

Abstract
Background
The direct role of coronary vessels in defibrillation, although hypothesized to be important, remains to be elucidated. We investigate how vessel-induced virtual-electrode polarizations assist reentry termination.
Methods and Results
A highly anatomically-detailed rabbit ventricular slice bidomain computer model was constructed from 25μm MR data, faithfully representing both structural and electrical properties of blood vessels. For comparison, an equivalent simplified model with intramural cavities filled-in was also built. Following fibrillation induction, 6 initial states were selected and biphasic shocks (5–70V) applied using a realistic Implanted Cardioverter Defibrillator electrode configuration. A fundamental mechanism of biphasic defibrillation was uncovered in both models, involving successive break excitations (after each shock phase) emanating from opposing myocardial surfaces (in septum/LV), which rapidly closed-down excitable gaps. The presence of vessels accelerated this process, achieving more rapid and successful defibrillation: defibrillation failed in 5 cases (all due to initiation of new activity), compared to 8 with the simplified model (5/8 failures due to surviving activity). At stronger shocks, virtual-electrodes formed around vessels, rapidly activating intramural tissue due to break excitations, assisting the main defibrillation mechanism and eliminating all activity <15ms of shock-end in 60% of successful shocks (36% in simplified model). Subsequent analysis identified only vessels approximately >200um diameter participated via this mechanism. Consequently, wavefronts could survive intramurally in the simplified model, leading to reentry and shock-failure.
Conclusions
We provide new insight into defibrillation mechanisms by showing how intramural blood vessels facilitate more effective elimination of existing wavefronts, rapid closing-down of excitable gaps and successful defibrillation, and give guidance towards the required resolution of cardiac imaging and model generation endeavors for mechanistic defibrillation analysis.
Keywords: electrophysiology, vasculature, defibrillation, fibrillation
Introduction
Although application of a strong defibrillation shock to the heart remains the only reliable means of terminating ventricular fibrillation (VF), many aspects underlying its success still remain a mystery. Specifically, the exact mechanisms by which externally-applied electric fields interact with cardiac tissue structure to successfully activate a sufficient mass of myocardium[1] and achieve defibrillation lack comprehensive understanding.
Experimentally, much of our current understanding of defibrillation mechanisms has been derived from whole-ventricle optical mapping investigations[2-4] which only probe polarization patterns from tissue layers close to the epicardial surface[5]. However, fine-scale discontinuities in tissue structure, known to be present throughout the myocardial wall, have been postulated to play a crucial role in defibrillation through the creation of virtual-electrodes, as current induced by the applied field is redistributed, assisting in the bulk activation of myocardium. More recent transmural optical mapping recordings from excised left-ventricular (LV) wedge preparations[6], alongside similar high-resolution computer bidomain simulations[7,8], have confirmed the existence of intramural virtual-electrodes following shocks applied during diastole. However, experimental limitations in optical resolution and distortion due to photon scattering[5] have thus far prevented direct correlation of virtual-electrodes with specific intramural structures. Furthermore, restrictions in model domain size of both experiments and simulations has limited examination of the direct implications of shock-induced intramural activation upon reentry termination and defibrillation.
Despite the theorized importance of intramural structures in virtual-electrode formation and defibrillation, existing knowledge of the effects of shocks at the whole-ventricle level has been obtained largely from more simplified computer models, lacking any form of intramural structures[9]. Nonetheless, close agreement has been found throughout with experimental observations of global defibrillation mechanisms made from whole-ventricular surface optical mapping experiments[3,5,10], questioning the importance of incorporating such fine-scale features within models and their relevance in defibrillation. Recent advances in MR imaging[11,12], however, have facilitated the inclusion of unprecedented levels of anatomical detail, including representations of the coronary vascular system, within whole-ventricular computational models[8,12], presenting an opportunity to perform such investigation.
As blood vessels represent the largest discontinuities within the myocardial wall, they thus suggest an important substrate for virtual-electrode formation. In a recent study[8], we demonstrated how fine-scale information regarding the coronary vasculature could be incorporated into a detailed computational LV wedge model from high-resolution MR data[12], showing how virtual-electrodes form around blood vessels following shocks applied during action potential plateau, assisting intramural myocardium activation. However, the study did not directly relate its findings to clinical defibrillation mechanisms.
In this study, we aim to investigate the causal link between vessel-mediated virtual-electrode formation during clinically-realistic defibrillation shock application and successful reentry termination. To facilitate our investigation, a highly anatomically-detailed rabbit ventricular slice bidomain model was constructed from 25μm MR data[12], representing both structural and electrical properties of blood vessels[8]. Biphasic defibrillation shocks were applied via an implanted cardioverter defibrillator (ICD) electrode set-up to a variety of different fibrillation episodes. Comparison of results to a simplified model (lacking any form of intramural structures) allowed dissection of the specific role played by the coronary vasculature, made possible through direct examination of transmembrane potential (Vm) dynamics throughout the full 3D volume of the myocardial walls, thus overcoming an inherent limitation of experimental investigations.
