

Abstract
Autophagy is essential for prolonging yeast survival during nutrient deprivation; however, this report shows that some autophagy proteins may also be accelerating population death in those conditions. While leucine starvation caused YCA1-mediated apoptosis characterized by increased annexin V staining, nitrogen deprivation triggered necrotic death characterized by increased propidium iodide uptake. Although a Δatg8 strain died faster than its parental strain during nitrogen starvation, this mutant died slower than its parent during leucine starvation. Conversely, a Δatg11 strain died slower than its parent during nitrogen starvation, but faster during leucine starvation. Curiously, although GFP-Atg8 complemented the Δatg8 mutation, this protein made ATG8 cells more sensitive to nitrogen starvation, and less sensitive to leucine starvation. These results were difficult to explain if autophagy only extended life but could be an indication that a second form of autophagy could concurrently facilitate either apoptotic or necrotic cell death.
Keywords: Apoptosis, Atg8, autophagy, CVT, necrosis, programmed cell death, starvation, yeast
Introduction
Studies of yeast have distinguished two sequences of events leading to growth arrest and to death. One sequence, typified by the process induced when yeast have been deprived of polyamines, culminates with the breakup of the plasma membrane so that previously excluded dyes like propidium iodide (PI) are able to enter the cell.1 Although some PI+ cells can repair this damage and survive if stress is removed,2 most cells that begin to take up PI continue to deteriorate until they succumb to necrosis. Other treatments including exposure to low levels of H2O2, produce a more complex series of changes involving chromatin condensation, DNA breakage,3 and membrane rearrangements that expose phosphatidylserine to exogenously provided annexin V.4 These events herald apoptotic death, but in most cases, cellular degeneration continues as cells undergo secondary necrosis.5
In the literature there is also a third pathway called autophagic cell death (ACD). Autophagy was initially defined as the cell’s primary response to nutrient deprivation or to the pharmacological agent, rapamycin. When these treatments set autophagy in motion, vesicles called autophagosomes harvested cytoplasmic proteins and organelles and delivered them to vacuoles or lysosomes to be disassembled into amino acids and lipids useful for preserving life until external nutrients are restored. However, in a handful of cases,6-8 these autophagosomes were the only visible responses to stress prior to the demise of the cell.9 Some have inferred that such cells, lacking any of the hallmarks of apoptosis or necrosis, died from excessive, nonspecific harvesting of organelles and proteins that compromised their ability to maintain essential homeostatic processes. However, persuasive arguments have been made that death might not have resulted from increased autophagic activity, but because autophagic activity failed to prevent it.10,11 For many, the resolution of this debate hinges on whether or not cells with defective autophagy genes are able to survive conditions that kill normal cells.12
A study using yeast recently presented evidence for just such a case.13 Cells treated with 13 mM Zn2+ died necrotically unless any one of seven autophagy genes was inactivated. At the same time, inactivation of other autophagy genes accelerated cell death, while the inactivation of the remaining genes had no effect on survival at all. Based on the mutants’ phenotypes, it was suggested that the autophagic proteins could be sorted into four classes representing four combinatorial modules. When acting on their own, two of these modules performed necrotic cell death. When joined with a third module, the machinery performed starvation-induced, nonselective autophagy. Finally, when all of the modules were functionally joined, autophagy harvested a few proteins selectively including the vacuolar protein aminopeptidase 1 and delivered them to the yeast vacuole. Yet, despite this evidence that autophagic proteins played an active part in cell death, the dying cells did not harvest typical reporters of autophagic activity like ROSELLA14 or Rpl25-GFP15 that would have been expected if indiscriminate autophagy caused ACD. It appeared instead that a selective autophagic process that could not be tracked with any of the tested autophagy reporters enabled cells to go necrotic.
This was by no means the first evidence indicating that autophagy contributed to necrotic cell death. Samara et al. found that the loss of the C. elegans gene homologous to ATG1 reduced the number of cells dying necrotically.16 More recently, Shin et al. found that inhibiting autophagy inhibited the necrotic death of macrophages infected with a Mycoplasm tuberculosis mutant.17 In at least one study, inhibiting the mouse equivalents of the yeast genes ATG7 and ATG8 blocked autophagic harvesting of catalase, and by doing so, prevented necrotic death resulting from a catastrophic increase in reactive oxygen-caused damage.18 The contribution of autophagy to zinc-induced necrotic cell death (ziNCD) in yeast13 might therefore not be as unusual as it first seems, but merely an extreme example of the cell’s response to a number of lethal treatments.
