

Abstract
Synucleinopathies like Parkinson disease and dementia with Lewy bodies (DLB) are characterized by α-synuclein aggregates within neurons (Lewy bodies) and their processes (Lewy neurites). Whereas α-synuclein has been genetically linked to the disease process, the pathological relevance of α-synuclein aggregates is still debated. Impaired degradation is considered to result in aggregation of α-synuclein. In addition to the ubiquitin-proteasome degradation, the autophagy-lysosomal pathway (ALP) is involved in intracellular degradation processes for α-synuclein. Here, we asked if modulation of ALP affects α-synuclein aggregation and toxicity. We have identified an induction of the ALP markers LAMP-2A and LC3-II in human brain tissue from DLB patients, in a transgenic mouse model of synucleinopathy, and in a cell culture model for α-synuclein aggregation. ALP inhibition using bafilomycin A1 (BafA1) significantly potentiates toxicity of aggregated α-synuclein species in transgenic mice and in cell culture. Surprisingly, increased toxicity is paralleled by reduced aggregation in both in vivo and in vitro models. The dichotomy of effects on aggregating and nonaggregating species of α-synuclein was specifically sensitive to BafA1 and could not be reproduced by other ALP inhibitors. The present study expands on the accumulating evidence regarding the function of ALP for α-synuclein degradation by isolating an aggregation specific, BafA1-sensitive, ALP-related pathway. Our data also suggest that protein aggregation may represent a detoxifying event rather than being causal for cellular toxicity.
Keywords: alpha-synuclein, dementia with Lewy bodies (DLB), Lewy body, lysosomal degradation, Parkinson disease (PD), protein aggregation
Introduction
Synucleinopathies including Parkinson disease (PD) and dementia with Lewy bodies (DLB) are characterized by intracellular inclusion [Lewy bodies (LBs) and neurites (LNs)] composed of misfolded and aggregated forms of α-synuclein.1,2 Although aggregated α-synuclein is the pathological hallmark of the diseases, increasing evidence suggests that oligomeric forms of α-synuclein represent the toxic species rather then aggregated structures. Alpha-synuclein can be degraded by the ubiquitin-proteasome degradation system (UPS), which is impaired in sporadic PD and clearly linked to familiar forms of PD.3 Complementary to the UPS, proteins can also be degraded by the autophagy-lysosomal pathway (ALP).4 ALP activation has been described within substantia nigra neurons of PD.5 Impaired UPS function leads to increased ALP degradation of α-synuclein suggesting a dynamic interplay between both intracellular degradation systems.6-8 Intracellular components are degraded by the ALP using two major pathways: chaperone-mediated autophagy (CMA) and macroautophagy.9 CMA involves chaperone-mediated targeting of identified proteins containing a KFERQ peptide motif via lysosomal-associated membrane protein (LAMP-1/2A) transporter into the lysosome.10 Macroautophagy comprises sequestration of larger cytosolic structures, such as aggregated proteins or organelles, into autophagosomes that fuse with lysosomes initiating the final degradation process.11 Alpha-synuclein contains a CMA target motif and is degraded by CMA in neural cell lines.7,12,13 In addition, macroautophagy is also involved in α-synuclein degradation.13 Components of the macroautophagy pathway are increased in human cerebral cortex tissue from DLB patients, and inhibition of macroautophagy increased α-synuclein accumulation in vitro.14
We and others have shown that chaperone proteins such as Hsp70 control proper folding and refolding of α-synuclein and its degradation.15-17 In transgenic mice overexpressing human α-synuclein, Hsp70 can induce degradation of insoluble α-synuclein species as a neuroprotective mechanism.15,18 Also, activation of autophagy by BECN1 ameliorates α-synuclein-induced changes.18,19 In addition, we have previously shown that the co-chaperone CHIP is able to switch between ALP and UPS mediated degradation of α-synuclein in the same cell culture model of α-synuclein aggregation used herein.20
To further explore the role of ALP mediated degradation of insoluble, aggregated α-synuclein species compared with soluble, nonaggregated forms, we (I) analyzed the expression of CMA and macroautophagy markers in temporal cortex tissue of DLB patients and controls, and (II) evaluated the role of ALP in both in vivo and in vitro models of synucleinopathy. Interestingly, we observed an upregulation of the CMA associated protein LAMP-2A and macroautophagy marker LC3-II both in the cortex from patients with DLB as well as in our in vivo and in vitro models of α-synuclein aggregation. Furthermore, modulation of ALP suggests a protective role of aggregation and indicates that a dissociation may exist between aggregation and toxicity of α-synuclein.
Results
Expression of ALP markers in human DLB cortex
ALP is able to degrade cytosolic proteins and their aggregates in lysosomes. Recently, increased expression of markers for macroautophagy14,21,22 and altered expression levels of regulatory molecules of the ALP were found in DLB.23 Here, we assessed lysosomal marker LAMP-1, CMA marker LAMP-2A, and macroautophagy associated LC3-II in temporal cortex of DLB patients compared with controls. Interestingly, LBs stained for LC3 and LAMP-2A (Fig. 1A and B), but not for LAMP-1. LAMP-2A was also found in Lewy neurites (Fig. 1B). Quantitative analysis of expression levels by SDS-PAGE and western blotting revealed a significant increase of about 30% for LC3-II (+30% ± 8.2% SEM), and LAMP-2A (+35% ± 10.3% SEM) levels within the lysosome-enriched fraction in DLB cases (Fig. 1C and D). No changes in LC3-II levels were found in the post-nuclear fraction (Fig. 1E and F), where probing for LAMP-2A did not reveal a substantial signal (data not shown). The lysosomal associated protein LAMP-1 was reduced in the lysosome enriched fraction (by 43% ± 7.0% SEM) (Fig. 1C and D) and increased in the cytoplasmic fraction (by 62% ± 16.0% SEM) (Fig. 1E and F) suggesting a potential intracellular redistribution from lysosomes to the cytosol. These changes in ALP markers in DLB suggest a pathophysiological relevant alteration of both CMA and macroautophagy associated pathways. We therefore continued our investigation into the role of ALP in α-synuclein aggregation and toxicity in models of synucleinopathy.

