbidity associated with delivery of these immunosuppresives. Indeed, incorporation of TNF antagonists into engineered, sustained release drug formulations may be a crucial aspect of the design of novel IVD therapeutics, in order to minimize the undesirable side effects of delivering potent TNF antagonists and other immunosuppresives systemically. Soluble TNF receptor-based therapies provide the benefits of high affinity and low molecular weight for designing locally delivered, sustained release systems for treating IVD pathologies[42,43].
TNFα is likely not the only mediator of pathology in the IVD, with some data indicating a more crucial role for IL-1β in initiating matrix catabolism and IVD degeneration[30]. However, the robust inflammatory response of pathologic IVD cells to TNFα reported herein supports previous findings[2,7,11,13,15,16] and motivates continued interest in targeting TNFα for treating IVD pathologies. In this study, nanomolar concentrations of sTNFRII conferred a potent antagonistic effect on TNFα-induced secretion of NO and PGE2 in vitro, but did not attenuate IL6 release. Interestingly, intracellular antagonism of TNFα with a p38 MAPK inhibitor[14] had a statistically significant effect on PGE2 and IL6, but had no effect on NO, whereas we observed effects on NO and PGE2, but not IL6 secretion, using an extracellular-acting antagonist. Blocking the TNFα signaling cascade before TNFR activation, as done here, would suggest that differences in intracellular signaling cascade upregulation for these mediators only partly contribute to differences in our findings for IL6. Rather, sTNFRII may be interacting with another membrane-bound component of pathologic IVD cells, such as membrane bound TNFα, and lead to the activation of cascades specific to IL6, but not NO or PGE2 upregulation. Future studies utilizing separate intracellular signaling pathway inhibitors may elucidate differences in regulatory mechanisms between NO, PGE2, and IL6, as well as the unexpected effect of sTNFRII on IL6. The effect of TNFα on extracellular matrix synthesis or protease expression for IVD cells was not measured in this study, but may be of tremendous interest in future studies as the effects of TNFα and sTNFRII on net catabolic activity as well as net matrix synthesis may be expected to differ.
NO, PGE2, and IL6 are known to have pro- and anti-inflammatory activities in osteoarthritis models and articular chondrocytes[44], but these cytokines’ exact roles in IVD pathologies require further definition. Herniated IVD specimens have been shown to produce NO, PGE2, and IL6[5,45], as well as a host of other inflammatory mediators[11,13], spontaneously in vitro or after stimulation with proinflammatory cytokines. Nitric oxide has been shown to mediate the change in proteoglycan synthesis in human IVD explants in response to hydrostatic pressure[46], as well as pain-related behavior in a rat model of radiculopathy[47]. PGE2 is a product of cyclooxygenase-2 (COX-2) activity, an enzyme implicated in the pathology of IVD herniation[48,49], and epidural injection of a COX-2 inhibitor for treating a rat radiculopathy model significantly alleviated mechanical allodynia[50]. IL6 may play a role in activating or attracting glial cells to sites of nerve injury[51], or may be a key inflammatory product of glial cells recruited to some nerve root injuries[52]. Some association between IL6 protein levels and painful manifestations of disc degeneration, but not normal disc physiology, has been demonstrated[11]. Interestingly, in one rat radiculopathy model, IL6 protein levels were observed to increase following local delivery of a pharmacologic cocktail of IL-1 receptor antagonist and sTNFRI[51]. This finding mirrors our own observations of increased IL6 induction by sTNFRII without TNFα by primary IVD cells and may imply a similar mechanism persists for other relevant cell types of this pathology. While the roles of NO, PGE2, and IL6 are not entirely clear, they each represent good measures of inflammation in response to TNFα. The substantially greater release of NO and PGE2 from our studies (Figure 1) suggests these two inflammatory mediators may be better readouts for evaluating new TNF antagonists in vitro.