Most of what we now understand about autophagy began with studies of how the process manifested itself during nitrogen starvation.19,20 We therefore proceeded to test whether the phenotypic differences between mutants during ziNCD correlated with phenotypic differences during nitrogen starvation, and with the less understood response to leucine starvation. Although the two forms of starvation might be expected to cause similar forms of damage, they, in fact, have been previously shown to elicit very different responses. Thus, leucine-starved cells accumulate almost as many autophagosomes as cells starved for all nitrogen and amino acid sources,19 yet based on the vacuole-dependent processing of GFP-Atg8 and pApe1, and on the upregulation of Atg4 and Atg8, amino acid-starved cells autophagically process less protein than nitrogen-starved ones.21 Defective autophagy may account for the observation that leucine-starved cells lose colony-forming ability faster22 than nitrogen-starved ones.23
The present study found additional ways that the two forms of starvation differed from each other. We show that, like zinc treatments, nitrogen starvation caused the vast majority of cells to become membrane-permeable to propidium iodine (PI), a trait associated with primary necrosis. Leucine-starved populations, on the other hand, consisted of a mixture of cells. Some only accumulated PI, some only stained with annexin V (a phenotype associated with early apoptosis), and some stained with both, like apoptotic cells undergoing secondary necrosis,5 or nonapoptotic cells undergoing severe necrosis.24 Leucine-starved populations failed to harvest autophagic reporter proteins efficiently yet at least some autophagic gene knockout mutations that extended the life of zinc-treated cells, extended the life of leucine-starved ones. Despite these contributions by autophagy proteins, we found no evidence for a unique form of death attributable to ACD. Based on these studies, it was concluded that autophagic processes aided both apoptotic and necrotic death, but did not bias which death pathway was used during each stress.
Results and Discussion
The loss of ATG8 and ATG11 had opposite effects on cell survival during leucine and nitrogen starvations
Previous studies showed that autophagic mutants displayed one of three different phenotypes when grown on excess zinc.13 Some mutants like Δatg29 had no effect on zinc tolerance. Others like Δatg8 were more resistant to zinc than the parental strain, while a handful like Δatg11 were more sensitive. These studies and others led to the suggestion that autophagic proteins participated in competing processes, some responsible for extending life, and some able to shorten it. The current set of experiments indicated that the colony-forming ability (CFA) of Δatg8 cells harboring the URA3 plasmid, YEp25 declined rapidly during nitrogen starvation while Δatg11 cells with the same vector maintained their viability longer than the other lines for the first 3 d (Fig. 1A). During leucine starvation, these phenotypes were reversed (Fig. 1D). In fact, the initial responses of Δatg8 and Δatg11 toward leucine starvation resembled the responses of these two mutants to zinc treatment, with the notable difference being that all genotypes eventually succumbed to amino acid deprivation whereas Δatg8 cells formed colonies in the presence of 13 mM Zn2+. One explanation for such phenotypes was that these autophagy proteins participated in two diametrically opposed pathways of which one reduced CFA.

Figure 1. Autophagy mutants did not respond uniformly to nutrient deprivation. (A and D) BY4741 and its derivatives bearing deletions of the indicated genes were transformed with a vector, YEp,25 and grown in SD medium to an OD600nm of approximately 0.4, harvested, washed, and then diluted to 0.04 OD600nm using either SD-leucine (SD-L) or SD-nitrogen sources (SD-N) as described in Materials and Methods. Samples were removed every 12 h (SD-L) or 72 h (SD-N), plated onto YPD medium and incubated 2 d at 30°C to assay for survivors. This recovery was normalized to the number of survivors at 0 h. Each point is the average and standard deviation of 3–4 experiments. (B–C and E–F) Cells were removed from each culture after 12 h (SD-L) and 24 h (SD-N) starvation, cell wall-permeabilized, treated simultaneously with PI and annexin V, and viewed microscopically at 520 nm (annexin-FITC) and 620 nm (PI).13 Each value is the average percentage (and standard deviation) of stained cells within a population of 100–200 cells based on 3–4 independent experiments. Results are grouped into two sets: the black bar shows cells staining with annexin V alone (interpreted as undergoing apoptosis), and the white and gray bars show cells staining with PI or PI and annexin (interpreted as undergoing different forms of necrosis). *The CFA (the percentage of cells surviving relative to the initial cell number) at the time of sampling is shown below each mutant. The classification noted at the top of (C and F) indicates how the CFA of each strain was scored 13 when grown on 13 mM Zn2+.
Autophagy mutations accelerated both necrotic and apoptotic modes of death
Previous studies reported that zinc-treated cells predominantly died through necrosis, based on PI accumulation, on their failure to bind annexin V, and on the fact that survival was not affected by the loss of any one of four genes involved in apoptosis.13 Even before the colony-forming ability of the population of nitrogen-starved cells began to decline (Fig. 1A), the percentage of PI-accumulating cells began to increase (Fig. S1). By comparison, leucine-starved populations of PS cells showed a complex spectrum of phenotypes after only 3 h (Fig. S1). At that time, some cells took up PI, but more than twice as many bound annexin V. By 12 h, an even larger number stained with both treatments (Fig. 1E). In this study we could not determine whether cells that stained with both dyes initiated a mixed cell death response from the very beginning of the stress,24 or instead deteriorated through an apoptotic process known as secondary necrosis26 that generated membrane holes large enough to let annexin V (MW = 35.8 kDa) as well as PI (MW = 0.67 kDa) enter. Mutational studies did little to distinguish between these alternatives. While the deletion of ATG8 had little effect on the number of cells initially undergoing primary necrosis, and instead reduced the number of cells staining with annexin V, or PI and annexin V (Fig. 1E), the deletion of ATG11 had its greatest effect on the number of cells staining with PI or with both reagents. This observation, that autophagic mutations had their greater impact on necrotic death in one case, and apoptotic death in another, implied that these proteins were not wedded to either cell death process, nor part of a unique death pathway on their own. Instead, the operation of these proteins impartially accelerated whichever mechanism killed cells during each particular stress.