Methods
Computational Model
A tetrahedral finite element ventricular slice model (thickness 1.5mm) was generated from a previously-published high-resolution rabbit MR data-set (resolution 25μm)[12] (Figure 1(a)), following prior segmentation and manual removal of free papillary muscles. The mesh (Figure 1(b)) consisted of both myocardial tissue and surrounding extracellular bath, filling ventricular and intramural cavities. Blood vessels within the slice model were identified and tagged. Mean vessel density was approximately 1.34 vessels/mm2 with minimum vessel cavity diameter represented ~100μm.
Figure 1.

(a) Axial-slice from rabbit MR data-set[12]. (b)Finite element ventricular slice model with highlighted region showing cardiac fiber orientation assigned using rule-based method[8].
Cardiac fiber architecture was assigned using a rule-based method[8], accounting for transmural variation in helix angle and continuous fiber negotiation around intramural structures seen in histology (Figure 1(b)). To dissect the specific role played by intramural structures, a simplified model was also produced in which all intramural cavities were filled-in during segmentation, prior to meshing. Identical stimulation protocols were applied to both vessel/simplified models throughout.
Simulating Electrical Activation
Electrical activation was simulated using the bidomain equations[13], solved with the Cardiac Arrhythmia Research Package[14]. Conductivities were based on experimentally-derived values[15], scaled to reduce conduction velocity by 25%, as occurs during heart failure. Electrical conduction through the connective tissue of the vessel lumen wall was reduced by assigning the experimentally derived conductivity[8] of 0.010S/m to extracellular bath elements that directly bordered the vessel cavity/myocardium interface. Bath conductivity, including within vessel cavities, was 1.0S/m. Membrane dynamics were represented by a recent rabbit ventricular cell model[16], slightly adjusted to produce sustained VF-like activity, and further augmented by two additional currents, activated at large potentials, to simulate membrane responses to strong shocks. See Supplemental Material for detailed descriptions of computational methods.
Simulation Protocol
Induction of Fibrillation
Fibrillation was induced as described in Supplemental Material. A selection of 6 initial states were chosen from the fibrillatory episodes, which acted as pre-shock states for defibrillation shock delivery, providing a range of possible initial conditions. As closely as possible, initial states were matched between vessel/simplified models, notwithstanding the inherent complexity of the fibrillatory-episodes (shown in Supplemental Material Figure 3).
Defibrillation Shock Application
An ICD-like electrode configuration for defibrillation shock delivery included a catheter placed in the right ventricle (RV) (diameter 6 French[17]) and active can in the bath near the posterior LV (shown in Figure 2(a)). Biphasic defibrillation shocks of shock-strength (SS) between 5–70V (leading-edge voltage) were delivered to the selected initial fibrillatory states. The RV catheter acted as the anode during the first shock phase[10] and the active can ground, with polarity reversed during the second phase with magnitude 50% of the first. Tilt and duration of each phase were 50% and 3.5ms, respectively. The distribution of extracellular potential (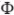 e) within the simplified model is shown in Figure 2(a), showing regions of high field-gradient concentrated primarily in the posterior LV and septum. Figure 2(b)&(c) then compare the response of the vessel and simplified models to a 40V shock applied during diastole, by showing a difference map (following mapping of data between respective meshes) of induced extracellular potential gradient e within the tissue (panel b) and induced Vm (panel c) 1ms into the shock. These difference maps highlight the significant magnitude of variation in both e and Vm within intramural myocardial tissue, focussed around vessel cavities and within regions of high field-strength (posterior LV and septum) between vessel/simplified models.
e) within the simplified model is shown in Figure 2(a), showing regions of high field-gradient concentrated primarily in the posterior LV and septum. Figure 2(b)&(c) then compare the response of the vessel and simplified models to a 40V shock applied during diastole, by showing a difference map (following mapping of data between respective meshes) of induced extracellular potential gradient e within the tissue (panel b) and induced Vm (panel c) 1ms into the shock. These difference maps highlight the significant magnitude of variation in both e and Vm within intramural myocardial tissue, focussed around vessel cavities and within regions of high field-strength (posterior LV and septum) between vessel/simplified models.
Figure 2.

(a) ICD electrode set-up and e distribution within simplified model 1ms into 40V defibrillation shock; Difference map of induced e (panel (b)) and Vm (panel (c)) between vessel and simplified models.
Data Analysis
Induced post-shock arrhythmias were defined as sustained if reentrant activity lasted for >100ms. Tissue was classified as excitable if Vm<−60mV (inactivation threshold of sodium current[10]). Intramural tissue was defined as points lying within 0.25<e<0.75, where e is the normalized transmural distance from endo- to epicardium. See Supplemental Material Section 1.4.3 regarding statistical significance of simulation data.
