

Abstract
C/EBPα (CEBPA) is mutated in approximately 8 % of AML in both familial and sporadic AML and, with FLT3 and NPM1, has received most attention as a predictive marker of outcome in patients with normal karyotype disease. Mutations clustering to either the N- or C-terminal (N-and C-ter) portions of the protein have different consequences on the protein function. In familial cases the N-ter form is inherited with patients exhibiting long latency period before the onset of overt disease, typically with the acquisition of a C-ter mutation. Despite the essential insights murine models provide the functional consequences of wild-type C/EBPα in human hematopoiesis and how different mutations are involved in AML development have received less attention. Our data underline the critical role of C/EBPα in human hematopoiesis and demonstrate that C/EBPα mutations (alone or in combination) are insufficient to convert normal human hematopoietic stem/progenitors (HSC/HPCs) into leukemic initiating cells, although individually each altered normal hematopoiesis. It provides the first insight into the effects of N- and C-terminal mutations acting alone and to the combined effects of N/C double mutants. Our results mimicked closely what happens in CEBPA mutated patients.
Keywords: Leukemic stem cell, oncogene, AML, xenotransplantation, C/EBPα
Introduction
Acute Myeloid Leukemia (AML) is defined by an accumulation of immature myeloblasts in the periphery and bone marrow (BM). Like other cancers, AML is a multistep process characterized by an alteration of different pathways affecting cell proliferation and myeloid differentiation (1-3). Although the specific relationship between altered proliferation and differentiation in AML remains elusive, a two-hit model has been proposed (4) in which both an uncontrolled proliferation (and/or apoptosis) and a block of differentiation are required to progress to AML with neither of these alone sufficient to cause leukemia. Recent studies suggests that the disruption of CCAAT/enhancer-binding protein-α (C/EBPα) transcription factor may perturb both differentiation and cell cycle (5) due to its role in cell cycle arrest, repression of self-renewal and myeloid differentiation during normal hematopoiesis (6-10) and the fact that the gene encoding this factor, CEBPA, is frequently mutated or hypermethylated in sporadic cases of AML (5, 11, 12).
CEBPA/C/EBPα is a member of the C/EBP family of bZIP transcription factors encoding two different translational isoforms of 42 kDa and 30 kDa (p42 and p30 respectively) by use of alternative AUG codons within the same open reading frame. The shorter form retains the C-terminal domain but lacks part of the N-terminal trans-activation domain and is unable to block cell cycle and induce granulocytic differentiation (11, 13-15). The effects of p42 on cell cycle control are complex due, at least in part, to the upregulation of p21WAF1 (16, 17), its interaction with cyclin-dependent kinases 2 and 4 (CDK2 and CDK4) (18), and its repression of E2F (14, 19, 20). Myeloid differentiation is equally dependent on collaboration with other specific transcription factors to regulate cell fate (7-11; 21). C/EBPα (as homo-, heterodimers) binds directly to lineage specific promoters (18, 22, 23) with a requirement for p42 to activate the transcriptional machinery (TBP/TFIIB and CBP/p300) (24, 25). It has also been speculated that both cell growth arrest and differentiation may be coupled via its link with the chromatin remodelling protein SWI/SNF (26, 27).
CEBPA is mutated in both familial and in approximately 8% of sporadic AML with a predilection for the normal karyotype sub-group (28-39). Mutations fall into two groups N-terminal truncating mutations that lead to preferential expression of p30 and an alteration in the balance between p42 and p30 isoforms and C-terminal mutations, which typical locate at the junction between the basic region and the leucine-zipper and disrupt DNA-binding and dimerization. The pattern of CEBPA mutations observed are also variable with single C-ter or N-ter mutations, rarer cases of homozygous mutation as a result of mitotic recombination (39, 40) or most frequently as biallelic mutations with the simultaneous occurrence of both N- and C- ter mutations (28, 31, 32). This is further complicated by cases where both mutations are present on the same allele. Although it has been assumed that CEBPA mutations favour a good outcome, more recent data suggest that this is confined to patients with biallelic mutations (39, 41, 42). Our group and others have identified families in which affected members have inherited a predisposing N-ter germline mutation, with the acquisition of an additional somatic C-ter mutation (36; 43-45) coinciding with the onset of disease (12, 36) leading to the hypothesis that N-ter plus C-ter mutations cooperate to induce a full blown leukemia.