We then extended this comparison of zinc-induced and leucine starvation-induced death to include other mutants. During leucine-starvation, each strain was monitored at the time when PS cells showed a 30% loss in CFA (Fig. 1). However, rather than focusing on the change in CFA over time, we analyzed the pathway by which the mutants died based on PI uptake and annexin V binding at a single time point. For the most part, the additionally tested autophagic mutants responded as they did on zinc-supplemented media. Two knockout mutations that reduced necrotic death during zinc treatment (Δatg7 and Δatg15), delayed the loss of CFA and reduced annexin V staining during leucine starvation. One that had no effect on zinc tolerance (Δatg19), had no effect on survival during leucine starvation. Yet there were notable exceptions to this correlation. The loss of the yeast metacaspase gene, YCA1, had no effect on zinc-induced necrotic cell death, but increased CFA from the PS’s 69% ± 4 viable cells to the Δyca1 CFA of 93% ± 3 during leucine-starvation induced apoptotic death (Fig. 1F). In addition, Δtor1 and Δatg29 mutants behaved like the parental strain on zinc-rich medium, but like Δatg8 during leucine starvation suggesting, perhaps, that nutrient recycling was only relevant to surviving the latter stress.
Different pro-life processes operated during leucine and nitrogen starvation
The responses of mutants to nitrogen-starvation were generally mirror-images of their responses to the other stresses. Whereas leucine-starved cells died quickly over the course of hours (Fig. 1D), cells that were concurrently deprived of all other potential nitrogen donors including leucine, died slowly over the course of days (Fig. 1A). While the Δatg11 mutation accelerated death during leucine starvation (Fig. 1A), this mutation sustained population growth during the first few days of nitrogen starvation (Fig. 1D). Further studies indicated Δatg7, Δatg19 and Δyca1 mutations neither benefited nor harmed nitrogen starved cells during the first 24 h (Fig. 1C). Consideration of all these phenotypes suggested that the kind of autophagy that sustained life during zinc treatment and leucine starvation worked against the survival of PS cells during the onset of nitrogen starvation. After day 3, this pro-death activity was either suppressed or overshadowed by the contribution of Atg11 (as well as Atg8, 15 and 29) to survival.
Nitrogen-starved cells were also assayed for PI accumulation and annexin V staining. As was done to study leucine starvation, we wanted to produce a snapshot of the initial responses to stress and so assayed phenotypes after 24 h, when most members of populations of mutants like Δatg8 were still viable. At this time, most cell death was accompanied by necrosis (Fig. 1B and C). This PI+ phenotype continued to predominate at 6 d (Fig. S1), when the CFA of PS cells had dropped to 86 ± 0.9% (Fig. 1A). At 6 d, this PI+ phenotype also dominated populations of starving Δatg11 cells (PI+ cells made up 15 ± 1.1%, PI+/ annexin V+ made up 2.5 ± 0.7%, and annexin V+ ones made up 0.9 ± 0.2% of the population; data not shown). The fact that PI+/ annexin V+ cells remained scarce throughout this time course suggested that the onset of apoptosis or severe necrosis was being delayed because cells were mounting a more successful response to nitrogen-starvation than they were able to mount against leucine starvation, possibly because of the way that autophagy was being performed during each stress.
Nitrogen starvation-induced autophagy (also known as macroautophagy) has been the model for uncovering most of what we know about the contribution that this process makes to cell survival.27 This pathway is negatively regulated by the TOR complex.28 Neither the deletion of TOR1 (Fig. 1C), nor simultaneous treatment with rapamycin (Table 1), a chemical inhibitor of Tor1,28 affected cell death during nitrogen starvation indicating that the kinase was already fully inhibited by nitrogen starvation. On the other hand, both the deletion of TOR1 and treatments with rapamycin halved the rate of death during leucine starvation (Fig. 1F and Table 1). This confirmed what others have noted:21 leucine-starved cells survive longer if nonselective autophagy is induced, but are unable to induce this form of autophagy effectively without pharmacological help.