Figure 1. Immunohistochemistry for LC3 (A) and LAMP-2A (B) in α-synuclein positive Lewy bodies (LBs) in midbrain of DLB patients. Co-staining for both ALP markers was found in LBs. In addition, LAMP-2A was found in Lewy neurites (B, lower panel). Scale bars: 10 µm. Protein levels of LAMP-1, LAMP-2A and LC3 in cell lysates (lysosome enriched fraction, C and D; cytosolic fraction, E and F) of temporal cortex tissue of DLB cases and controls. Western blot analysis of ALP marker is shown for two representative DLB cases and controls (C and E). Quantitative densitometry revealed increased levels of LC3-II and LAMP-2A in the lysosome enriched fraction (D; DLB n = 12, controls n = 9), but not in the post-nuclear fraction (F; DLB n = 15, controls n = 11; D, upper panel). A LAMP-2A specific signal could only be detected in the lysosome enriched fraction. LAMP-1 was reduced in the lysosome-enriched fraction and increased in the post-nuclear fraction. (Student’s t-test, *p < 0.05).
Alteration of ALP markers in in vitro and in vivo α-synuclein aggregation models
Co-expression of a C-terminal modified α-synuclein (SynT) and synphilin-1 leads to α-synuclein aggregation (SynT-aggregation model) in transfected cells (Fig. 2A).15,24 It is important to note that the in vitro SynT-aggregation model differs from other available in vitro models because it enables the visualization of larger α-synuclein aggregates (2–5 µm diameter) in contrast to unmodified wild-type α-synuclein (WT-Syn) transfected cells that do not form large intracellular aggregates (Fig. 2A). In addition, α-synuclein species in the SynT-aggregation model are detergent resistant and are positive for ThioS labeling, suggesting the presence of fibrillar, aggregated α-synuclein (Fig. 2A). Smaller α-synuclein positive structures (< 2 µm) are found in both models, but are not ThioS positive (Fig. 2A). Structural electron microscopy images revealed electron-dense structures associated to autophagic vesicle formations that are not present in untransfected cells (Fig. 2B, EPON-embedded sections). These intracellular aggregates are α-synuclein positive after immunogold-labeling for α-synuclein (Fig. 2B, α-synuclein immunogold). Interestingly, α-synuclein aggregates in the SynT-aggregation model co-label LAMP-1, LAMP-2A and LC3 (Fig. 3A), whereas WT-Syn transfected cells did not show any changes in LAMP-1/2A or LC3 expression pattern compared with untransfected neighboring cells (Fig. 3B).

Figure 2. SynT-aggregation model and WT-Syn control. (A) Transient co-transfection of H4 neuroglioma cells with an 93 aa long C-terminal tag fused to α-synuclein (SynT) co-expressed with synphilin-1 resulted in larger intracellular α-synuclein aggregation.15,24 Immunostaining for α-synuclein revealed several aggregates of 2–5 µm diameter that were resistant to RIPA (Ri) and urea (U) extractions and ThioS-positive. Smaller aggregates (< 2 µm) were ThioS negative. Transfection with untagged human WT α-synuclein (WT-Syn) did not result in larger α-synuclein immunopositive aggregates and was found only in the TBS-soluble fraction 24 h after transfection. Electron microscopy of untransfected (1st image) or SynT/Synphilin1 transfected H4 neuroglioma cells (2nd–4th image, B) revealed electrodense structures (2nd image and higher magnification in 3rd image) in transfected cells only, that were associated with vesicular structures which resemble autophagic vesicles. Aggregates were α-synuclein positive (immunogold-labeling for α-synuclein, 4th image). Scale bar: 1 µm.

Figure 3. (A and B) ALP marker expression pattern in transfected H4 cells showed a co-staining of LAMP-1, LAMP-2A and, LC3 and α-synuclein positive aggregates in the SynT-aggregation model (A; indicated by arrows, representative α-synuclein aggregates are magnified), whereas no changes of ALP marker expression pattern were observed in WT-Syn control transfected cells compared with nontransfected neighboring cells (B). (C) In temporal cortex tissue of α-synuclein transgenic animals LAMP-2A co-labeled mainly the outer rim of α-synuclein positive inclusions (arrows), whereas co-labeling of LC3 and α-synuclein could be detected either at the rim (arrows), or homogenously distributed. LAMP-1 was not found in α-synuclein inclusions in vivo (data not shown). Scale bars: 10 µm.
Similar results were obtained in α-synuclein transgenic mice overexpressing WT α-synuclein that show α-synuclein immunopositive inclusions in the cortex (Fig. 3C).18 Likewise, the inclusions colabel with LAMP-2A and LC3 (Fig. 3C), however no costaining for LAMP-1 was detected (data not shown). It is interesting to note that LAMP-2A was mainly found at the outer rim of the inclusions, whereas LC3 was found either homogenously distributed within or at the rim only (Fig. 3C). It remains unclear if this observation is due to incomplete antibody penetration or different pools of aggregates in terms of ALP marker colabeling. Nevertheless, this pattern of expression supports the association between α-synuclein aggregation and ALP marker expression.
Quantification of ALP marker expression in α-synuclein transgenic animals and in the SynT-aggregation model using quantitative western blot analysis (Fig. 4A and B) revealed similar changes to the human DLB samples: LAMP-2A and LC3-II were significantly increased in temporal cortex tissue of α-synuclein transgenic mice compared with nontransgenic mice where no α-synuclein inclusions were present (Fig. 4A and C). In the SynT-aggregation model LC3-II levels were also significantly increased compared with empty vector transfected H4 cells (Fig. 4B and D). A nonsignificant increase for LAMP-2A (p = 0.12) was also observed in the SynT model. No significant changes for LAMP-1 levels were found in both in vivo and in vitro models. Transfection with WT-Syn controls did not lead to a change in ALP marker expression (Fig. 4D), supporting the association of ALP specifically to the α-synuclein aggregation process in both models.

Figure 4. Expression levels of LAMP-1/2A and LC3-II in α-synuclein transfected H4 cells and in α-synuclein transgenic mice. Representative western blots and intensity quantification of ALP marker normalized to actin are shown for in vivo (4 animals/group, A and C) and in vitro (5 independent experiments, B and D). Quantification of ALP marker expression in α-synuclein transgenic animals revealed significant increased LAMP-2A and LC3-II levels compared with non-transgenic littermates (C). LC3-II levels were also increased in the SynT-aggregation model (SynT) compared with WT-Syn control transfected H4 cells (WT-Syn, D). LAMP-2A levels were increased, but did not reach significance (p = 0.12 compared with WT-Syn, D). ALP marker expression in α-synuclein aggregation models were either compared with nontransgenic animals or to WT-Syn transfected cells (after normalization to empty vector transfected cells). Student’s t-test, *p < 0.05.