The mechanism by which CEBPA mutants induced leukemogenesis has been addressed in mice using homozygous knock-in studies that express either p30 or p42 c/ebpα alone or both mutated forms (46, 47) or more recently by overexpression of the mutated forms in mouse hematopoietic stem/progenitor cells (48). Despite the importance of these knock-in models, the extent to which these mirror the process of naturally occurring human disease is still uncertain. With the recent success of transforming primary human HSC/HPCs with viral delivery of MLL-ENL fusion oncogenic protein, we decided to investigate the role of the different CEBPA mutations in human stem/progenitors using both in vitro and in vivo assays (49, 50).
Experimental Procedures
Lentiviral vectors
Genomic DNA from normal BM and an AML patient (38), who had a four base pairs GGCC insertion at base 363 (N-ter mutation) and an in frame internal tandem duplication of 27 base pairs at 1096 (C-ter mutation) (Genbank Y11525), were used as template to generate carboxy-terminal FLAG tagged WT CEBPA, N-ter, C-ter and NC-ter mutated CEBPA, in the pCR2.1-TOPO vector by TOPO TA Cloning (Invitrogen, Paisley, UK). Every FLAG-tagged CEBPA form was cloned downstream of an IRES-EGFP cassette, previously inserted into the pEntr1A vector (Invitrogen, Paisley, UK) with the exception of the C-ter mutated form which was cloned next to an IRES-mCherry cassette. Different CEBPA-IRES-EGFP or C-ter CEBPA-IRES-mCherry were introduced by recombination into pHR’SIN-SEW Lentiviral Vector, which had been modified for compatibility with the Gateway System (Invitrogen, Paisley, UK).
Lentiviral supernatant was produced after cotransfecting 293T cells with pMD.2 VSV-G envelope plasmid, pCMV-dR8.74 helper plasmid, and CEBPA lentiviral vectors by Calcium-Phosphate Transfection protocol (51). Virus suspension was collected, filtered and concentrated by ultracentrifugation. Lentiviral titers were determined by GFP or mCherry analysis of transduced Hela cells on a LSR II flow cytometer (BD Pharmingen, Oxford Science Park, UK). Protein lysates of transduced human Lin negative CB cells were used to identify exogenous expression of Flagged-C/EBPα by standard Western-blot using an anti-FLAG antibodies (M2, Sigma, Gillingham, UK).
Purification and Transduction of Mouse and Human Hematopoietic Progenitors
Mouse Hematopoietic Progenitors (mLin−) were purified from BM mononuclear cells of C57BL6/6J mice by Lineage Cell Depletion (MACS, MiltenyiBiotec, Germany). The mouse cells were then prestimulated in StemSpam SFEM (serum-free expansion medium, StemCells Technologies, Canada) with 50ng/mL mouse Stem Cell Factor (mSCF) (R&D Systems, Minneapolis, USA), 100ng/mL human Interleukin-11 (hIL-11), 100ng/mL human Flt-3 Ligand (hFlt-3L) and 10ng/mL human Interleukin-3 (hIL-3) (PeproTech, Rocky Hill, NJ, USA) for 4-6 hours. After which, mLin− cells were transduced with the different lentivirus. On the other hand, cord blood (CB) was collected from mothers attending the Royal London Hospital, London, UK, after informed consent and via a protocol approved by the Local Research Ethics Committees. Mononuclear cells (MNC) were obtained by Ficoll density centrifugation and ammonium chloride red cell lysis. Density-separated CB MNCs were depleted for lineage marker positive cells via the StemSep™ system (Stem Cell Technologies, Canada) according to the manufacturer’s instructions to generate Lineage negative (Lin−) cells. Lin− cells were pre-stimulated in StemSpam SFEM and supplemented with 50ng/mL human Stem Cell Factor (hSCF), 50ng/mL human Flt-3 Ligand (hFlt-3L), 20ng/mL human Thrombopoietin (hTPO) and 10ng/mL human Interleukin-6 (hIL-6) (PeproTech) for 4-6 hours. Lentiviral supernatants were added at a multiplicity of infection of 30 (for single transduction) or 20 (for each lentivirus during the double transduction). All the transductions were carried out over-night in the presence of 4mg/mL of Polybrene (R&D Systems, Abingdon, UK). The efficiency of transduction was analyzed at four days by eGFP or / and mCherry expression.