Table 1. Rapamycin reduces the symptoms of death during leucine starvation.
| |
SD-N |
SD-L |
||
|---|---|---|---|---|
| Rapamycin | − | + | − | + |
| PS::YEp |
0.7 ± 0.5 |
1.0 ± 0.4 |
8.9 ± 1.4 |
3.0 ± 1.0 |
| |
4.0 ± 1.2 |
5.0 ± 1.0 |
17.7 ± 2 |
8.0 ± 1.5 |
|
Δatg8::YEp |
0.5 ± 0.3 |
0.8 ± 0.7 |
4.0 ± 1.1 |
2.0 ± 0.5 |
| |
12.6 ± 3 |
14.2 ± 1 |
9.3 ± 1.9 |
5.0 ± 1.0 |
|
Δatg11::YEp |
1.1 ± 0.6 |
0.8 ± 0.3 |
10 ± 2.5 |
8.0 ± 2.3 |
| |
4.0 ± 1.3 |
4.0 ± 0.8 |
35.6 ± 4 |
18.4 ± 2 |
|
Δatg29::YEp |
0.8 ± 0.3 |
0.5 ± 0.4 |
3.9 ± 0.7 |
4.1 ± 1.0 |
| 13.5 ± 3 | 12.1 ± 2 | 8.7 ± 2.1 | 7.6 ± 2.5 | |
The table shows the percent of cells (± standard deviation) staining with annexin V (upper rows) or with PI and both annexin V and PI (lower rows) for PS and mutant cultures starved for nitrogen for 24 h, or leucine for 12 h with (+) and without (−) 0.22 μM rapamycin as indicated. Each experiment was repeated 3 times. Note that Δatg29::YEp did not respond to rapamycin during this treatment and that rapamycin did not reduce apoptotic death in Δat11::YEp cells.
Rapamycin treatments provided further evidence that autophagy proteins operated differently during the two deprivation conditions. As shown in Table 1, the life-extending process induced during nitrogen starvation and rapamycin treatment depended upon Atg8 and Atg29, and not on Atg11. This verified that the pro-life process operating throughout this treatment was almost certainly synonymous with canonical macroautophagy.27 In contrast, the life-extending process that was activated during leucine starvation to prevent PI accumulation depended on Atg11 suggesting that this type of autophagy operated selectively,29 like the one that protected cells from excess zinc.13 Also like zinc-stressed cells, leucine-starved ones appeared to simultaneously carry out a death-promoting process, although in this case, one dependent not only on Atg8, but also on Atg29. Both proteins are considered vital for macroautophagy.29
The autophagy reporter protein GFP-Atg8 perturbed cell death during starvation
Unexpectedly, independent evidence for the existence of competing autophagic pathways came from studies employing a common reporter protein, GFP-Atg8, under the control of the native ATG8 promoter.30 This reporter is incorporated into autophagosomes, and processed in the vacuole to release free GFP and functional Atg8.30 Before using this construct to monitor autophagy, we performed a routine analysis to verify that the additional Atg8 would not affect the cell death processes of the transformants. Surprisingly, it did. During nitrogen starvation, a parental strain with the reporter developed 2.5-fold more PI+, and fewer surviving, cells than a strain with an unaugmented vector (Fig. 2). Conversely, leucine-starved PS cells expressing GFP::Atg8 showed 25% less PI staining, and 25% more CPA, than cells carrying the vector alone. We then assayed Δatg8 cells with the same construct. Here, the reporter merely returned both cell necrosis and cell survival to PS levels indicating that the chimeric protein compensated for the loss of native Atg8. If Atg8 was involved in a single process, it would be difficult to accommodate the similar effects that adding GFP-Atg8 and deleting the endogenous gene had on cell survival. An alternative interpretation for what was found was that excess Atg8 protein, coming from both the original and the introduced genes, reduced competition for a rate-limiting molecule needed by two or more opposing forms of autophagy. Leveling this playing field, in turn, benefited a life-promoting pathway in leucine-starved cells, and a death-promoting process in nitrogen-starved ones.
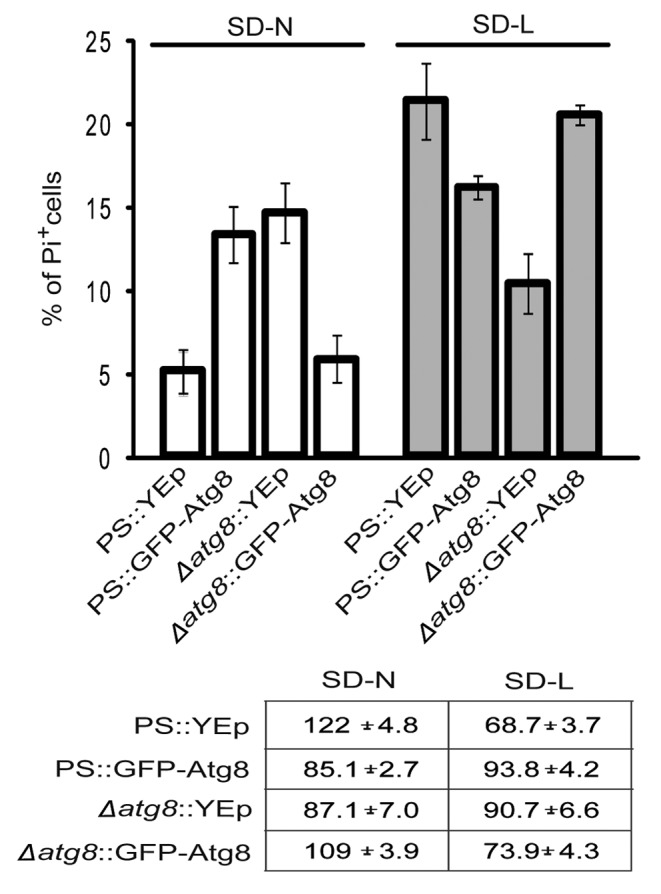
Figure 2. Expressing GFP-Atg8 in PS cells changed the percentages of cells dying during starvation. PS and Δatg8 cells were transformed with YEp or a plasmid expressing GFP-Atg8, starved and stained with PI, and scored. Because of the presence of GFP, it was not possible to score for annexin V-staining cells in these experiments. Each average (and standard deviation) was derived from 3–4 independent experiments, each counting 100–200 cells. The table shows the CFA of each strain at the time of sampling.