Results
Role of Vessels in Shock Success
Defibrillation shocks of SS 5/10/20/40/70V were applied to the 6 initial fibrillatory states of the two models. Analysis of post-shock activation patterns of the 30 episodes revealed defibrillation failed in just 5 cases in the vessel model, compared to 8 in the simplified model. Of those failed shocks, reentry was reinitiated due to new activation wavefronts induced by the shock in 4/5 of the failed vessel model shocks. In the simplified model, however, 5/8 failures were due to existing activity which failed to be extinguished by the shock.
The presence of vessels also affected the time-course over which successful defibrillation occurred. Of those successful shocks, all wavefronts throughout the model were entirely extinguished within 15ms of shock-end in 15 (/25, 60%) cases in the vessel model, compared to just 8 (/22, 36%) in the simplified case.
Uncovering a Common Biphasic Defibrillation Mechanism
Analysis of activation patterns during successful defibrillation episodes revealed a common mechanism of biphasic defibrillation, witnessed throughout all initial states and SS. Importantly, the fundamental basis of this mechanism was found to be common to both models, independent of the presence of vessels.
Figure 3(a) demonstrates how successful defibrillation is achieved via this mechanism, showing evolution of activity throughout a 40V shock in the simplified model. Large excitable gaps exist in the LV and septum prior to the shock (0ms). The first shock phase strongly depolarizes the anterior LV endocardial wall and large regions of the LV-septum endocardium (3.5ms). At the end of this first phase, break excitations are elicited from these strongly polarized walls which are free to propagate into the large intramural excitable gaps in the LV and septum. However, the second phase of the shock now strongly depolarizes the RV-septum endocardium, as well as the anterior LV epicardium (7ms). As witnessed previously[4], the energy delivered in the second phase of the shock is sufficient only to reverse the negative polarization previously established by the shock, whilst partially preserving positive polarization. At shock-end, a second set of break excitations is thus elicited which propagate into the intramural excitable gaps of the LV and septum, in the opposite direction to the first set following phase 1. Consequently, these successive break excitations from opposing myocardial surfaces, elicited at the end of each shock phase, very rapidly close-down intramural excitable regions in the LV and septum, like drawing a curtain (10ms).
Figure 3.

Common mechanism of successful biphasic defibrillation. Vm distribution pre-, during and post- 40V shock (panel (a)) and at shock-end for SS5/10/20V (panel (b)) applied to simplified model state I4. (c) Post-shock Vm distribution of 40V shock applied to corresponding vessel model state I4.
Although this biphasic defibrillation mechanism was clearly evident for strong shocks (including 70V, not shown), its specific operation was strongly dependent upon applied SS. Figure 3(b) shows corresponding shock-end (7ms) Vm distributions following 5/10/20V shocks in the simplified model. For weak shocks (5V), the second shock phase (P2) lacks sufficient strength to reverse the negative polarization set-up by phase 1 (P1). Thus, only a single excitation from the first phase exists at shock-end, only occurring in regions of relatively high field-strength in the LV/septum. For intermediate shocks (10V), both phases have sufficient strength to elicit strong surface depolarizations and induce two separate break excitations, however, their extent is not as widespread as for stronger shocks, leading to an asymmetrical (and slower) closing-down of excitable gaps in the LV/septum. At 20V, the break excitation pattern from the shock becomes more similar to that seen at 40V (Figure 3(a)), although the speed at which the break excitations propagate and close-down the excitable gaps increases with SS (discussed below).
Finally, Figure 3(c) shows the corresponding post-shock 10ms image following a 40V shock in the vessel model, demonstrating a very similar mechanism to Figure 3(a). However, here less of the excitable gap remains, particularly in the septum, suggesting the presence of vessels assists in closing-down the excitable gap via this biphasic mechanism, which we now investigate further below.
Role of Vessel-Mediated Virtual-Electrode Formation in Defibrillation
During all strong shocks (20–70V), virtual-electrodes were seen to form around blood vessel structures in all defibrillation episodes in the vessel model, largely focussed around regions of high field-strength (septum/posterior LV), and being more significant around larger vessel structures. Following the shock, break excitations were elicited from these virtual-electrodes, most evident in intramural regions containing excitable areas, which themselves were relatively unaffected by the direct action of the shock.
Interaction with Main Biphasic Defibrillation Mechanism
Figure 4(a) shows an example of vessel-mediated virtual-electrode formation following a 40V shock applied to state I1 of the vessel model. Here, vessel-induced break excitations produce new wavefronts, which propagate through excitable intramural tissue between the larger break excitation wavefronts (originating from myocardial surfaces) brought about by the main defibrillation mechanism. Consequently, the intramural excitable gap, located predominantly within the septum, is more rapidly closed-down than in the corresponding 40V post-shock simplified model case (Figure 4(b)) where large excitable gaps remain, most noticeable in the septum.
Figure 4.