ALP modulates α-synuclein aggregation and toxicity in vitro
Changes in ALP markers in human DLB tissue and in models of α-synuclein aggregation suggest that ALP modulation is a potential target for the treatment of synucleinopathies. Thus, we examined whether modulation of ALP affects α-synuclein aggregation and toxicity (Fig. 5). It is important to note that even though 40–50% of SynT transfected cells develop α-synuclein aggregates (Fig. 5A and C), toxicity is not significantly increased compared with nonaggregating WT-Syn control transfected cells (Fig. 5B and D). In order to inhibit ALP, BafA1 was used at a 200 nM concentration which is known to specifically block the vacuolar ATPase. This results in blocked acidification of lysosomes consecutively preventing fusion between autophagosomes and mature lysosomes,25 paralleled by an increase of LC3 levels in H4 cells (Suppl. Figs.). Intriguingly, inhibition of autophagy using BafA1 decreased the percentage of aggregate bearing cells from 43.5% (+4.72% SEM) to 8.3% (± 0.75% SEM, p < 0.001) (Fig. 5E). Unexpectedly, this was paralleled by a significant increase of toxicity by 3.4 fold (± 1.09SEM) (Fig. 5F). Control transfection with WT-Syn did not result in aggregate formation (either with or without BafA1) or in a significant toxic effect (Fig. 5F). Thus, the dichotomy effect of BafA1 on aggregation and toxicity was specific for aggregating α-synuclein species.

Figure 5. Transient transfection with the SynT aggregation model leads to α-synuclein positive aggregate formation (green, A) in 40–50% of transfected cells (C), whereas transfection with WT-syn did not result in aggregation (green, B and C) and served as control. Note that transfected cells (either with SynT or WT-syn) were not caspase-3 positive (red, A and B). Nuclei were stained in blue (A and B). (D) Toxicity as measured by the release of adenylate kinase in transfected cells (fold of mock transfected cells) showed no difference between the SynT-aggregation model (n = 9) or WT-Syn control (n = 7). Inhibition of ALP using BafA1 reduced the number of cells with SynT-aggregates (E, *p < 0.001) paralleled by increased toxicity of SynT (F, *p < 0.05). Treating WT-Syn transfected cells with BafA1 did not result in aggregation and its toxicity was not affected by ALP modulation. ALP activation by rapamycin did not alter toxicity or aggregation in transfected cells. Scale bars: 10 µm.
Rapamycin inhibits the mammalian target of rapamycin (mTOR) pathway and thereby inducing ALP.26 However, rapamycin (200ng/ml) did not significantly alter the number of cells with aggregates (52.1% ± 4.19% SEM) and had no effect on toxicity (Fig. 5E and F). Likewise, ALP induction did not result in aggregate formation or significant changes of toxicity (1.19 fold ± 0.27) in WT-Syn controls (Fig. 5F).
Modulation of ALP in α-synuclein transgenic mice
In order to expand on our in vitro findings, we treated transgenic human WT α-synuclein expressing mice18 with BafA1 or rapamycin. Overexpression of α-synuclein results in a reduction of dendritic (MAP2) and synaptic markers (SY38) compared with nontransgenic animals (Fig. 6). Similar to the effects of BafA1 on aggregation and toxicity in the in vitro α-synuclein aggregation model, we observed an augmented dendritic and synaptic damage in BafA1 treated α-synuclein transgenic mice compared with saline treated animals. Furthermore, inclusion-like α-synuclein immunoreactivity was reduced by BafA1. However, this was not paralleled by a general reduction of α-synuclein expression levels, since punctated diffuse immunoreactivity was significantly increased. Rapamycin also reduced inclusion-like α-synuclein immunoreactivity with no change of punctated α-synuclein expression levels (Fig. 6), suggesting that α-synuclein metabolism is not exclusively regulated by mTOR dependent activation of ALP. Comparable with the in vitro findings, no change in MAP2 or SY38 immunoreactivity was observed after rapamycin treatment. In addition, no changes were observed in treated non-tg littermates.

Figure 6. Neuropathological alterations in the temporal cortex of α-synuclein transgenic mice. Immunocytochemical analysis with an antibody against α-synuclein (red, top panel) showed that transgenic mice (B) displayed α-synuclein positive inclusions and punctated expression patterns compared with nontransgenic control (A). Alpha-synuclein positive inclusions were reduced by BafA1 treatment (C) compared with saline treated animals. The dendritic marker MAP2 and the synaptic marker SY38 were reduced in α-synuclein transgenic mice (F and J) compared with non-transgenic mice (E and I). ALP inhibition with BafA1 aggravated MAP2 and SY38 immunoreactivity levels (G and K). Quantitative analysis of inclusion- and punctated α-synuclein (D), MAP2 (H) and SY38 (L) immunoreactivity are displayed in the right panel. Significant changes are indicated (*) as compared with saline-treated animals (for α-synuclein immunoreactivity) or compared with saline-treated non-tg animals (for MAP2 and SY38 immunoreactivity). Scale bars: 10 µm.
To further investigate the difference between α-synuclein inclusion and punctated immunoreactivity, we analyzed detergent soluble and insoluble expression levels.2,15,27 The increase of punctated α-synuclein staining after BafA1 treatment (Fig. 6C) was paralleled by an increase of insoluble α-synuclein levels assessed by SDS-PAGE and western blotting (Fig. 7), whereas soluble levels of α-synuclein were significantly reduced following BafA1 treatment. Thus, whereas BafA1 reduced the number of α-synuclein inclusions as assessed by immunofluorescence (Fig. 5), insoluble α-synuclein levels were increased. By contrast, rapamycin led to an overall reduction of α-synuclein levels both for soluble and insoluble species compared with vehicle-treated animals (Fig. 7).