Despite genetic and biochemical analyses indicating that at least two forms of autophagy operated in GFP-Atg8 expressing cells, we could not detect changes in the localization of the reporter in the Δatg8 strain (Fig. 3). While nitrogen-starved cells acquired green-fluorescing vacuoles (Fig. 3A) and showed enhanced GFP-Atg8 processing (Fig. 3B), the vacuoles of leucine-starved ones were nearly as dark as those in cells during normal growth (Fig. 3A). The appearance of these vacuoles differed from those in a previous report.21 However, those studies were performed in an ATG8 strain. In PS cells with that genotype, we also found fluorescence accumulated in vacuoles during leucine-starvation (Fig. S2). In fact, had we only analyzed the PS strain, we might have concluded that leucine-starvation had induced macrophagy. After considering the phenotypes of Δatg8 cells, a more nuanced explanation might be that the combined Atg8 and GFP-Atg8 level artificially augmented the pro-life pathway, allowing it either to eliminate mistranslated or misfolded proteins, or to recycle the limiting amino acid more quickly.

Figure 3. GFP-Atg8 was not harvested or processed by Δatg8 cells during leucine starvation. (A) The photos show representative cells that were pre-grown in SD and then stained for 30 min with FM4–64. Stained cells were then washed and starved for 12 h (SD-L) or 24 h (SD-N) and examined. (B) Proteins were extracted from cultures harvested at the same times as in (A), separated by PAGE, transferred to duplicate membranes, and treated with antibodies to visualize GFP or actin. The amounts of GFP-Atg8 and GFP relative to actin are shown in the lower table.
Leucine-starved cells were less sensitive to rapamycin
After failing to see evidence for GFP-Atg8 processing in Δatg8 cells, we attempted to visualize autophagic events with an alternative reporter, HttQ25-GFP, an approximately 34 kD reporter built from the first 17 amino acids of the human protein, huntingtin, with 25 glutamine residues fused to a FLAG-tagged GFP, that was expressed in yeast from a GAL1 promoter.31 The polyglutamine sequence on this reporter enables it to form unstable aggregates in vitro.32 Preliminary experiments in galactose medium showed this protein was not toxic to yeast (data not shown), nor did it alter the percentages of cells that stained with PI during nitrogen or leucine starvation (compare strains with this reporter shown in Fig. S3 with the percentages of PI+ cells in Fig. 1B and E). Although this reporter was harvested during nitrogen starvation (Fig. S4A), it too failed to accumulate in vacuoles, or to show evidence of increased proteolytic degradation, during leucine starvation (Fig. S4B).
We then turned to Ape1-RFP,33 a substrate for nonspecific autophagy, as well as for one very selective process called cytoplasm-to-vacuole targeting.34 As shown in Figure 4A, this protein was harvested constitutively during nutrient-replete conditions, but vacuolar accumulation increased 20–30-fold more during either rapamycin treatment or nitrogen starvation (Fig. 4B). The amount of processed Ape1 protein also increased during these conditions, although this increase was not dramatic (Fig. 4C).

Figure 4. Leucine-starved cells harvested basal levels of Ape1. (A) Representative cells in cultures expressing Ape1-RFP after 12 h (SD-L) and 24 h (SD-N) starvation in the presence or absence of 0.22 µM rapamycin. (B) Average percentage of the population (± standard deviations) showing the indicated phenotypes. Each value was derived from looking at 100–200 cells in 3 independent experiments. (C) Proteins were extracted from PS cultures harvested at the times indicated above, separated by PAGE, transferred to duplicate membranes, and treated with antibodies to visualize Ape1 or actin. The amounts of Ape1 relative to actin are shown in the lower table.