CFC and LTC-IC assays
Infected Lin− cells were plated in triplicate in methylcellulose media (Methocult H4434 and Methocult M3434 for human and mouse respectively, StemCells Technologies, Vancouver, Canada) to assess Colony Forming Units (CFU). Cells from primary plates were transferred to secondary and tertiary plates in new methylcellulose media. Transduced and total colonies were scored at 7 days and 14 days for mouse and human respectively, following plating and classified according to their morphology.
Long-Term Culture-Initiating Cell assays (LTC-IC) were performed according to the manufacturer’s instructions. Briefly, Lin− cells were plated after transduction on irradiated M2-10B4 stromal cells for five weeks. The cells were then plated in methylcellulose media (Methocult H4435, StemCells Technologies, Canada) with colonies scored 14 days later.
Liquid Cultures assays
Two types of liquid culture were used one for maintaining stem/progenitors and the second for inducing myeloid differentiation. For the maintenance of HSC/progenitors, transduced Lin− cells were cultured in IMDM (Gibco, Invitrogen, Paisley, UK) / 10% Fetal Calf Serum (FCS) supplemented with 20ng/mL SCF, 50ng/mL Interleukin-3 (IL-3), 20ng/mL IL-6 and 10ng/mL Granulocyte-Colony Stimulating Factor (G-CSF) (PeproTech). Fresh media was added every week. For Myeloid promoting differentiation, transduced cells were cultured as previously described for two weeks (49) in IMDM/15% FCS supplemented with 2ng/ml IL-3 and 20ng/ml SCF.
Immunodeficient mouse transplantations
All animal experiments were performed in compliance with Home Office and institutional guidelines. NOD/SCID/β2 microglobulin null mice (NOD/SCID/β2) were bred at Charles Rivers Laboratories (Moorgate, UK), housed in micro-isolators and fed sterile food and acidified water. Mice aged 8–12 weeks were sub-lethally irradiated at 375 rads (Cesium 137 source) up to 24 hours before intra-venous (i.v.) injection of cells. NOD/SCID/β2 mice were transplanted i.v. with 0.5-1×105 Human Lin− cells after transduction. The animals were sacrificed after 4, 7 or 8 weeks, then femurs, pelvis and tibias were flushed and all cells were pooled for analysis. In most animals, BM aspirations were performed four weeks after transplantation. When secondary transplants were performed, pooled BM cells from primary recipients, sacrificed at 7 to 8 weeks, were depleted of mouse cells by Mouse/Human Chimera Enrichment kit (StemCells Technologies, Vancouver, Canada) to a >90% purity of human cells. 1×106 human enriched cells from 7 weeks primary mice were transplanted intra-bone (i.b.) and analyzed after 5 weeks, or 0.9-5×106 human cells from 8 weeks primary mice were transplanted i.v. and analyzed 6 weeks later.
Flow Cytometry
Cell suspensions were analyzed in PBS/2% FCS on a FACS LSR II (BD Pharmingen, Oxford Science Park, UK), where transduced cells were identified based on their expression of eGFP and/or mCherry. Surface markers were detected with fluorescent human specific antibodies from BD Pharmingen (anti-CD14 APC-H7, anti-CD14 PE-Cy7, anti-CD15 APC, anti-CD19 APC, anti-CD19 PE, anti-CD33 PE, anti-CD34 PE-Cy7, anti-CD34 APC and anti-CD45 PE-Cy7). Apoptosis was analyzed in cell suspensions in Binding Buffer (BD Pharmingen, Oxford Science Park, UK) after Annexin V-AlexaFluor647 (Molecular Probes-Invitrogen, Paisley, UK) and DAPI incubation. Intracellular staining was performed to study cell cycle. Cells were fixed by PBS / 2% Paraformaldehyde (PFA) and permeabilized with PBS / 0.2% Triton X-100 before adding anti-Ki67 AlexaFluor647 (Molecular Probes-Invitrogen, Paisley, UK). Fixed cells were resuspended in PBS / 2% FCS solution with DAPI with the data analyzed using FlowJo software (Tree Star, Oten, Switzerland).