Vessel-mediated virtual-electrode formation. (a) Shock-end and post-shock Vm distributions following 40V shock applied to vessel model state I1. (b) Corresponding post-shock simplified model Vm distribution. (c) Variation in percentage of excitable tissue within intramural septum throughout shock for all SS applied to states I1 of vessel (solid-lines) and simplified (dashed-lines) models.
Figure 4(c) quantifies this effect, showing how the percentage of intramural excitable septal tissue changes pre- and post-shock for all SS applied to states I1. In the vessel model, as SS increases, less excitable tissue exists within the intramural septum both at shock-end and post-shock. For higher SS, the virtual-electrodes formed around vessel cavities become stronger and larger, increasing the amount of intramural tissue depolarized at shock-end. These larger virtual-electrodes are then more able to elicit break excitations upon cessation of the shock, depolarizing larger amounts of tissue post-shock as they propagate away from cavities.
In contrast, the amount of intramural septal excitable tissue within the simplified model remains relatively constant throughout the shock for all SS in the absence of intramural cavities around which virtual-electrodes may form and because the direct effect of the shock decays exponentially from the tissue surface, not affecting intramural tissue. Although there is a decrease in excitable tissue post-shock, as break excitations from either side of the septal wall (due to the main defibrillation mechanism) invade the intramural area, the extent of the excitable area is still significantly greater than the corresponding area in the vessel model. Note, however, that such an effect is most significant where regions of large pre-shock excitable gaps coincide with areas of high field-strength.
Overall Quantitative Effect of Vessels
Above, we focussed on one particular defibrillation episode, demonstrating how vessel-mediated virtual-electrodes assist defibrillation by closing-down intramural excitable gaps. Here, we quantify this trend across all episodes, investigating how applied SS affects the witnessed mechanism.
Figure 5(a) shows that the mean decrease in LV/septum intramural excitable tissue (relative to pre-shock), both at shock-end (left) and post-shock (right), is greater in the presence of vessels; a difference which increases with SS as vessel-mediated virtual-electrodes increase in size, strength and number, most evident at shock-end. For example, the simplified model shows a relatively small decrease even at high SS: 28.5% at 70V compared to 63.8% decrease in the vessel model. Post-shock, differences between the models are less, due to propagation of surface-mediated break excitations from the main mechanism into intramural regions. Overall, for all 30 pairs of defibrillation episodes (5SS, 6 initial states), the greater decrease seen in the vessel model was statistically significant for both shock-end (p<0.0003) and post-shock (p<0.0025) cases, assessed using a Wilcoxon signed-rank test (see Supplementary Methods for specific details). Note, the RV was not included as the direct action of the shock strongly affected all intramural regions.
Figure 5.

(a) Mean percentage decrease in volume of excitable intramural tissue within LV/septum, averaged over all 6 defibrillation episodes, as a function of SS at shock-end (left) and post-shock (right) for vessel (blue solid-line, squares) and simplified (red dashed line, circles) models. (b) Percentage of successful defibrillation episodes in which all activity is entirely eliminated <15ms of shock-end as function of SS for vessel (blue solid-line, squares) and simplified (red dashed-line, circles) models.
Such a vessel-assisted reduction in intramural excitable regions following the shock also affected the overall time-course over which successful defibrillation occurred. Figure 5(b) shows that the percentage of defibrillation shocks to achieve entire elimination of all wavefronts <15ms following the shock is greater in the vessel model over all SS. This difference is again statistically significant considering this binary outcome over all 30 matched pairs of defibrillation episodes with p<0.03, assessed using a McNemar test to compare proportions in the 2 models (see Supplemental Material for details). However, differences between models become most noticeable at strong SS due to the more widespread formation of virtual-electrodes surrounding vessels which also become larger and stronger.
Finally, in addition to vessels interacting via break excitation wavefronts formed through the main defibrillation mechanism, they were also frequently seen to play an important role through their interaction with existing activation wavefronts, described in the Supplemental Material.
Defibrillation Failure due to Intramural Wavefront Survival
In one particular strong shock episode (40V), lack of activation of intramural areas due to the absence of vessels in the simplified model resulted in defibrillation failure, shown in Figure 6; comparatively, defibrillation succeeded in all such strong shock episodes in the vessel model. Here, pre-existing refractory tissue in the anterior LV wall prevents propagation of break excitations mediated from epicardial/endocardial surfaces via the main defibrillation mechanism into intramural tissue (7ms). Furthermore, the lack of vessels means that the existing wavefront within the septum (0ms) is largely unaffected by the shock, which thus continues to propagate through the, now recovered, excitable region in the anterior LV wall (35ms). Consequently, this sole-existing intramural wavefront continues its progression, eventually breaks-up and causes defibrillation failure.
Figure 6.

Vm distribution during 40V episode applied to simplified model state I3 showing intramural wavefront leading to defibrillation failure.