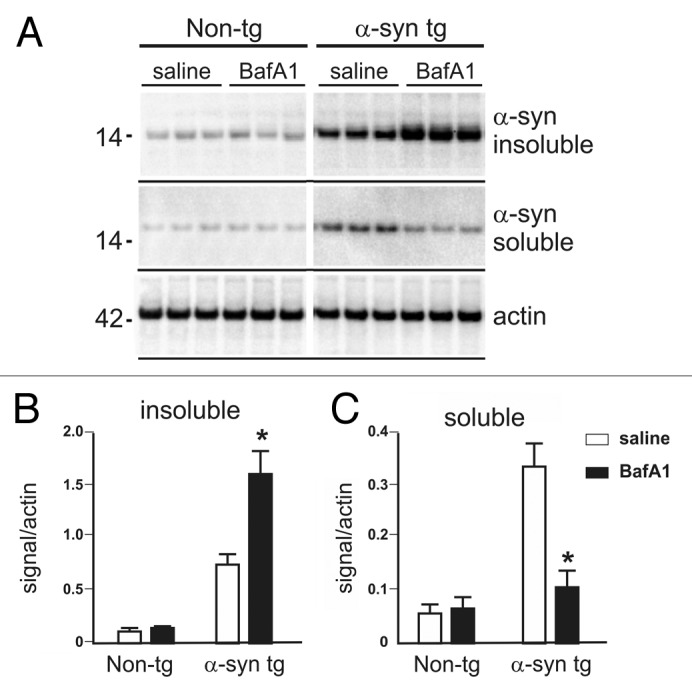
Figure 7. Detergent soluble and insoluble α-synuclein levels in BafA1 and rapamycin- treated transgenic and nontransgenic mice (A). The amount of detergent insoluble α-synuclein fraction was increased by BafA1 (B), to the expense of soluble α-synuclein levels (C). Significant changes are indicated as BafA1 treatment. (*) compared with vehicle treatment p < 0.05, Student’s t-test.
Molecular pathways of α-synuclein aggregation
To further decipher the role of the BafA1-sensitive ALP for α-synuclein aggregation we evaluated other inhibitors of ALP. Similar to BafA1, the lysosomotropic agent choloroquine compromises autophagy by neutralizing lysosomal pH and thereby blocking V-ATPase function.25,28 Interestingly, chloroquine was also able to reduce the number of SynT aggregate bearing cells, in a similar way as BafA1 treatment (Fig. 8A). However, in contrast to BafA1, the SynT-aggregation model did not show increased toxicity after chloroquine treatment (Fig. 8B). Treatment with 3-MA as an inhibitor of ALP mainly affecting macroautophagy19 led to a smaller reduction of SynT aggregation, whereas no increase of toxicity was observed compared with nonaggregating WT-syn (Fig. 8). To summarize neither 3-MA nor chloroquine was able to mimic the BafA1-mediated effect on α-synuclein aggregation and toxicity.

Figure 8. Aggregation and toxicity of transfected H4 cells after treatment with different ALP modulating compounds. The effect of BafA1 on SynT-aggregation (A,B) could not be resembled by 3-MA that did not reduce SynT-aggregation (B). Interestingly, blocking V-ATPase function using chloroquine (CQ) could substantially reduce aggregation, similar to BafA1 treatment (B), however did not increase toxicity of SynT (C). Here, increase of SynT toxicity over WT-syn by blocking ALP was only present in BafA1 treated cells. Treatment with 3-MA even reduced toxicity of SynT compared with BafA1. Toxicity was measured by quantifying activated caspase-3 immunoreactivity in the SynT-aggregation model compared with nonaggregating WT-Syn transfected H4 cells. Significant changes are indicated as treatment (*) compared with control treatment or treatment compared with BafA1. #p < 0.05, Student’s t-test. Scale bars: 10 µm.
Discussion
The current study analyzed the role of the ALP on aggregated forms of α-synuclein in vivo (in temporal cortex tissue of DLB patients and transgenic mice) and in vitro in a cell culture model of α-synuclein aggregation. We have identified an aggregate-specific effect of ALP modulation by BafA1 both in vivo and in vitro which (1) expands the evidence that intracellular protein aggregation may be a detoxifying event rather than causal for cellular toxicity; and (2) suggest that aggregation/inclusion formation depends rather on a BafA1-sensitive ALP pathway that is distinct from other modifications of the ALP. Also, our data suggests that different pools of α-synuclein (soluble, insoluble, aggregated) are metabolized by distinct cellular degradation pathways supporting the importance of a detailed characterization of soluble, oligomeric and aggregated forms of α-synuclein in different models of synucleinopathies.
Autophagy has been reported to be impaired in PD brain as revealed by ultrastructural analysis in nigral neurons.5 In addition, regulatory molecules of ALP such as mTOR and Atg7 are altered in DLB brain tissue.23 Here we show increased CMA and macroautophagy markers in the temporal cortex of DLB patients, as well as in in vivo and in vitro α-synuclein aggregation models. This is in agreement with elevated levels of markers for macroautophagy such as LC3-II in DLB patients14,21 in the substantia nigra in PD,29 and in transgenic mouse models overexpressing α-synuclein.14,30 In PD, the CMA marker LAMP-2A is reduced in the substantia nigra of PD patients,31 similar to other lysosomal components including LAMP-1, cathepsin D and Hsc70.29,32 Likewise, we detected a reduction of LAMP-1 in the temporal cortex tissue of DLB patients (lysosome enriched fraction), however, we also found an increase of LAMP-1 in the cytoplasmic fraction. Intracellular trafficking and translocation of LAMP-1 has been linked to melanosome generation, and treatment with quinolones prevents melanogenesis, also altering intracellular LAMP-1 localization.33 Nevertheless, we could not observe changes of LAMP-1 expression in α-synuclein aggregation models. Thus, the distinct role of LAMP-1 for α-synuclein aggregation in DLB cannot be addressed in the present model. Taken together, ALP metabolism may differ in cortical synucleinopathy (as in DLB) from substantia nigra pathology in PD. In the present study an induction of both macroautophagy marker LC3-II and CMA-associated LAMP-2A levels in the lysosome-enriched fractions from temporal cortex tissue in 12 DLB patients was observed, matching recent findings by Tanji et al. and Higashi et al.21,22 LC3 and LAMP-2A also co-labeled LB, suggesting an activation of autophagy including CMA and macroautophagy in DLB. Larger patient studies using comprehensive analysis of regional specific (e.g., cortical, striatal structures, substantia nigra) and subcellular neuronal and non-neuronal ALP activities may help to better understand the topographical pattern of ALP and its contribution to the disease progression in synucleinopathies.