Quantitative real-time PCR analysis
RNA was isolated using RNAeasy kit (Qiagen, Crawley,UK) from transduced cells after sorting on MoFlo cell sorter (Beckman Coulter, High Wycombe, UK). RNA was reversed transcribed with SuperScript III (Invitrogen) according to the manufacturer’s instructions. The cDNA was subjected to Quantitative real-time PCR (QPCR) using Fast SYBR Green Master Mix (Applied Biosystems, Cheshire, UK) and gene specific primers (Table S1). QPCRs were run on 7500 Fast System and analyzed on SDS software (Applied Biosystems, Cheshire, UK). Results were normalized to GADPH expression and expression of control samples according to the 2ΔΔCt method.
Statistical Analysis
Data are presented as the mean ± standard deviation (SD). Statistical analysis was performed using One-Way ANOVA test with Statgraphics Plus software (Statistical Graphics Corp.). In Vivo experiments were compared according a Negative Binomial Distribution with R software (R Foundation for Statistical Computing, http://www.R-project.org/).
Results
In order to investigate the effect of the different CEBPA mutations, genomic DNA from normal BM and an AML patient (38), with a four base pairs GGCC insertion at base 363 (N-ter mutation) and an in frame internal tandem duplication of 27 base pairs at 1096 (C-ter mutation) (Genbank Y11525) were used as template to generate lentiviral vectors with WT CEBPA, N-ter, C-ter and N- and C-ter mutated CEBPA (see material and methods) (Figure S1A). The expression of the corresponding C/EBPα forms was confirmed by Western-blot in Transduced Lin− cells (Figure S1B).
Effect of the transduction of different forms of C/EBPα on human hematopoietic progenitor compartment (CFU-C/LTC-IC)
In the first instance, we examined the effect of over-expressing individual mutations (N-ter or C-ter), WT and control (empty vector) to determine the potential consequence of each mutation alone on human hematopoietic progenitor development. CB derived Lin− cells were transduced (Fig S1B and S1C) and the hematopoietic progenitor compartment tested by Colony Forming Unit (CFU) assay.
Surprisingly, using human cells, over-expression of WT C/EBPα as well as C-ter mutation (Fig. 1A) led to a significant reduction in the total number of CFUs compared to control whereas the total number of CFUs was not modify by the N-ter mutation.
Figure 1. Effect of different mutated forms of C/EBPα on human progenitors.

A) Analysis of effect of expression of WT C/EBPα , mutated C/EBPα on N-terminal and on C-terminal on Human Hematopoietic progenitors by Colony Forming Units assay. 2×103 human hematopoietic progenitors (Lin− cells) after transduction were plated in H4434 medium; two weeks later number, type of CFUs and expression or not of GFP were determinate using an inverted fluorescent microscope. The number of erythroid (grey) and myeloid (black) colonies per transduced cells (GFP+) are reported. (B) Maintenance of Human Hematopoietic Progenitors by expression of C/EBPα mutant. Serial replating from primary or Secondary CFU assays were done to evaluate Secondary or Tertiary CFUs respectively. Each CFU assays was plated in H4434 medium and quantify after two weeks. Numbers of transduced CFU for Control, WT C/EBPα, N-terminal and C-terminal are represented. (C) Differences in Hematopoietic Stem Cells after transduction were determinate by Long Term Culture Initiating Cells (LTC-IC) assay. 1×104 Human Lin− cells, after transduction, were plated on irradiated stromal cell layer (M2-10B4 cell line) for five weeks, then preserved progenitors was evaluated by CFU assay in H4435 medium. Results are shown as mean ± standard deviation (SD), with significant differences of p ≤ 0.05, p ≤ 0.01 or p ≤ 0.001 are indicating by (*), (**) or (***) respectively. All the data are from a minimum of triplicates of two independent experiments.