Effect of Vessels During Weak Defibrillation Shocks
Although most evident during strong shocks, vessels were also seen to have an important impact during weaker shocks (5–10V). Figure 7 shows such an example, demonstrating the occurrence of conduction block in the vessel model due to the formation of a weak, but noticeable, depolarization around a large blood vessel in the RV free-wall (highlighted), insufficient to induce break excitation itself. In contrast, the RV wall in the simplified model remains unaffected by the shock; thus, the wavefront (present in a similar location to that in the vessel model), can propagate freely through the wall assisting the sustenance of the arrhythmia.
Figure 7.

Vm distributions during 5V shock applied to state I3 of vessel model (top) and simplified model (bottom) demonstrating conduction block due to mild virtual-electrode formation around large vessel in RV (highlighted) terminating the wavefront.
Identification of Minimum Vessel Cavity Size of Importance
Having identified that vessels play their most important role in defibrillation by providing the nucleus for break excitations which help close down local excitable gaps, we now identify the minimum vessel cavity size which supports this mechanism. A series of highly-simplified models representing vessel cavities of radii 50-500μm within a wedge of ventricular wall were created (see Supplemental Material) and biphasic shocks of SS 2.5-75V/cm applied. Figure 8(a) shows Vm distributions 1 & 7ms into the shock for a selection of stronger shocks applied to smaller cavity models (50-100μm).
Figure 8.

(a) Vm distributions 1/7ms into shocks applied to wedge models with different radii cavities. (b) Minimum SS required to induce break-excitation as a function of cavity radii. (c) Vm distributions 1ms into shock for models with increased vessel packing densities.
The smaller cavities act primarily like insulators, causing virtual-electrode patterns as current redistributes between intra-/extracellular domains (due to respective differences in fiber/cross-fiber conductivities) as it is diverted around the cavity. However, intriguing to note is that larger cavity sizes act primarily like conductors, causing different virtual-electrode patterns as current passes through the extracellular space of the cavity (see Supplemental Material).
Figure 8(b) plots the minimum SS at each cavity radius required to induce break excitations within the tissue. These Figures together show that even the strongest 75V/cm shock cannot induce a break-excitation within the 50μm model, and is just strong enough to induce one in the 75μm model, but that the 100μm model has an excitation threshold of just 40V/cm. Such cavity dimensions at which an appreciable effect is witnessed is of the same order-of-magnitude as the transverse length constant (~0.2mm) as expected from theoretical considerations.
To replicate potential electrotonic interactions of increased packing density of smaller vessels (50-100μm radii) the protocol was repeated to 4 vessels of each size evenly packed within a 0.81mm2 square (4.9mm−2). This increased packing density did not change the minimum SS predicted by Figure 8(b) for the 50-100μm radius cavities. Finally, the packing density was increased further for the 50μm vessels to 9 (11.1mm−2) which still did not induce a break excitation at 75V/cm. Vm distributions 1ms into 75V/cm shocks are shown in Figure 8(c) for the 50μm cavity for 4 and 9 vessels.
Discussion
In this study, we highlight the importance of the coronary vasculature during clinically-realistic defibrillation using an MR-derived high-resolution rabbit ventricular bidomain model, which facilitated unprecedented access to intramural polarization distributions during and following biphasic shocks. Specifically, we elucidate the mechanisms by which vessel-mediated virtual-electrode formation assist termination of existing reentrant activity, acting synergistically with the main biphasic defibrillation mechanism also uncovered (driven by excitation of external myocardial walls), and identify a minimum cavity size necessary for this mechanism.
Common Mechanism for Biphasic Defibrillation
At the tissue/organ level, the main experimentally-driven theories for the increased efficacy of biphasic (as opposed to monophasic) defibrillation shocks have centered on how biphasic waveforms fail to produce a substrate for re-initiation of fibrillation[2,4]. However, unlike our modelling approach, such surface measurement techniques cannot elucidate how biphasic shocks effectively eliminate existing intramural activity.
Our explicit analysis of intramural polarization levels during and after both shock phases allowed us to uncover a fundamental mechanism by which successful biphasic defibrillation was achieved over a range of SS and initial states (Figure 3). The mechanism involved the rapid closing-down of excitable gaps via successive break excitations from exterior myocardial walls (set-up after each shock-phase). Strong shocks induced stronger and more complete break excitations, extending more fully along myocardial walls than weaker shocks, eliminating excitable tissue more efficiently (Figure 3(b)). Although a similar mechanism of biphasic defibrillation has been suggested previously in a 1D fiber model[18], here we present novel elucidation of the mechanistic analysis of its operation during clinically-relevant defibrillation at the ventricular level.
Although assisted by the presence of vessels, the fundamental basis of the mechanism was seen to be common to both vessel and simplified models (Figure 3(a)&(c)). Such a finding provides an explanation for the lack of a significantly large difference in defibrillation success rates between the two models as well as the close comparison between previous simulations studies using more simplified models[3,10] with experimental findings.