Several studies using different cell culture models of synucleinopathies have shown that the ALP participates in α-synuclein degradation and its alteration may support α-synuclein mediated neurodegeneration.13,20,34,35 Initial reports showed that WT and mutant α-synuclein,6,7 as well as oligomeric forms of α-synuclein12,14 are targeted by ALP. Most of the previous studies report increased accumulation of α-synuclein by inhibiting ALP, or reduced α-synuclein levels by activating ALP, respectively. Induction of ALP using BECN1 overexpression attenuated α-synuclein accumulation which could be reversed by BafA1.19 Likewise, we observed increased levels of α-synuclein in insoluble fractions after BafA1 treatment of α-synuclein transgenic mice, as well as increased punctated expression pattern of α-synuclein (Figs. 6 and 7). Knockdown of ALP-related genes Becn1 or Atg5 at an early stage of initiating autophagy, or inhibiting macroautophagy by 3-MA resulted in increased α-synuclein oligomer formation in another in vitro model supporting the role of macroautophagy for α-synuclein degradation.14 Thus, pre-aggregated oligomeric forms of α-synuclein may be directly targeted by ALP mediated degradation pathways as previously suggested.14,36,37
However, our data suggest that aggregated α-synuclein species are targeted by ALP differently from pre-aggregated oligomeric forms: Here, we observed a reduction of α-synuclein aggregation by BafA1 in vitro. The cell culture aggregation model is characterized by ThioS-positive α-synuclein aggregates localized in autophagosomal structures (Fig. 2). Thus, it differs form other cell culture models by the presence of higher aggregated α-synuclein species depending on the C-terminal modification of α-synuclein (SynT). They seem to react differently from smaller intracellular inclusions generated by untagged α-synulcein (Figs. 2, 5 and 8).20 Thus, our data suggest that BafA1 is not only inhibiting ALP-dependent protein degradation, but also modulates protein aggregation specifically. Even though we see a clear effect of BafA1 treatment on LC3 levels, and a similar effect on aggregation could be induced by chloroquine, we cannot exclude ALP independent mechanisms related to BafA1 on aggregation and toxicity of α-synuclein. The macrolide BafA1 displays a broad spectrum of biological activities including inhibition of the vacuolar ATPases (V-ATPase) thereby blocking acidification and protein degradation in lysosomes.25 However, BafA1 can also modulate ALP by affecting the plasma membrane ATPase (P-ATPase), ATP-binding cassette transporters,38,39 and by functioning as a potassium ionophore impairing mitochondrial functions.40 Thus, BafA1 may modify a whole array of cellular responses to aggregation than only blocking lysosomal degradation. This might explain why other ALP inhibitors could not recapitulate the effect of BafA1 completely. Here, blocking ALP with 3-MA did not result in reduced SynT aggregation, and did not increase toxicity. Likewise, chloroquine did not increase toxicity of SynT, even though aggregation was reduced (Fig. 8). Similarly, chloroquine blocks lysosomal function by raising lysosomal pH and thereby inhibiting lysosomal function.25,28 Both agents were able to reduce SynT aggregates, but only BafA1 increased SynT toxicity. This is in line with the findings, that BafA1 is able to rescue chloroquine-induced apoptosis, potentially independent from its V-ATPase inhibition.41
In addition to its effect on α-synuclein aggregation, modulation of ALP by BafA1 leads to increased α-synuclein dependent toxicity both in vivo and in vitro. This suggests that α-synuclein aggregation may be not toxic by itself, but rather reflects a protective intracellular response regulated by a BafA1 sensitive mechanism, potentially involving parts of the ALP. In agreement with our findings, a cytoprotective effect of ALP induction can be blocked by BafA1 or LAMP-2 knockdown, which is reversible at an initial stage only, whereas at later stage cells become apoptotic.42 Similarly BECN1 induced ALP activation ameliorated synaptic and dendritic pathology, which was accompanied by reduced α-synuclein levels.19
ALP activation in vivo and in vitro by rapamycin has been shown to be protective in models for neurodegenerative disorders.29,37 In rapamycin-treated mice we observed a reduction of α-synuclein inclusion, suggesting that ALP induction by rapamycin might lead to degradation of α-synuclein species (supplementary figures). However, we did not observe a conclusive protective effect of rapamycin neither in vivo nor in vitro. In another study, the protective effect of rapamycin required the presence of rotenone to additionally impair mitochondrial activity, suggesting an interplay between protein aggregation, toxicity and mitochondrial activity. Thus, it is unclear if the protective effect of rapamycin involves ALP mediated neuronal α-synuclein degradation or aggregation. Future experiments using more specific small-molecule enhancers (SMERs) of mammalian autophagy acting downstream of mTOR,43 or via mTOR independent activation of ALP44 may help to decipher this pathway.
Taken together, our in vitro and in vivo data link the effect of BafA1 to a novel pathway, potentially involving ALP and α-synuclein aggregation processes. We propose that this novel BafA1 sensitive pathway may represent a detoxification mechanism which could be targeted for therapeutic interventions.
Materials and Methods
Human brain samples
Human temporal cortex tissue was obtained through the Massachusetts Alzheimer Disease Center (ADRC) brain bank, the Harvard Brain Tissue Resource Center and the University of Maryland brain bank as described previously.2 The pathological diagnosis of DLB was performed according to established criteria.45 Control brains did not demonstrate any neuropathological alterations. For the quantitative western blot analysis of ALP markers a post-nuclear fraction was derived from temporal cortex tissue of 15 DLB patients [age: 80.9 y ± 5.7 SD; sex: 9 male (M), 4 female (M); post-mortem interval (PMI): 13.5 h ± 5.8 SD, 2 patient data missing] and 11 control cases (age: 71.9 y ± 12.6 SD; sex: 3 M, 7 F; PMI: 13.5 h ± 5.8 SD, 1 patient data missing); Tissue of 12 DLB patients (age: 82.4 y ± 3.4; sex: 8 M, 3 F; PMI: 14.0 h ± 6.6 SD, 1 patient data missing) and 9 control cases (age: 72.4 y ± 14.2 SD; sex: 1 M, 7 F; PMI: 14.6 h ± 5.7, 1 patient data missing) was available for generation of lysosome-enriched fractions.