When looking at the percentage of erythroid and myeloid progenitors present in each group, we show that overexpression of the N-ter favor myeloid differentiation (Fig. 1A) whereas WT and C-ter mutation block colony formation with a predominant effect on erythroid colony formation. This blockage of erythroid colony formation has been documented for WT on human CD34+ previously(53). When trying to dissect which myeloid colonies was mostly affected by the N-ter mutation, we did not see any difference in the ratio of CFU-G, CFU-M or CFU-GM in the N-ter compare to control (Figure S4). Serial replating experiments demonstrate that cells overexpressing C-ter have a lower self-renewal potential than control, N-ter or WT (Fig. 1B). The LTC-IC capacity of the transduced cells also showed distinctive patterns. The different CEBPA mutations had opposing effects with N-ter slightly increasing the number of LTC-IC whereas the C-ter as with the WT overexpression did not give rise to any LTC-IC (Fig. 1C).
Our data does not follow the results of Kato et al., obtained in mice where the C-ter was able to increase self-renewal (48). To determine whether it was due to differences in the constructs used or due to the intrinsic differences between human and mouse cells, we decided to overexpress our constructs in mouse stem/ progenitors hematopoietic cells. As show in Figure 2, we show an increase in the number of colonies with both N-ter and C-ter with the C-ter cells and to a lesser degree the N-ter being able to give rise to serial colonies indicative of the increase of self-renewal of these cells. Thus it appears that intrinsic properties between mouse and human are responsible for the differences observed and not due to the difference in mutant constructs used. We thus went back using human cells for the rest of the analysis.
Figure 2. Effect of different mutated forms of C/EBPα on mouse progenitors.
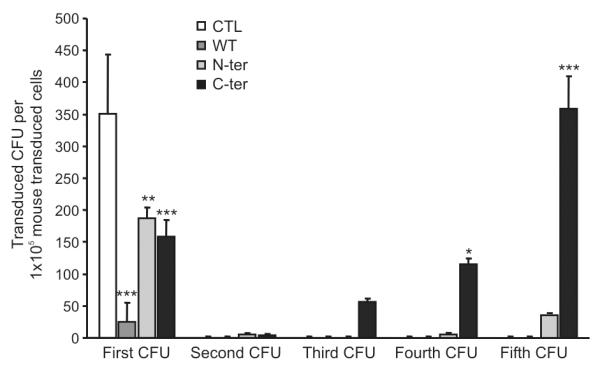
Effect of expression of WT C/EBPα (dark grey bars), mutated C/EBPα on N-terminal (light grey) and on C-terminal (black) was evaluated on mouse hematopoietic progenitors by Colony Forming Units assay. 2×103 mouse hematopoietic progenitors (mLin− cells) after transduction were plated in M3434 medium; one week later number and type of CFUs were determinate. Serial replating was performed to evaluate secondary, tertiary, fourth or fifth CFUs respectively. Numbers of transduced CFU by Control (white bar), WT C/EBPα (dark grey), N-terminal (light grey) and C-terminal (black) are represented. The increase in colonies after serial replating observed could be explained by the presence of live immature cells in the methylcellulose, which were able after replating to mature and give rise to CFUs. Results are shown as mean ± standard deviation (SD), with significant differences of p ≤ 0.05, p ≤ 0.01 or p ≤ 0.001 are indicating by (*), (**) or (***) respectively.
These first results apportioned distinct functions for N- and C-ter CEBPA mutations and that overexpression of the N-ter mutation alone is sufficient to induce myeloid differentiation while maintaining immature progenitors. This is in line with its potential role as pre-leukemic event favoring proliferation of myeloid precursor cells and thus enhancing their chance to acquire further mutations, consistent with its predisposing function in familial AML.