CONCLUSION
Human primary IVD cells responded to TNFα in vitro by releasing large, statistically significant levels of NO, PGE2, and IL6 compared to controls. We report in vitro evidence of the effects of sTNFRII on pathologic IVD cells stimulated with TNFα, wherein nanomolar doses of sTNFRII were able to significantly attenuate TNFα-induced secretion of key inflammatory mediators. These findings highlight the importance of targeting TNFα in inflammatory pathologies of the IVD and emphasize the value of using soluble TNF receptors, and their analogues, to treat such pathologies. Any future TNF antagonists and related drug delivery formulations for treating IVD pathologies may benefit from the IC50 data reported herein as a metric for evaluating new drug performance and bioactivity before proceeding to pre-clinical animal models.
Acknowledgments
This work was funded by the NIH (R01AR047442, P01AR050245, R01AR057410) and the Duke BME Howard Clark Pre-Doctoral Fellowship.
References
- 1.Le Maitre C, Hoyland J, Freemont A. Catabolic cytokine expression in degenerate and herniated human intervertebral discs: IL-1 and TNF expression profile. Arthritis Research & Therapy. 2007;9:R77. doi: 10.1186/ar2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Séguin CA, Pilliar RM, Roughley PJ, Kandel RA. Tumor necrosis factor [alpha] modulates matrix production and catabolism in nucleus pulposus tissue. Spine. 2005;30:1940. doi: 10.1097/01.brs.0000176188.40263.f9. [DOI] [PubMed] [Google Scholar]
- 3.Olmarker K, Rydevik B. Selective inhibition of tumor necrosis factor-[alpha] prevents nucleus pulposus-induced thrombus formation, intraneural edema, and reduction of nerve conduction velocity: possible implications for future pharmacologic treatment strategies of sciatica. Spine. 2001;26:863. doi: 10.1097/00007632-200104150-00007. [DOI] [PubMed] [Google Scholar]
- 4.Peng B, Hao J, Hou S, Wu W, Jiang D, Fu X, Yang Y. Possible pathogenesis of painful intervertebral disc degeneration. Spine. 2006;31:560. doi: 10.1097/01.brs.0000201324.45537.46. [DOI] [PubMed] [Google Scholar]
- 5.Kang JD, Georgescu HI, McIntyre-Larkin L, Stefanovic-Racic M, Donaldson WF, III, Evans CH. Herniated lumbar intervertebral discs spontaneously produce matrix metalloproteinases, nitric oxide, interleukin-6, and prostaglandin E2. Spine. 1996;21:271. doi: 10.1097/00007632-199602010-00003. [DOI] [PubMed] [Google Scholar]
- 6.Olmarker K, Larsson K. Tumor Necrosis Factor [alpha] and Nucleus-Pulposus-Induced Nerve Root Injury. Spine. 1998;23:2538. doi: 10.1097/00007632-199812010-00008. [DOI] [PubMed] [Google Scholar]
- 7.Weiler C, Nerlich AG, Bachmeier BE, Boos N. Expression and distribution of tumor necrosis factor alpha in human lumbar intervertebral discs: a study in surgical specimen and autopsy controls. Spine. 2005;30:44. doi: 10.1097/01.brs.0000149186.63457.20. [DOI] [PubMed] [Google Scholar]
- 8.Nygaard Ø, Mellgren SI, Østerud B. The inflammatory properties of contained and noncontained lumbar disc herniation. Spine. 1997;22:2484. doi: 10.1097/00007632-199711010-00004. [DOI] [PubMed] [Google Scholar]
- 9.Haro H, Shinomiya K, Komori H, Okawa A, Saito I, Miyasaka N, Furuya K. Upregulated expression of chemokines in herniated nucleus pulposus resorption. Spine. 1996;21:1647. doi: 10.1097/00007632-199607150-00006. [DOI] [PubMed] [Google Scholar]
- 10.Matsui Y, Maeda M, Nakagami W, Iwata H. The involvement of matrix metalloproteinases and inflammation in lumbar disc herniation. Spine. 1998;23:863. doi: 10.1097/00007632-199804150-00005. [DOI] [PubMed] [Google Scholar]