Antibodies
For immunofluorescence microscopy analysis: LAMP-1: (H4A3, 1:100, Developmental Studies Hybridoma Bank—DSHB, University of Iowa; H4A3, 1:500, Abcam, ab25630); LAMP-2A (1:200, Abcam, ab18528); LC3 (2G6, 1:500, nanoTools, 0260-100); α-synuclein (H3C, 1:3000; kindly gift from J.M. George, University of Illinois; 1:500, Millipore, AB5464; 15G7, 1:250, Alexis/Enzo Life Sciences, ALX-804-258); MAP2 (1:100; Millipore, MAB3418); synaptophysin (Sy38; 1:500, Millipore, MAB5258) cleaved caspase-3 (Asp175, 1:500, Cell Signaling Technology, 9661). For western blot analysis: LAMP-1 (H4A3, 1:200, DSHB); LAMP-2A (1:200, Abcam, ab18528); LC3-I/II (1:500, Novus Biologicals, NB100-2331SS; 2G6, 1:500, nanoTools, 0260-100); α-synuclein (Syn-1, 1:1000, BD Transduction Laboratories, 610787); β-actin (AC40, Abcam, ab1103), GAPDH (Millipore, MAB374), α-tubulin (clone DM1A, Sigma, T6199). Secondary antibodies: goat anti-mouse (IR800CW, 1:5000, Li-Cor Biosciences, 926-32210; Tyramide Signal Amplification-Direct (Red) system, NEN Life Science, 1553461); goat anti-rabbit (IR680LT, 1:5000, Li-Cor Biosciences, 926-68021), goat anti-mouse/rabbit (FITC, 1:100, Vector Laboratories, W0405, W0320); donkey anti-mouse/rabbit (Alexa488, 1:3000, Invitrogen, A21202, A21206; Alexa568, 1:3000, Invitrogen, A10037, A10042); donkey anti-rabbit (Cy3, 1:200, Jackson Immunoresearch, 711-165-152).
Immunohistochemistry of human brain samples
For LB detection 40 µm midbrain sections from selected DLB patients were PFA fixed, washed in TRIS buffered saline (TBS), permeabilized using 1% triton X-100 and blocked in 1% normal goat serum + 0.5% milk in TBS (blocking solution) for 1h at RT. Primary antibodies against LAMP-1, LAMP-2A or LC3 were incubated overnight at 4°C, followed by washing in TBS and secondary antibody application for 1hr at room temperature. Tissue sections were mounted and coverslipped using Vectashield® containing DAPI (Vector Laboratories, H1200). Confocal images were taken using a 63X water-immersion objective on a Zeiss LSM510 microscope (Thornwood).
Alpha-synuclein transgenic mice and treatments
Heterozygous transgenic mice expressing human wild-type (WT) α-synuclein under the control of the PDGF-β promoter show abnormal detergent-insoluble α-synuclein in the neocortex and limbic system and develop α-synuclein immunoreactive cytoplasmic aggregate-like inclusions in the brain.18,46 Nine months old mice were infused with BafA1 or rapamycin (10 mg/kg 0.5 ul/h for 2 weeks) into the lateral ventricle (osmotic minipump, Alzet, 1007D model) as described.47 Mice were euthanized 2 weeks later. Brains were removed, post-fixed in phosphate-buffered 4% PFA, pH 7.4, at 4°C for 48 h and sectioned at 40 µm with a vibratome 2000 (Leica). All procedures were completed under the specifications set forth by the Institutional Animal Care and Use Committee.
Immunohistochemistry of transgenic mice
Analysis of neuronal synaptic and dendritic integrity and ALP marker co-labeling was performed with 40 µm-thick vibratome sections immunostained for synaptophysin (Sy38), MAP2, α-synuclein, LAMP-2A, or LC3 followed by a secondary antibody incubation and laser scanning confocal microscopy (Bio-Rad). The synaptophysin and α-synuclein immunoreactive structures were detected with the Tyramide Signal Amplification™-Direct (Red) system while MAP2 was detected with the horse anti-mouse fluorescein isothiocyanate (FITC) antibody (1:75, Vector Laboratories, FI-2000) and mounted under glass coverslips with anti-fading media (Vector Laboratories, H-1200). All sections were processed under the same standardized conditions. Digitized immunolabeled blind-coded sections images were serially imaged with the laser scanning confocal microscope and analyzed with the NIH Image program 1.63 to determine the percentage of the area of the neuropil occupied by MAP2-, α-synuclein, or Sy38-immunoreactive structures as described.19 The Quantimet 570C (Leica) was used to quantify α-synuclein immunoreactive inclusions in the neocortex. For each mouse, a total of three sections were analyzed and for each section, four fields in the frontal cortex and hippocampus were examined.
Cell culture model of α-synuclein aggregation
The constructs for human WT untagged α-synuclein (WT-Syn) and its C-terminal tagged version (SynT) have been described.15,24,48 Transient transfection of H4 cells (HTB-148, ATCC) with SynT and synphilin-1 resulted in aggregation and detergent insoluble α-synuclein fraction (Fig. 2).24 H4 cells were maintained in Opti-MEM (Invitrogen, 51985-042) supplemented with 10% fetal bovine serum, passaged, plated on chamber slides (Labtek-II, Nalgen-Nunc, 154526) or coverslips 24 h prior to transfection. Equi-molar ratios of plasmids were transfected using Superfect (Qiagen, 301305) according to the manufacturers' protocol (2 h transfection + 4 h recovery time). ALP modifiers were incubated during the last 18h prior fixation. In an additional set of experiments H4 cells were transiently transfected using the calcium-phosphate method.49 2× BES-buffered saline solution containing phosphate ions (50 mM BES, 280 mM NaCl, 1.5 mM Na2HPO4xH2O, pH 6.98) were mixed with calcium chloride solution (2.5 M) containing the plasmids. Cells were incubated with plasmid-calcium-phosphate coprecipitates for 17 h followed by media change and recover time (7 h). ALP modifiers were incubated during the last 24 h before fixation and processing for immunocytochemistry and toxicity assessment. Co-transfection with an empty backbone-vector [pcDNA3.1(+), Invitrogen, V790-20] and mock transfection was used as control. Bafilomycin A1 (200 nM, BafA1, Sigma Aldrich, B1793) or rapamycin (200 ng/ml, Rap, Sigma Aldrich, RO395) were prepared in DMSO, 3-methyladenine (5 mM, 3-MA, Sigma Aldrich, M9281), and chloroquine diphosphate salt (50 µM, CQ, Sigma Aldrich, C6628) in water.