Effect of the different forms of C/EBPα on cell cycle and apoptosis
In order to test the effect of the different forms of C/EBPα on the maintenance of HSC/progenitors, we cultured the cells in vitro in a supportive stem cell medium for up to 14 days and cell cycle was analyzed by DNA content and Ki67 expression (Fig 3A and S2A). WT and C-ter C/EBPα led to a block in cell cycle, maintaining transduced cells in G0 (Fig 3A), whereas the N-ter promoted the transition from G0 to G1 with a potential block in G1 phase. These results were in concordance with the expression analysis of cell cycle regulator genes (p21WAF (p21), INK4C (p18)) (Fig S2B). Indeed, p18 and p21, which are known inhibitors of cell cycle progression show elevated expression in WT, as p18 in C-ter whereas the potential block in G1 phase seen in N-ter is less clear. We also could not exclude that the block in G0 observed here in WT and C-ter was also due at least in part to their effect in inducing differentiation.
Figure 3. Expression of WT and mutants C/EBPα modifies Proliferation and Apoptosis of Human Hematopoietic Progenitors.

(A) Analysis of Cell Cycle of transduced cells at 2 weeks in culture. Different Cell Cycle Stages were identified by FACS according expression of Ki67 and DNA amount by DAPI. Cell Cycle profile of Human Lin- transduced by Control (white bars), WT C/EBPα (dark grey), N-terminal (light grey) and C-terminal (black) is shown. Data are from three independent experiments done for triplicate. (B) Transduction by different C/EBPα forms increases Human Lin- cell survival. Representative Dot-Plot representations of DAPI and Annexin V distinguish alive (left square) and dead cells (right square). Means ± SD are indicated. (C) Quantitative real-time PCR analysis of Bcl-2 in transduced Human Cells. Sorted transduced cells were analyzed in two independent experiments. Results are expressed as mean ± SD, with significant differences of p ≤ 0.05, p ≤ 0.01 or p ≤ 0.001 are indicating by (*), (**) or (***) respectively.
The disappearance of WT and C-ter transduced cells appears to be Bcl-2 independent and not due to apoptosis, as the percentage of apoptotic cells in the transduced population of human hematopoietic progenitors determined by Annexin-V staining and DAPI permeability was decreased in WT, C-ter and N-ter cells (Fig 3B), consistent with the anti-apoptotic role of C/EBPα (52). These results were confirmed by QPCR (Fig 3C), where an increase of BCL2 expression in WT, N-ter and C-ter was observed, indicating that neither N-ter nor C-ter mutations affected the regulation of BCL2 .
Effect of double mutations in human progenitors
The patterns of CEBPA mutations in AMLs are varied with patients having N-ter or C-ter alone or having both N- and C-ter mutations either on the same allele or more frequently as biallelic mutations. In order to examine the role of the biallelic versus mono-allelic mutation, CB Lin− cells were transduced with either N-ter/C-ter together or a corresponding NC-ter construct. Firstly, we assessed the effect of N-ter/C-ter and NC-ter expression on CFCs. As previously shown for WT and C-ter alone (Fig 1A) the N-ter/C-ter combination reduced the number of total CFUs (Fig. 4A). The total number of CFC in NC-ter was similar to control. The combination of N-ter/C-ter induced a reduction in erythroid colonies as for WT and C-ter (Fig. 4A) whereas NC-ter had no effect. To examine the effect of these mutations on self-renewal, we performed secondary and tertiary replating from primary CFUs (Fig. 4B). No hematopoietic progenitors were detected upon secondary replating for the N-ter/C-ter C/EBPα whilst the NC-ter slightly increase the self-renewal potential of the cells as evidenced by an increase in secondary colonies and the presence of tertiary colonies. Accordingly, in LTC-IC assays (Fig. 4C) the N-ter/C-ter transduced cells had no primitive progenitor potential while the NC-ter cells, demonstrating a six-fold increase in LTC-ICs number compared to control. Despite the presence of the N-ter mutation, an arrest in cell cycle as previously demonstrated for both WT and C-ter C/EBPα was detected in the N-ter/C-ter cells (Fig 4D). Both double mutation combinations showed a similar cell survival to the other C/EBPα constructs tested (data not shown).
Figure 4. Biallelic and Double monoallelic mutations on C/EBPα modifies differently human progenitor compartment and Cell Cycle.