Interaction Between Vessel-Mediated Virtual-Electrodes and Defibrillation
Fine-scale intramural discontinuities in tissue structure have been postulated to play a crucial role in defibrillation through creation of virtual-electrodes, assisting bulk activation of a critical myocardial mass. For the first time, our high-resolution bi-ventricular model has allowed investigation of how fine-scale structures, such as blood vessels, impact the process of defibrillation using a clinically-realistic ICD set-up.
This study has demonstrated that virtual-electrodes form around intramural vessel cavities, helping eliminate pre-shock intramural excitable gaps and extinguish existing reentrant activity. Such shock-induced vessel-mediated effects were seen to increase, and thus depart further from the simplified model, with SS; as virtual-electrodes become larger, stronger and more wide-spread with increasing SS (as field strength is sufficiently high in more areas), a larger volume of intramural tissue is directly activated by the shock, and quicker eradication of post-shock excitable gaps by faster propagating break excitations occurs. Although in our ventricular slice model, the more rapid closing-down of post-shock excitable gaps due to vessels did slightly increase defibrillation success, this mechanism may be of more importance at the whole ventricle level.
Even at weak shocks, mild shock-induced depolarizations around large vessels were seen to result in conduction block, assisting reentry termination. In addition, such mild effects can interact more subtly, slightly slowing intramural wavefronts both through direct interaction with the wavefront itself (Supplemental Material Figure 5), and the creation of mild depolarizations in excitable gaps in its path (not shown). Consequently, both these effects demonstrate how vessels can assist in the disruption of existing (intramural) fibrillatory activity, even at weak shocks, suggesting an explanation for the overall defibrillation success of the vessel model.
Finally, previous conceptual studies in 2D sheet models have suggested the importance of small-scale virtual-electrodes formed by microscopic fluctuations in tissue conductivity in preventing re-initiation of reentry during defibrillation[19,20]. Although here we focus primarily on extinguishing existing fibrillatory activity, we suggest that virtual-electrodes induced around vessels (representing larger heterogeneities) could also play an important role via this mechanism.
Virtual-Electrode Patterns Produced by Vessels
The wedge model in Figure 8 allowed careful analysis of how the shock-induced break excitation mechanism scaled with vessel size. It also uncovered an intriguing fundamental biophysical interaction between shock-induced current flow and the vessel cavity. Specifically, the insulating effects of the low conductivity lumen wall combined with the highly-conducting bath within the cavity leads to size-dependent polarization. Small diameter vessels act as insulators since the resistance of crossing the lumen is large relative to the added intracellular resistance encountered negotiating the vessel. With larger diameters, intracellular path length increases while increased surface area reduces lumen resistance, making the vessel act more like a conductor. Conductor and insulator effects differ in their induced polarity since one will promote intra- to extracellular current flow while the converse occurs in the other. However, the most important issue identified in this study is that virtual-electrodes, regardless of polarity and underlying mechanism, form around vessels and initiate break excitations which help defibrillate. Overall, the results from this study qualitatively strongly agree with a recent experimental study identifying the importance of vessels during low-energy defibrillation therapies[21], and reinforce their findings by providing important 3D knowledge regarding the intramural behaviour of the vasculature during such protocols. Uncovering such an important biophysical mechanism could have important applications in helping understand similar interactions of electric fields with other heterogeneities such as infarct scars or myolaminar sheets[7].
Implications for Simplified Models
In the simplified model, even at stronger SSs where the direct activation of the shock extends further into the mid-wall (due to virtual-electrode effects as a result of fiber curvature and bi-ventricular geometry, for example), large intramural excitable regions still exist at shock-end, which were seen to be present to a lesser extent in the vessel model. Consequently, in Figure 7 we uncovered a case in which defibrillation failed in the simplified model at a strong (40V) shock, compared to the vessel model which successfully defibrillated all episodes. Here, the absence of vessel-induced effects in the mid-wall provided an excitable avenue in the LV/septum junction through which a wholly intramural wavefront could continue to propagate, sustaining reentry. As this mechanism is similar to that of tunnel propagation (shown recently in a study using a simplified whole-ventricular model[10]), the results from our study may suggest that such tunnel propagation is suppressed in the presence of vessels. However, we believe that the high non-uniformity of the ICD-field (particularly within a full ventricular model) will in fact leave many regions of the thick LV wall with an insufficient field-strength to produce virtual-electrodes around vessels, thus leaving intramural excitable gaps through which tunnel propagation may occur. Nevertheless, we could expect the presence of vessels within the model to attenuate the degree of potential tunnel propagation avenues, and possibly help explain differences in their location witnessed between simulations and experiment[10].