Immunocytochemistry and toxicity assessment in vitro
One day after transfection, cells were fixed with 4% PFA (20 min RT), washed with TBS and blocked in fish skin gelatin buffer + 0.1% triton X-100 for 1 h, cells were incubated with primary antibody (α-synuclein; LAMP-2A; LAMP-1; LC3-I/II) for 4 h at RT or overnight at 4°C, followed by secondary antibody incubation for 1h. Cells were analyzed by fluorescence microscopy using a Nikon Eclipse TE300 (Nikon Instruments). For ThioS labeling, fixed cells were incubated prior to immunofluorescence labeling for 5 min in 0.5 mg/ml thioflavin-S, and washed in 80% ethanol. The number of cells containing α-synuclein-immunopositive aggregates were assessed by an observer blind to the transfection conditions (i.e., the co-transfected plasmid) according random sampling as described.15 At least 400 cells from two wells were assessed for each experiment. A positively transfected cell was scored based on the presence of α-synuclein immunostaining compared with background (which in all cases was negligible). A transfected cell containing aggregates was scored based on the presence of at least one detectable aggregate of α-synuclein immunostaining. The percentage of cells containing aggregates compared with the total number of transfected cells was recorded. Toxicity was analyzed by measuring the release of adenylate kinase into the media according to the manufacturer’s protocol (ToxiLight; Lonza, LT07-117), or counting cells immunopositive for the apoptotic marker cleaved caspase-3 (mean percentage of positive cells from a minimum of 400 DAPI-positive cells/well).
Electron microscopy of the SynT aggregation model in H4 cells
Electron microscopy of the SynT aggregation model in H4 cells was performed as described.50 Cell pellets were fixed in 2.5% glutaraldehye containing PBS-buffer. After washing, the remaining pellet was embedded in 2% agar, and fixed overnight. After washing, cells were incubated in 2% osmium tetroxide for 2 h, dehydrated serially to ethanol at 20°C, followed by an incubation in propylenoxid twice for 30 min, in 1:3 epoxyresin (EPON812) / ethanol for 5 h, and 100% EPON 812 overnight. The EPON block was polymerized at 50°C for 48 h. For post-embedding immunogold labeling, cells were pelleted and fixed in 4% paraformaldehyde, 0.1% glutaraldehyde and 0.1 M cacodaylat buffer (pH 7.4) for 1 h at 4°C. After washing, the pellet was embedded in 2% agar, washed in 3.7% sucrose/0.1 M cacodaylat buffer, dehydrated serially to 70% ethanol, incubated in 1:1 London resin white (LRW) / 70% ethanol for 1 h, followed by incubation in LRW overnight. Polymerisation was performed at 45°C for 24 h. For transmission electron microscopy (EM), ultra-thin sections were generated, treated with uranyl acetate and lead citrate (EPON-embedded tissue), and processed for electron microscopy (Carl Zeiss AG, LEO 906E). For immunogold labeling, ultrathin sections were blocked in 0.5% ovalbumine, 0.5% fish gelatine containing TBS for 30 min, followed by anti-α-synuclein (1:2000, Millipore, AB5464) incubation (in 0.5% ovalbumine containing TBS) overnight at 4°C. After washing, secondary antibody (goat-anti rabbit conjugated 10 nm gold, 1:30, British Biocell International Ltd.) was incubated for 1 h. Sections were contrasted in uranyl acetate for 3 min and examined with a LEO 906E transmission EM.
SDS-PAGE and western blotting
For SDS-PAGE and western blot analysis human brain lysates were generated as described.15 A cytosolic fraction was obtained by manual homogenization and incubation in ice-cold lysis buffer containing 25 mM TRIS-HCl pH 7.4, EDTA 1 mM, protease inhibitor cocktail (Complete Mini, Roche, 11836170001) + 0.1% SDS for 1 h, followed by centrifugation at 13000 rpm for 10 min. The resulting supernatant was taken as a post-nuclear, cytosolic fraction. To generate a fraction enriched in lysosomal membranes, samples were suspended in sucrose buffer (0.32 M sucrose, 10 mM TRIS-HCl pH 7.4, EDTA 1mM, protease inhibitor cocktail) and homogenized using 27-gauge needles, followed by centrifugation at 1000 rcf for 10 min. The supernatant was collected and again centrifuged at 20,000 rcf for 10 min. The resulting pellets were redissolved in sucrose buffer and used as a lysosome-enriched fraction.
Soluble and insoluble fractions from mouse brains were generated as described.51 Briefly, mouse brain samples were homogenized in PBS (pH 7.4), 0.32 M sucrose, 50 mM HEPES, 25 mM MgCl2, 0.5 mM DTT, 200 µg/ml PMSF, 2 µg/ml pepstatin A, 4 µg/ml leupeptin, 30 g/ml benzamidine hydrochloride and centrifuged at 1,000 × g for 10 min at 4°C. Supernatant was collected and centrifuged at 100,000 × g for one hour at 4°C. The final supernatant was collected (soluble fraction) and the remaining pellet was re-homogenized in the same buffer (insoluble fraction) and processed for SDS-PAGE.
Homogenates from transfected cell culture models were generated in homogenization buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, protease inhibitor cocktail, pH 8.0) by sonication and freeze-thawing followed by centrifugation at 4°C for 10 min with 800 rcf as described.2,15,31 Equal amounts of protein (15–40 µg) were separated on 4–12% or 12% Bis-Tris gels (Invitrogen, NP0336, NP0343), transferred to a PVDF membrane (Immobilon-P, Millipore, IPVH00010) and blocked for 1 h. Incubation with primary antibodies for α-synuclein, LAMP-1, LAMP-2A or LC3-I/II was followed by signal detection using fluorescent-labeled secondary antibodies. Immunoblots were processed and fluorescent signals were quantified. Blots were also probed for actin or α-tubulin.
Statistical analysis
Statistical analysis for comparison of groups in the in vitro experiments was performed with Student’s t-test. The in vivo experiments were conducted in triplicate on blind-coded samples. After the results were obtained, the code was broken and data were analyzed with the StatView 5.0 program (SAS Institute, Inc.). Differences among means were assessed by one-way ANOVA followed by post hoc Dunnett’s testing.