- 11.Burke JG, Watson RWG, McCormack D, Dowling FE, Walsh MG, Fitzpatrick JM. Intervertebral discs which cause low back pain secrete high levels of proinflammatory mediators. Journal of Bone & Joint Surgery, British Volume. 2002;84:196–201. doi: 10.1302/0301-620x.84b2.12511. [DOI] [PubMed] [Google Scholar]
- 12.Demircan MN, Asir A, Cetinkal A, Gedik N, Kutlay AM, Colak A, Kurtar S, Simsek H. Is there any relationship between proinflammatory mediator levels in disc material and myelopathy with cervical disc herniation and spondylosis? A non-randomized, prospective clinical study. European Spine Journal. 2007;16:983–986. doi: 10.1007/s00586-007-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burke JG, Watson RWG, Conhyea D, McCormack D, Dowling FE, Walsh MG, Fitzpatrick JM. Human nucleus pulposis can respond to a pro-inflammatory stimulus. Spine. 2003;28:2685. doi: 10.1097/01.BRS.0000103341.45133.F3. [DOI] [PubMed] [Google Scholar]
- 14.Studer RK, Aboka AM, Gilbertson LG, Georgescu H, Sowa G, Vo N, Kang JD. p38 MAPK inhibition in nucleus pulposus cells: a potential target for treating intervertebral disc degeneration. Spine. 2007;32:2827. doi: 10.1097/BRS.0b013e31815b757a. [DOI] [PubMed] [Google Scholar]
- 15.Séguin CA, Bojarski M, Pilliar RM, Roughley PJ, Kandel RA. Differential regulation of matrix degrading enzymes in a TNF -induced model of nucleus pulposus tissue degeneration. Matrix Biology. 2006;25:409–418. doi: 10.1016/j.matbio.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Séguin CA, Pilliar RM, Madri JA, Kandel RA. TNF-[alpha] Induces MMP2 Gelatinase Activity and MT1-MMP Expression in an In Vitro Model of Nucleus Pulposus Tissue Degeneration. Spine. 2008;33:356. doi: 10.1097/BRS.0b013e3181642a5e. [DOI] [PubMed] [Google Scholar]
- 17.Abe Y, Akeda K, An HS, Aoki Y, Pichika R, Muehleman C, Kimura T, Masuda K. Proinflammatory cytokines stimulate the expression of nerve growth factor by human intervertebral disc cells. Spine. 2007;32:635. doi: 10.1097/01.brs.0000257556.90850.53. [DOI] [PubMed] [Google Scholar]
- 18.Aoki Y, Rydevik B, Kikuchi S, Olmarker K. Local application of disc-related cytokines on spinal nerve roots. Spine. 2002;27:1614. doi: 10.1097/00007632-200208010-00004. [DOI] [PubMed] [Google Scholar]
- 19.Igarashi T, Kikuchi S, Shubayev V, Myers RR. Exogenous tumor necrosis factor-alpha mimics nucleus pulposus-induced neuropathology: molecular, histologic, and behavioral comparisons in rats. Spine. 2000;25:2975. doi: 10.1097/00007632-200012010-00003. [DOI] [PubMed] [Google Scholar]
- 20.Goupille P, Mulleman D, Chevalier X. Is interleukin-1 a good target for therapeutic intervention in intervertebral disc degeneration: lessons from the osteoarthritic experience. Arthritis Research & Therapy. 2007;9:110. doi: 10.1186/ar2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korhonen T, Karppinen J, Paimela L, Malmivaara A, Lindgren KA, Bowman C, Hammond A, Kirkham B, Järvinen S, Niinimäki J. The treatment of disc herniation-induced sciatica with infliximab: one-year follow-up results of FIRST II, a randomized controlled trial. Spine. 2006;31:2759. doi: 10.1097/01.brs.0000245873.23876.1e. [DOI] [PubMed] [Google Scholar]
- 22.Genevay S, Stingelin S, Gabay C. Efficacy of etanercept in the treatment of acute, severe sciatica: a pilot study. Annals of the Rheumatic Diseases. 2004;63:1120–1123. doi: 10.1136/ard.2003.016451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen SP, Bogduk N, Dragovich A, Buckenmaier CC, III, Griffith S, Kurihara C, Raymond JL, Richter PJ, Williams N, Yaksh TL. Randomized, double-blind, placebo-controlled, dose-response, and preclinical safety study of transforaminal epidural etanercept for the treatment of sciatica. Anesthesiology. 2009;110:1116. doi: 10.1097/ALN.0b013e3181a05aa0. [DOI] [PubMed] [Google Scholar]