(A) Effect of C-terminal with N-terminal mutated C/EBPα (biallelic mutations) or NC-terminal mutated C/EBPα (double monoallelic mutation) in Human Hematopoietic Progenitors were determines by CFU assays. The number of erythroid (grey) and myeloid (black) CFUs in the tranduced cells are presented. (B) Serial replating of Control, N-ter+C-ter or NC-ter mutated C/EBP transduced progenitors are presented. (C) LTC-IC assay to evaluate effect in HSC after expressing C-ter+N-ter or NC-ter C/EBPα. (D) Cell Cycle profile of control, N-ter + C-ter or NC-ter expressing after 14 days of culture. Results are shown as mean ± standard deviation (SD), with significant differences of p ≤ 0.05, p ≤ 0.01 or p ≤ 0.001 are indicating by (*), (**) or (***) respectively. All the data are from a minimum of triplicates of two independent experiments.
These results document differences between biallelic and mono-allelic mutations. For the NC-ter, the inclusion of the mutation on C-ter seems to damper the effects observed with N-ter alone as despite having an increase in self-renewal and LTC-IC, no difference was observed at the induction of the myeloid differentiation contrary to what was observed for the N-ter alone. The N-ter/C-ter double mutant behaves similarly to the C-ter alone with blockage of cell cycle and a decrease in self-renewal despite the presence of the N-ter mutation. These results provide the first experimental data to explain the favorable clinical outcome observed exclusively in bi-allelic mutated patients and suggest a potential role for NC-ter favoring an increase in self-renewal. Nevertheless, we could not also exclude that differences in the transcription factor level, (N-ter/C-ter mutants having almost as twice the level as the NC-ter) might have also on impact on the different phenotype observed.
Effect of different C/EBPα forms in myeloid differentiation
To determine the effects of the different forms of C/EBPα on myeloid differentiation, the production of monocytes, granulocytes and granulocytes precursors were assessed following the induction of myeloid differentiation (Fig 5A). A large proportion of granulocytic precursors could be detected in controls with some mature monocytes and granulocytes. In comparison, WT C/EBPα promoted granulocytic differentiation whereas the C-ter mutation blocked the terminal differentiation of monocytes and to a lesser degree of granulocytes. The N-ter has no effect on granulocytic precursors but seems to induce terminally differentiated granulocytes at the expense of monocytes. Interestingly, NC-ter was similar to control with only a slight decrease in monocytes. In comparison N-ter/C-ter double transduced cells were reminiscent of C-ter mutants. We further examined the level of endogenous C/EBPα and PU.1 transcript key regulator of myeloid differentiation. It appears that the overexpression of N-ter/C-ter induced a 32-fold increase in the endogenous level of C/EBPα whereas other constructs have little effect. Both N-ter and C-ter alone and to a lesser extend NC-ter induce a significant increase in PU.1 whereas with the combination of N-ter/C-ter, this effect disappears (Fig S3).
Figure 5. Modification of Myeloid Differentiation by of WT and mutants C/EBPα.

(A). Human Lin− cells were cultured in Myeloid Conditions for two weeks. FACS analysis evaluated percent of different populations of Myeloid Lineage in transduced cells: Monocytes (hCD33+/hCD14+, grey), Granulocytes (hCD33low/hCD15+, white) and Granulocytic precursors (hCD33+/hCD15+, black). (B) Percentage of CD34+CD38− (white) and CD34+CD38+ (black) in transduced cells after two weeks of culture in myeloid conditions. Percent of each subset is indicated. Data show triplicates of three independent experiments. Significant differences of p ≤ 0.05 or p ≤ 0.001 are indicating by (*) or (**) respectively.
Effect of different C/EBPα forms in Hematopoietic stem/progenitor cells
To determine the effect of the different forms of C/EBPα on hematopoietic stem/progenitor cells (HSPCs) maintenance, the percentage of CD34+CD38− and CD34+CD38+ were estimated (Fig 5B). We observed a significant reduction of the both CD34+CD38− and CD34+CD38+ compartment in WT, N-ter, C-ter and N-ter/C-ter, whereas in NC-ter group we observed a maintenance of the CD34+CD38− compare to control with a concomitant 2 fold increase in CD34+CD38+ cells.