Implications for High-Resolution Cardiac Imaging and Model Generation
We have identified that only vessel cavities greater than approximately 100μm radius play an important role in defibrillation by providing the nucleus for shock-induced break excitations which rapidly close-down excitable gaps in the vicinity of the cavity (Figure 8). The specific relationship between cavity size and required SS agrees qualitatively with that predicted in a recent analytic study using a simplified linearized bidomain approximation[21] and quantitatively predicts a similar minimum cavity radius (~150μm). This finding has significant implications for high-resolution cardiac image acquisition and computational model development[11,12] for mechanistic defibrillation investigation, providing an important lower limit on the required resolution of a particular imaging modality used to generate the model. Identifying features >200μm across suggests that lower resolution (~100μm) MR and DTMRI may be sufficient for model construction as opposed to the potential use of histological data which presents many more challenges both for imaging and model generation[7,11]. Finally, coarser computational models may facilitate the future use of finite element meshes with fewer degrees of freedom, lowering the burden of such highly computationally intensive simulations, widening the scope of what is tractable to simulate.
Study Limitations
Although our model incorporates an unprecedented level of structural detail, it is somewhat limited by its slice geometry. Primarily, although the bi-ventricular geometry still faithfully represents many important reentry pathways, the nature of the sustained arrhythmias pre-shock, as well as post-shock activation sequences, may differ when scaled-up to the whole ventricular level. However, throughout the study, conclusions were drawn following comparisons made between the vessel and simplified models, and care was taken to match the initial complexity of fibrillation episodes. Thus, any limitations in arrhythmia dynamics due to model size restrictions would have impacted both models equally. Finally, our model represents important heterogeneity in ICD-field gradient in x,y-directions (Figure 2(a)). Shock-induced virtual-electrode polarizations on epi-/endocardial surfaces and vessel cavities, and subsequent propagation of break excitations, also then occurr primarily in this plane, thus captured well by our model.
Despite using a state-of-the-art cardiac simulation environment[14], the large, highly-detailed mesh combined with small time-steps required for strong ICD shocks resulted in significant computational demands on each simulation (10ms bidomain shock episode required ≈20hrs on 32 cores). Consequently, the number of initial fibrillatory states, maximum SS and number of SSs used were limited, preventing computation of a full DFT-90 plot[10] and assessment of whether our results of overall defibrillation success were of statistical significance.
Although our MR-derived model contains intramural vessel cavities, absent from our model is fine-scale detail regarding inter-myolaminar cleavage planes, seen in histological reconstruction models[7]. The histological pre-processing stage used to acquire such data involves dehydrating the tissue, reducing myocyte volume, and exacerbating apparent cleft sizes. However, such structures are not visible in lower resolution (fully hydrated) MR data. Whilst simulations[7] and experiments[22] have identified the potential importance of clefts as a substrate for virtual-electrode formation, it remains an open question to what extent this is affected by accentuated cleft size.
Supplementary Material
Acknowledgements
The authors acknowledge the Oxford 3D Heart Project for access to MR data, and Oxford Supercomputing Centre.
Funding Sources: MJB is supported by the Wellcome Trust (Sir Henry Wellcome Postdoctoral Fellowship), GP by Austrian Science Fund FWF grant (F3210-N18) and EJV by Natural Sciences and Engineering Research Council of Canada and MITACS.
Footnotes
Conflict of Interest Disclosures: Cardiosolv LLL, Baltimore, Maryland (EJV & GP).
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zipes D, Fischer J, King R, Nicoll A, Jolly W. Termination of ventricular fibrillation in dogs by depolarizing a critical amount of myocardium. Am J Cardiol. 1975;36:37–44. doi: 10.1016/0002-9149(75)90865-6. [DOI] [PubMed] [Google Scholar]
- 2.Ripplinger CM, Efimov IR. Cardiac Bioelectric Therapy. Springer; 2008. Ch 4.4: The virtual electrode hypothesis of defibrillation. [Google Scholar]