Despite this sensitivity to increases in autophagic activity, the Ape1-RFP reporter failed to reveal increased protein harvesting during leucine starvation (Fig. 4A–C). In fact, leucine starvation partially de-sensitized cells to rapamycin treatment so that there were nearly 3-fold fewer-than-expected cells with intensely fluorescent vacuoles, and nearly half as much processed Ape1 protein produced. Interestingly, leucine-starved melanoma cells behave much like leucine-starved yeast: both fail to induce autophagy significantly, both die from caspase-dependent apoptosis, and neither is induced to full autophagy capacity by rapamycin.35
Overall, these experiments demonstrated that mutational studies were able to reveal what our current set of reporter proteins could not. Without the autophagy mutants, we would have no reason to suppose autophagy operated during either zinc treatment or leucine starvation since none of the standard reporters showed extensive accumulation in the vacuoles of the treated cells. The genetic studies, by contrast, revealed that cells performed two different processes in each of these conditions. One process protected cells from damage, and one process acted to accelerate cell death. The balance between these pathways could be shifted toward extending life by treating cells with rapamycin, or in either direction by expressing GFP-Atg8. However, at this time, we still cannot say which proteins operate in each pathway. Based on the small survey reported here, Atg29 played a much more important role protecting cells from the effects of leucine starvation than it did protecting cells from the effects of zinc.13 Moreover, while Atg11 operated in processes that protected cells during zinc treatment and leucine starvation, it appeared to interfere or compete with life-extending processes during nitrogen starvation. More subtly, the loss of Atg15, a vacuolar lipase that extends the life span of cells during caloric restriction,36 switched the primary pathway of death during nitrogen starvation from necrosis to apoptosis. It is possible that cells that are unable to breakdown their autophagosomes in the vacuole, and thus unable to recycle the autophagosome contents, are dying like cells starved for leucine.
The second outcome of these studies is to add to the growing evidence that autophagy operates differently, if at all, during leucine starvation.21,35 This deviation from the pathway operating during nitrogen starvation might have hastened the process that caused so many cells to show signs of late apoptosis/secondary necrosis. A complete understanding of the basis for this defect, and of how pro-life and pro-death autophagic pathways differ, must await the identification of new reporters, ones capable of tracking what appear to be highly selective operations. Until such time, our current interpretation of the results presented here is that cells are able to use different combinations of autophagy proteins to harvest different cell constituents, and by so doing, affect the factors that specifically trigger either necrotic or apoptotic death.
Materials and Methods
Yeast strains, plasmids and media
Saccharomyces cerevisiae strains and plasmids used in this study are listed in Table S1. Each mutant obtained from an Open BiosystemsTM collection of yeast deletion mutants was derived from BY4741 (MATa his2 leu2 met15 ura3) and has a kanMx cassette in place of the indicated open reading frame. All cells were grown in minimum medium (either SD or SG; 0.67% yeast nitrogen base without amino acids, 2% glucose (SD) or 2% galactose (SG) and strain-specific amino acids and nucleotides), or rich medium (YPD; 1% yeast extract, 2% peptone, 2% dextrose), or SD or SG medium without leucine (SD-L or SG-L), or in nitrogen-depleted minimum media [SD-N or SG-N; 0.17% yeast nitrogen base without amino acids and ammonium sulfate, 2% glucose (SD-N) or 2% galactose (SG-N)] at 30°C. When necessary, medium was supplemented with 1.5% agar and/or 0.22 µM rapamycin (Alexis Biochemicals, R-5000). The plasmid carrying the GFP-ATG8 chimera was a generous gift from Dr. Y. Ohsumi,37 Ape1-RFP was generously provided by Dr. M. Thumm,33 and HTTQ25-GFP was kindly provided by Dr. M. Sherman.31 Each was transformed into appropriate yeast strains using standard protocols.38
Viability assay, annexin and propidium iodide staining
In order to ensure all cultures were in comparable stages of growth in each experiment, each strain was grown to mid-log phase (approximately 0.4 OD600nm) in SD medium supplemented with appropriate amino acids and nucleotides. At that time, samples were taken, washed twice with TE buffer pH 8.0 (10 mM TRIS-HCl, 1 mM EDTA) and diluted to 0.04 OD600nm using fresh SD-L or SD-N for leucine or total nitrogen starvation experiments, respectively. Every 12 h (SD-L) or 72 h (SD-N), 100 µL of each culture were taken and diluted three and 4-fold in TE buffer and 100 µL were spread onto YPD plates. The inverted plates were incubated at 30°C for two days before colonies were counted in order to calculate the CFA which was defined here as the ratio of viable cells mL−1 at tn relative to the number of viable cells mL−1 at t0 expressed as a percentage. To estimate the mode of death, samples from appropriate strains were taken after 12 h (SD-L) and 24 h (SD-N) incubation. Cell wall-permeabilized cells were then stained with 2 μl annexin-FITC (BioLegend, 640905) and 2 μl (500 μg/ml) PI (195458, MP Biomedicals, LLC) following our previously described protocol.13 In these studies, approximately 90% of the annexin V-binding cells also accumulated PI indicating that they were either in secondary26 or severe necrosis. Cell walls were not permeabilized when only propidium iodide uptake was tested. Each average (and its standard deviation) represents the percentage of stained cells within a population of 100–200 cells based on 3–4 independent experiments.