Supplementary Material
Acknowledgments
This study was supported by the Bavarian State Ministry of Sciences, Research and the Arts (ForNeuroCell), Hamburg Foundation for International Research and Studies (fellowship to D.E.F), Federal Ministry of Education and Research (01GN0979), the Albert-Raps Foundation, grants of the University Hospital, Erlangen (ELAN No. 08.11.05.1; IZKF No. TP9), Bavaria California Technology Center (BaCaTeC), Parkinson’s Disease Foundation (fellowship to D.E.F.), and the NIH grants (AG5131, NS057096, AG03197, AG10435, AG18440, AG022074 and NS038372.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/19371
References
- 1.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 2.Klucken J, Ingelsson M, Shin Y, Irizarry MC, Hedley-Whyte ET, Frosch MP, et al. Clinical and biochemical correlates of insoluble alpha-synuclein in dementia with Lewy bodies. Acta Neuropathol. 2006;111:101–8. doi: 10.1007/s00401-005-0027-7. [DOI] [PubMed] [Google Scholar]
- 3.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–22. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 4.Jaeger PA, Wyss-Coray T. All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Mol Neurodegener. 2009;4:16. doi: 10.1186/1750-1326-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 6.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 7.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 8.Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J Neurosci. 2011;31:14508–20. doi: 10.1523/JNEUROSCI.1560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 10.Dice JF. Chaperone-mediated autophagy. Autophagy. 2007;3:295–9. doi: 10.4161/auto.4144. [DOI] [PubMed] [Google Scholar]
- 11.Dunn WA., Jr. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol. 1990;110:1935–45. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. 2004;24:1888–96. doi: 10.1523/JNEUROSCI.3809-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, et al. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol. 2009;175:736–47. doi: 10.2353/ajpath.2009.080928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 reduces alpha-synuclein aggregation and toxicity. J Biol Chem. 2004;279:25497–502. doi: 10.1074/jbc.M400255200. [DOI] [PubMed] [Google Scholar]
- 16.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–8. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 17.Klucken J, Outeiro TF, Nguyen P, McLean PJ, Hyman BT. Detection of novel intracellular alpha-synuclein oligomeric species by fluorescence lifetime imaging. FASEB J. 2006;20:2050–7. doi: 10.1096/fj.05-5422com. [DOI] [PubMed] [Google Scholar]
- 18.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–9. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 19.Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009;29:13578–88. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem. 2005;280:23727–34. doi: 10.1074/jbc.M503326200. [DOI] [PubMed] [Google Scholar]
- 21.Higashi S, Moore DJ, Minegishi M, Kasanuki K, Fujishiro H, Kabuta T, et al. Localization of MAP1-LC3 in vulnerable neurons and Lewy bodies in brains of patients with dementia with Lewy bodies. J Neuropathol Exp Neurol. 2011;70:264–80. doi: 10.1097/NEN.0b013e318211c86a. [DOI] [PubMed] [Google Scholar]
- 22.Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol Dis. 2011;43:690–7. doi: 10.1016/j.nbd.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 23.Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.McLean PJ, Kawamata H, Hyman BT. Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience. 2001;104:901–12. doi: 10.1016/S0306-4522(01)00113-0. [DOI] [PubMed] [Google Scholar]
- 25.Shacka JJ, Klocke BJ, Roth KA. Autophagy, bafilomycin and cell death: the “a-B-cs” of plecomacrolide-induced neuroprotection. Autophagy. 2006;2:228–30. doi: 10.4161/auto.2703. [DOI] [PubMed] [Google Scholar]
- 26.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 27.Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem. 2003;278:44405–11. doi: 10.1074/jbc.M308041200. [DOI] [PubMed] [Google Scholar]
- 28.Shao E, Forgac M. Involvement of the nonhomologous region of subunit A of the yeast V-ATPase in coupling and in vivo dissociation. J Biol Chem. 2004;279:48663–70. doi: 10.1074/jbc.M408278200. [DOI] [PubMed] [Google Scholar]
- 29.Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010;30:12535–44. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem. 2010;285:13621–9. doi: 10.1074/jbc.M109.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67:1464–72. doi: 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]
- 32.Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis. 2009;35:385–98. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 33.Ni-Komatsu L, Tong C, Chen G, Brindzei N, Orlow SJ. Identification of quinolines that inhibit melanogenesis by altering tyrosinase family trafficking. Mol Pharmacol. 2008;74:1576–86. doi: 10.1124/mol.108.050633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rockenstein E, Schwach G, Ingolic E, Adame A, Crews L, Mante M, et al. Lysosomal pathology associated with alpha-synuclein accumulation in transgenic models using an eGFP fusion protein. J Neurosci Res. 2005;80:247–59. doi: 10.1002/jnr.20446. [DOI] [PubMed] [Google Scholar]
- 35.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–60. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–88. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, et al. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 38.Hunke S, Döse S, Schneider E. Vanadate and bafilomycin A1 are potent inhibitors of the ATPase activity of the reconstituted bacterial ATP-binding cassette transporter for maltose (MalFGK2) Biochem Biophys Res Commun. 1995;216:589–94. doi: 10.1006/bbrc.1995.2663. [DOI] [PubMed] [Google Scholar]
- 39.Sharom FJ, Yu X, Chu JW, Doige CA. Characterization of the ATPase activity of P-glycoprotein from multidrug-resistant Chinese hamster ovary cells. Biochem J. 1995;308:381–90. doi: 10.1042/bj3080381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teplova VV, Tonshin AA, Grigoriev PA, Saris NE, Salkinoja-Salonen MS. Bafilomycin A1 is a potassium ionophore that impairs mitochondrial functions. J Bioenerg Biomembr. 2007;39:321–9. doi: 10.1007/s10863-007-9095-9. [DOI] [PubMed] [Google Scholar]
- 41.Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE, et al. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol. 2006;69:1125–36. doi: 10.1124/mol.105.018408. [DOI] [PubMed] [Google Scholar]
- 42.González-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquère S, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 43.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–8. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–52. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 45.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–24. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 46.Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, et al. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–78. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- 47.Veinbergs I, Van Uden E, Mallory M, Alford M, McGiffert C, DeTeresa R, et al. Role of apolipoprotein E receptors in regulating the differential in vivo neurotrophic effects of apolipoprotein E. Exp Neurol. 2001;170:15–26. doi: 10.1006/exnr.2001.7684. [DOI] [PubMed] [Google Scholar]
- 48.Kawamata H, McLean PJ, Sharma N, Hyman BT. Interaction of alpha-synuclein and synphilin-1: effect of Parkinson’s disease-associated mutations. J Neurochem. 2001;77:929–34. doi: 10.1046/j.1471-4159.2001.00301.x. [DOI] [PubMed] [Google Scholar]
- 49.Pedraza CE, Bassett DC, McKee MD, Nelea V, Gbureck U, Barralet JE. The importance of particle size and DNA condensation salt for calcium phosphate nanoparticle transfection. Biomaterials. 2008;29:3384–92. doi: 10.1016/j.biomaterials.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 50.Van Agtmael T, Schlötzer-Schrehardt U, McKie L, Brownstein DG, Lee AW, Cross SH, et al. Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum Mol Genet. 2005;14:3161–8. doi: 10.1093/hmg/ddi348. [DOI] [PubMed] [Google Scholar]
- 51.Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010;277:3051–67. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.