- 3.Rodriguez B, Li L, Eason J, Efimov IR, Trayanova NA. Differences between left and right ventricular chamber geometry affect cardiac vulnerability to electric shocks. Circ Res. 2005;97:168–175. doi: 10.1161/01.RES.0000174429.00987.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Efimov IR, Cheng Y, Yamanouchi Y, Tchou PJ. Direct evidence of the role of virtual electrode-induced phase singularity in success and failure of defibrillation. J Cardiovasc Electrophysiol. 2000;11:861–868. doi: 10.1111/j.1540-8167.2000.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 5.Bishop MJ, Rodriguez B, Qu F, Efimov IR, Gavaghan DJ, Trayanova NA. The role of photon scattering in optical signal distortion during arrhythmia and defibrillation. Biophys J. 2007;93:3714–3726. doi: 10.1529/biophysj.107.110981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fast VG, Sharifov OF, Cheek ER, Newton JC, Ideker RE. Intramural virtual-electrodes during defibrillation shocks in left ventricular wall assessed by optical mapping of membrane potential. Circulation. 2002;106:1007–1014. doi: 10.1161/01.cir.0000027103.54792.9c. [DOI] [PubMed] [Google Scholar]
- 7.Hooks DA, Tomlinson KA, Marsden SG, LeGrice IJ, Smaill BH, Pullan AJ, Hunter PJ. Cardiac microstructure: implications for electrical propagation and defibrillation in the heart. Circ Res. 2002;91:331–338. doi: 10.1161/01.res.0000031957.70034.89. [DOI] [PubMed] [Google Scholar]
- 8.Bishop MJ, Boyle PM, Plank G, Welsch D, Vigmond EJ. Modeling the role of the coronary vasculature during external field stimulation. IEEE Trans Biomed Eng. 2010;57:2335–2345. doi: 10.1109/TBME.2010.2051227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trayanova NA, Plank G, Rodriguez B. What have we learned from mathematical models of defibrillation and post-shock arrhythmogenesis? Application of bidomain simulations. Heart Rhythm. 2006:1232–1235. doi: 10.1016/j.hrthm.2006.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Constantino J, Long Y, Ashihara T, Trayanova NA. Tunnel propagation following defibrillation with ICD shocks: Hidden postshock activations in the left ventricular wall underlie isoelectric window. Heart Rhythm. 2010;7:953–961. doi: 10.1016/j.hrthm.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plank G, Burton RA, Hales P, Bishop MJ, Mansoori T, Bernabeu MO, Garny A, Prassl AJ, Bollensdorff C, Mason F, Mahmood F, Rodriguez B, Grau V, Schneider JE, Gavaghan DJ, Kohl P. Generation of histo-anatomically representative models of the individual heart: tools and application. Phil Trans Roy Soc A. 2009;367:2257–2292. doi: 10.1098/rsta.2009.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bishop MJ, Plank G, Burton RA, Schneider JE, Gavaghan DJ, Grau V, Kohl P. Development of an anatomically-detailed MRI-derived rabbit ventricular model and assessment of its impact on simulations of electrophysiological function. Am J Physiol Heart Circ Physiol. 2010;298:H698–718. doi: 10.1152/ajpheart.00606.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plonsey R. Bioelectric sources arising in excitable fibers (ALZA Lecture) Ann Biomed Eng. 1988;16:519–546. doi: 10.1007/BF02368014. [DOI] [PubMed] [Google Scholar]
- 14.Vigmond EJ, Hughes M, Plank G, Leon L. Computational tools for modeling electrical activity in cardiac tissue. J Electrocardiol. 2003;36:69–74. doi: 10.1016/j.jelectrocard.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Clerc L. Directional difference of impulse spread in trabecular muscle from mammalian heart. J. Physiol. 1976;255:335–346. doi: 10.1113/jphysiol.1976.sp011283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahajan A, Shiferaw Y, Sato D, Baher A, Olcese R, Xie L-H, Yang M-J, Chen P-S, Restrepo JG, Karma A, Garfinkel A, Qu Z, Weiss JN. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys J. 2008;94:392–410. doi: 10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matula MH, Brooks MJ, Pan Q, Pless BD, Province RA, Echt DS. Biphasic waveforms for ventricular defibrillation: optimization of total pulse and second phase durations. Pacing Clin Electrophysiol. 1997;20:2154–2162. doi: 10.1111/j.1540-8159.1997.tb04231.x. [DOI] [PubMed] [Google Scholar]
- 18.Trayanova NA, Bray MA. Membrane refractoriness and excitation induced in cardiac fibers by monophasic and biphasic shocks. J Cardiovasc Electrophysiol. 1997;8:745–757. doi: 10.1111/j.1540-8167.1997.tb00833.x. [DOI] [PubMed] [Google Scholar]
- 19.Plank G, Leon LJ, Kimber S, Vigmond EJ. Defibrillation depends upon conductivity fluctuations and the degree of disorganization of reentry patterns. J Cardiovasc Electrophysiol. 2005;16:205–216. doi: 10.1046/j.1540-8167.2005.40140.x. [DOI] [PubMed] [Google Scholar]
- 20.Mazeh N, Roth BR. A mechanism for the upper limit of vulnerability. Heart Rhythm. 2009;6:361–367. doi: 10.1016/j.hrthm.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luther S, Fenton FH, Kornreich BG, Squires A, Bittihn P, Hornung D, Zabel M, Flanders J, Gladuli A, Campoy L, Cherry EM, Luther G, Hasenfuss G, Krinsky VI, Pulmir A, Gilmour RF, Bodenschatz E. Low-energy control of electrical turbulence in the heart. Nature. 2011;475:235–239. doi: 10.1038/nature10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fast VG, Rohr S, Gillis A, Kleber A. Activation of Cardiac Tissue by Extracellular Electrical Shocks: Formation of Secondary Sources at Intercellular Clefts in Monolayers of Cultured Myocytes. Circ Res. 1998;82:375–385. doi: 10.1161/01.res.82.3.375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.