Fluorescent microscopy and immunoblot analysis
Tested strains were grown in SD medium to an OD600nm = 0.4, incubated for 30 min at 30°C in YPD containing 20 µg/ml FM 4-64 (Invitrogen, T-13320) (for vacuole membrane visualization), washed twice with TE buffer and diluted to OD600nm = 0.04 in fresh appropriate medium. Unless otherwise stated, all cultures were then grown for 12 h (SD-L) or 24 h (SD-N) before 4 µL of each culture were spotted onto microscope slides previously coated with poly l-lysine (Sigma-Aldrich, P-4707). Cells expressing HTTQ25-GFP were starved in the presence of 1.0 mM phenylmethylsulfonyl fluoride (PMSF) when being prepared for microscopic analyses. Images were taken using a Nikon i80 Eclipse fluorescent microscope equipped with an Lambda LS unit and an Photometrix Cool SNAP ES digital camera with appropriate filters. The excitation time for GFP-Atg8 was 300 ms (Fig. 3A) and 2 sec and 500 ms for PS and Δatg8, respectively (Fig. S1); for Ape1-RFP (Fig. 3), the exposure was 1 sec; for HttQ25-GFP (Fig. S3), 400 ms.
Proteins were extracted from 5 OD units of cells following the method previously described.13 Obtained samples and a Color Plus Protein Ladder (BioLabs, P-7711S) were separated using an 8.5% SDS-PAGE gel and transferred to a Protran® nitrocellulose membrane (Whatman, 10402452). The membrane was blocked with blocking buffer (1.3% gelatin from cold water fish skin (Sigma, G7041) in PBS pH 7.0) overnight at 4°C and developed with primary rabbit polyclonal anti-GFP antiserum (Clonetech, 632377) for proteins extracted from Δatg8 GFP::ATG8 and PS HTTQ25-GFP, goat polyclonal anti-Ape1 (Santa Cruz Biotechnology, SC-26740) antibody for PS, and primary rabbit polyclonal anti-actin antibody (Sigma-Aldrich, A2066) for all three strains. Secondary antibodies for GFP-tagged proteins were IR700-labeled goat anti-rabbit (Rockland, 611-130-122). Secondary antibodies for Ape1 were alkaline phosphatase-conjugated donkey anti-goat (Santa Cruz Biotechnology, SC-2037). Actin was detected with primary rabbit polyclonal anti-actin antibody (Sigma-Aldrich, A2066) followed by IR700-labeled goat anti-rabbit (Rockland, 611-130-122) secondary antibody. Membranes were scanned using a Li-Cor Odyssey Infrared Imaging System (Li-Cor Bioscience) and analyzed using ImageJ (http://rsb.info.nih.gov/ij/index.html). The intensity of the band representing the protein of interest was normalized to that of actin. Each western has been replicated at least twice with similar results.
Supplementary Material
Acknowledgments
The authors would like to acknowledge that financial assistance for this research and its publication was provided by NIH Grant Number P20 RR016448 from the COBRE Program of the National Center for Research Resources, the Program for Microbiology, Molecular Biology, and Biochemistry, the Department of Plant, Soil, and Entomological Sciences, and by the UI Student Grant Programs awarded to Slawomir Dziedzic. The authors would especially like to express their gratitude to Dr. Patricia Hartzell for her critical advice, exceptional encouragement and support, and for the use of her microscope throughout the course of this project.
Glossary
Abbreviations:
- ACD
autophagic cell death
- CFA
colony-forming ability
- CVT
cytoplasm-to-vacuole targeting
- PI
propidium iodide
- PMSF
phenylmethylsulfonyl fluoride
- PS
parental strain
- Rap
rapamycin
- SD
synthetic minimal medium with 2% dextrose and all required nutrients unless indicated
- SG
synthetic minimal medium with 2% galactose and all required nutrients unless indicated
- YPD
yeast peptone medium with 2% dextrose
- ziNCD
zinc-induced necrotic cell death
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/19314
References
- 1.Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–14. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 2.Davey HM, Hexley P. Red but not dead? Membranes of stressed Saccharomyces cerevisiae are permeable to propidium iodide. Environ Microbiol. 2011;13:163–71. doi: 10.1111/j.1462-2920.2010.02317.x. [DOI] [PubMed] [Google Scholar]
- 3.Madeo F, Herker E, Maldener C, Wissing S, Lächelt S, Herlan M, et al. A caspase-related protease regulates apoptosis in yeast. Mol Cell. 2002;9:911–7. doi: 10.1016/S1097-2765(02)00501-4. [DOI] [PubMed] [Google Scholar]
- 4.Madeo F, Fröhlich E, Fröhlich KU. A yeast mutant showing diagnostic markers of early and late apoptosis. J Cell Biol. 1997;139:729–34. doi: 10.1083/jcb.139.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Büttner S, Bitto A, Ring J, Augsten M, Zabrocki P, Eisenberg T, et al. Functional mitochondria are required for alpha-synuclein toxicity in aging yeast. J Biol Chem. 2008;283:7554–60. doi: 10.1074/jbc.M708477200. [DOI] [PubMed] [Google Scholar]
- 6.Bursch W, Ellinger A, Kienzl H, Török L, Pandey S, Sikorska M, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis. 1996;17:1595–607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- 7.Chera S, Buzgariu W, Ghila L, Galliot B. Autophagy in Hydra: a response to starvation and stress in early animal evolution. Biochim Biophys Acta. 2009;1973:1432–43. doi: 10.1016/j.bbamcr.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 8.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Nomenclature Committee on Cell Death 2009 Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–66. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- 10.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–10. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7:457–65. doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- 12.Thorburn A. I think autophagy controls the death of my cells: what do I do to get my paper published? Autophagy. 2011;7:455–6. doi: 10.4161/auto.7.5.14797. [DOI] [PubMed] [Google Scholar]