- 24.Tobinick E, Davoodifar S. Efficacy of etanercept delivered by perispinal administration for chronic back and/or neck disc-related pain: a study of clinical observations in 143 patients. Current Medical Research and Opinion®. 2004;20:1075–1085. doi: 10.1185/030079903125004286. [DOI] [PubMed] [Google Scholar]
- 25.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Han J. Cellular responses to tumor necrosis factor. Current Issues in Molecular Biology. 2001;3:79–90. [PubMed] [Google Scholar]
- 27.Akeda K, An HS, Gemba T, Okuma M, Miyamoto K, Chujyo T, Kitahara S, Masuda K. P89 The intra-discal injection of naked NF [kappa] B decoy oligonucleotide is effective in restoring disc degeneration in the rabbit annular needle puncture model. The Spine Journal. 2005;5:S152–S153. [Google Scholar]
- 28.Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. Journal of Pharmacology and Experimental Therapeutics. 2003;306:624. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- 29.Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 30.Hoyland JA, Le Maitre C, Freemont AJ. Investigation of the role of IL-1 and TNF in matrix degradation in the intervertebral disc. Rheumatology. 2008;47:809. doi: 10.1093/rheumatology/ken056. [DOI] [PubMed] [Google Scholar]
- 31.Olmarker K, Nutu M, Størkson R. Changes in spontaneous behavior in rats exposed to experimental disc herniation are blocked by selective TNF-alpha inhibition. Spine. 2003;28:1635–1642. doi: 10.1097/01.BRS.0000083162.35476.FF. [DOI] [PubMed] [Google Scholar]
- 32.Onda A, Yabuki S, Kikuchi S. Effects of neutralizing antibodies to tumor necrosis factor-alpha on nucleus pulposus-induced abnormal nociresponses in rat dorsal horn neurons. Spine. 2003;28:967. doi: 10.1097/01.BRS.0000061984.08703.0C. [DOI] [PubMed] [Google Scholar]
- 33.Chen PCH, DuBois GC, Chen MJ. Mapping the domain (s) critical for the binding of human tumor necrosis factor-alpha to its two receptors. Journal of Biological Chemistry. 1995;270:2874. doi: 10.1074/jbc.270.6.2874. [DOI] [PubMed] [Google Scholar]
- 34.Engelmann H, Novick D, Wallach D. Two tumor necrosis factor-binding proteins purified from human urine. Evidence for immunological cross-reactivity with cell surface tumor necrosis factor receptors. Journal of Biological Chemistry. 1990;265:1531–1536. [PubMed] [Google Scholar]
- 35.Hale KK, Smith CG, Baker SL, Vanderslice RW, Squires CH, Gleason TM, Tucker KK, Kohno T, Russell DA. Multifunctional regulation of the biological effects of TNF-by the soluble type I and type II TNF receptors. Cytokine. 1995;7:26–38. doi: 10.1006/cyto.1995.1004. [DOI] [PubMed] [Google Scholar]
- 36.Edwards CK. PEGylated recombinant human soluble tumour necrosis factor receptor type I (r-Hu-sTNF-RI): novel high affinity TNF receptor designed for chronic inflammatory diseases. British Medical Journal. 1999;58:73–81. doi: 10.1136/ard.58.2008.i73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abraham E, Laterre PF, Garbino J, Pingleton S, Butler T, Dugernier T, Margolis B, Kudsk K, Zimmerli W, Anderson P. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: A randomized, double-blind, placebo-controlled, multicenter phase III trial with 1,342 patients. CRITICAL CARE MEDICINE-BALTIMORE- 2001;29:503–510. doi: 10.1097/00003246-200103000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Peppel K, Crawford D, Beutler B. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. Journal of Experimental Medicine. 1991;174:1483–1489. doi: 10.1084/jem.174.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baer AE, Wang JY, Kraus VB, Setton LA. Collagen gene expression and mechanical properties of intervertebral disc cell-alginate cultures. Journal of Orthopaedic Research. 2001:19. doi: 10.1016/S0736-0266(00)00003-6. [DOI] [PubMed] [Google Scholar]