- 13.Dziedzic SA, Caplan AB. Identification of autophagy genes participating in zinc-induced necrotic cell death in Saccharomyces cerevisiae. Autophagy. 2011;7:490–500. doi: 10.4161/auto.7.5.14872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosado CJ, Mijaljica D, Hatzinisiriou I, Prescott M, Devenish RJ. Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy. 2008;4:205–13. doi: 10.4161/auto.5331. [DOI] [PubMed] [Google Scholar]
- 15.Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10:602–10. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- 16.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15:105–12. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 17.Shin DM, Jeon BY, Lee HM, Jin HS, Yuk JM, Song CH, et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010;6:e1001230. doi: 10.1371/journal.ppat.1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–7. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–11. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schworer CM, Mortimore GE. Glucagon-induced autophagy and proteolysis in rat liver: mediation by selective deprivation of intracellular amino acids. Proc Natl Acad Sci U S A. 1979;76:3169–73. doi: 10.1073/pnas.76.7.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ecker N, Mor A, Journo D, Abeliovich H. Induction of autophagic flux by amino acid deprivation is distinct from nitrogen starvation-induced macroautophagy. Autophagy. 2010;6:879–90. doi: 10.4161/auto.6.7.12753. [DOI] [PubMed] [Google Scholar]
- 22.Boer VM, Amini S, Botstein D. Influence of genotype and nutrition on survival and metabolism of starving yeast. Proc Natl Acad Sci U S A. 2008;105:6930–5. doi: 10.1073/pnas.0802601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klosinska MM, Crutchfield CA, Bradley PH, Rabinowitz JD, Broach JR. Yeast cells can access distinct quiescent states. Genes Dev. 2011;25:336–49. doi: 10.1101/gad.2011311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawai H, Domae N. Discrimination between primary necrosis and apoptosis by necrostatin-1 in Annexin V-positive/propidium iodide-negative cells. Biochem Biophys Res Commun. 2011;411:569–73. doi: 10.1016/j.bbrc.2011.06.186. [DOI] [PubMed] [Google Scholar]
- 25.Ma H, Kunes S, Schatz PJ, Botstein D. Plasmid construction by homologous recombination in yeast. Gene. 1987;58:201–16. doi: 10.1016/0378-1119(87)90376-3. [DOI] [PubMed] [Google Scholar]
- 26.Eisenberg T, Carmona-Gutierrez D, Büttner S, Tavernarakis N, Madeo F. Necrosis in yeast. Apoptosis. 2010;15:257–68. doi: 10.1007/s10495-009-0453-4. [DOI] [PubMed] [Google Scholar]
- 27.Abeliovich H, Klionsky DJ. Autophagy in yeast: mechanistic insights and physiological function. Microbiol Mol Biol Rev. 2001;65:463–79. doi: 10.1128/MMBR.65.3.463-479.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–6. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 29.Lynch-Day MA, Klionsky DJ. The Cvt pathway as a model for selective autophagy. FEBS Lett. 2010;584:1359–66. doi: 10.1016/j.febslet.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prick T, Thumm M, Köhrer K, Häussinger D, Vom Dahl S. In yeast, loss of Hog1 leads to osmosensitivity of autophagy. Biochem J. 2006;394:153–61. doi: 10.1042/BJ20051243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walters RH, Murphy RM. Examining polyglutamine peptide length: a connection between collapsed conformations and increased aggregation. J Mol Biol. 2009;393:978–92. doi: 10.1016/j.jmb.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meiling-Wesse K, Epple UD, Krick R, Barth H, Appelles A, Voss C, et al. Trs85 (Gsg1), a component of the TRAPP complexes, is required for the organization of the preautophagosomal structure during selective autophagy via the Cvt pathway. J Biol Chem. 2005;280:33669–78. doi: 10.1074/jbc.M501701200. [DOI] [PubMed] [Google Scholar]
- 34.Kageyama T, Suzuki K, Ohsumi Y. Lap3 is a selective target of autophagy in yeast, Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2009;378:551–7. doi: 10.1016/j.bbrc.2008.11.084. [DOI] [PubMed] [Google Scholar]
- 35.Sheen JH, Zoncu R, Kim D, Sabatini DM. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell. 2011;19:613–28. doi: 10.1016/j.ccr.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang F, Watkins JW, Bermudez M, Gray R, Gaban A, Portie K, et al. A life-span extending form of autophagy employs the vacuole-vacuole fusion machinery. Autophagy. 2008;4:874–86. doi: 10.4161/auto.6556. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–81. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gietz RD, Woods RA. Yeast transformation by the LiAc/SS Carrier DNA/PEG method. Methods Mol Biol. 2006;313:107–20. doi: 10.1385/1-59259-958-3:107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.