- 40.Miles AM, Wink DA, Cook JC, Grisham MB. Determination of nitric oxide using fluorescence spectroscopy. Methods in enzymology. 1996;268:105–120. doi: 10.1016/s0076-6879(96)68013-6. [DOI] [PubMed] [Google Scholar]
- 41.Tomkins PKC, Webber D, Bowen G. The L929 cell bioassay for murine tumour necrosis factor is not influenced by other murine cytokines. J Immunol Methods. 1992;151:313–315. doi: 10.1016/0022-1759(92)90133-e. [DOI] [PubMed] [Google Scholar]
- 42.Shamji MF, Chen J, Friedman AH, Richardson WJ, Chilkoti A, Setton LA. Synthesis and characterization of a thermally-responsive tumor necrosis factor antagonist. Journal of Controlled Release. 2008;129:179–186. doi: 10.1016/j.jconrel.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shamji MF, Whitlatch L, Friedman AH, Richardson WJ, Chilkoti A, Setton LA. An injectable and in situ-gelling biopolymer for sustained drug release following perineural administration. Spine. 2008;33:748. doi: 10.1097/BRS.0b013e3181695773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loeser RF. Molecular mechanisms of cartilage destruction: mechanics, inflammatory mediators, and aging collide. Arthritis and rheumatism. 2006;54:1357. doi: 10.1002/art.21813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang JD, Stefanovic-Racic M, McIntyre LA, Georgescu HI, Evans CH. Toward a biochemical understanding of human intervertebral disc degeneration and herniation: contributions of nitric oxide, interleukins, prostaglandin E2, and matrix metalloproteinases. Spine. 1997;22:1065. doi: 10.1097/00007632-199705150-00003. [DOI] [PubMed] [Google Scholar]
- 46.Liu GZ, Ishihara H, Osada R, Kimura T, Tsuji H. Nitric oxide mediates the change of proteoglycan synthesis in the human lumbar intervertebral disc in response to hydrostatic pressure. Spine. 2001;26:134. doi: 10.1097/00007632-200101150-00005. [DOI] [PubMed] [Google Scholar]
- 47.Kawakami M, Tamaki T, Hashizume H, Weinstein JN, Meller ST. The role of phospholipase A2 and nitric oxide in pain-related behavior produced by an allograft of intervertebral disc material to the sciatic nerve of the rat. Spine. 1997;22:1074. doi: 10.1097/00007632-199705150-00004. [DOI] [PubMed] [Google Scholar]
- 48.Miyamoto H, Saura R, Harada T, Doita M, Mizuno K. The role of cyclooxygenase-2 and inflammatory cytokines in pain induction of herniated lumbar intervertebral disc. Kobe Journal of Medical Sciences. 2000;46:13–28. [PubMed] [Google Scholar]
- 49.Miyamoto H, Saura R, Doita M, Kurosaka M, Mizuno K. The role of cyclooxygenase-2 in lumbar disc herniation. Spine. 2002;27:2477. doi: 10.1097/00007632-200211150-00011. [DOI] [PubMed] [Google Scholar]
- 50.Kawakami M, Matsumoto T, Hashizume H, Kuribayashi K, Tamaki T. Epidural injection of cyclooxygenase-2 inhibitor attenuates pain-related behavior following application of nucleus pulposus to the nerve root in the rat. Journal of Orthopaedic Research. 2002;20:376–381. doi: 10.1016/S0736-0266(01)00114-0. [DOI] [PubMed] [Google Scholar]
- 51.Winkelstein BA, Rutkowski MD, Sweitzer SM, Pahl JL, DeLeo JA. Nerve injury proximal or distal to the DRG induces similar spinal glial activation and selective cytokine expression but differential behavioral responses to pharmacologic treatment. The Journal of Comparative Neurology. 2001;439:127–139. [PubMed] [Google Scholar]
- 52.Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Experimental Neurology. 1999;157:289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]