Expression of C/EBPα mutants alters human repopulating cells
Finally we assessed the effect of different CEBPA mutants on transplantation potential of human Lin− after transduction into NOD/SCID/β2. The level of human engraftment in all groups were quite similar ranging from 21.3 ± 4.8 in CTL, 28.9 ± 10.4 in WT, 26.6± 5.8 in N-ter, 28.8 ± 5.8 in NC-ter, 28.3 ± 13.4 in C-ter and 22.2 ± 9.2 in N-ter/C-ter after 8 weeks. The engraftment of the transduced human cells was analyzed at different times points (Fig 6A). In the control group the percentage of transduced cells fell from 38 to 15% between 4 and 8 weeks. WT CEBPA expressing cells were low at 4 weeks and 8 weeks (<1%) reflecting its tumor suppressor activity. The percentage of N-ter or C-ter transduced cells were comparable (15%) at 4 weeks but in both cases had decreased by 8 weeks (2.5%). The N-ter/C-ter cells were not detected (<0.01%) at either time point demonstrating that all mutants still have some tumor suppressor activities except potentially for the NC-ter. Indeed, only NC-ter transduced cells were detected at similar levels as the control group (42 - 22% at 4 and 8 weeks respectively).
Figure 6. WT and mutants C/EBPα Engraftment in Immunodeficient Mice.

(A) In vivo kinetic of transduced cells in Human Hematopoietic population. Sub-lethally irradiated NOD/SCID/β2 microglobulin null mice (NOD/SCID/β2) were transplanted with Human Lin- cells after transduction. Percent of Transduced cells in human engraftment was analyzed at different time points by FACS. Transduction efficiency of transplanted populations (white triangle), mean of transduced cells percentage at different time points (black dash) and transduced cells percentage of individual mice (black triangle or circle) are shown. Animal analyzed at 4 or 8 weeks by bone marrow aspirations are identified by black triangles; circles indicated that the mice have been culled first and bone marrow cells flushed for the analysis. Data from two independent experiments are shown. (B) Hematopoietic lineage analysis of transduced cells in vivo at different time points. Bar graph showing percent of Lymphoid (hCD19+, white bars) and Myeloid (hCD33+, black bars) transduced cells. Numbers represent Myeloid-Lymphoid ratio in transduced human cells. (C) Percentage of transduced monocytes, granulocytes and granulocyte precursors present in the myeloid compartment of engrafted mice 8 weeks post-transplant. Numbers represent the ratio of each fraction in transduced human cells. Results are shown as mean ± SD. Data from four independent experiments are shown. Significant differences of p ≤ 0.05, p ≤ 0.01 or p ≤ 0.001 are indicating by (*), (**) or (***) respectively. ND, not detected.
The phenotype of transduced cells was analyzed by FACS (Fig 6B and Table 1). In Control group, there was an equal proportion of myeloid and B cells at 4 weeks, which as expected shifted to lymphoid cells (hCD19+) at 8 weeks. WT C/EBPα expressing cells were mainly myeloid cells at both time points, consistent with the requirement of C/EBPα in inducing human myeloid differentiation in vivo. The N-ter C/EBPα demonstrated similar increase in myeloid production at 4 weeks, which disappeared at 8 weeks, contrary to the C-ter, which show no increase in myeloid at 4 weeks but an increase at 8 weeks. NC-ter behaves similarly to control in contrast to N-ter/C-ter construct where cells were not detectable.
Table 1.
Summary of Secondary Transplants with transduced human cells.
| Mouse | Group | transplanted hCD45+ (×106) cells from primary recipients |
Secondary recipients | ||
|---|---|---|---|---|---|
|
| |||||
| Doses of hCD45+(×106) cells |
GFP+ (%) | hCD45+ (%) | GFP+ in hCD45+ (%) | ||
| 1 | CTL | 1.30 | 42.3 | 0.00 | |
| 2 | CTL | 4.00 | 46.1 | 0.09 | 76.70 |
| 3 | CTL | 1.00 | 7.57 | 0.31 | 4.26 |
| 4 | CTL | 1.00 | 7.57 | 0.49 | 4.26 |
| 5 | CTL | 1.00 | 7.57 | 0.44 | 5.08 |
| 6 | WT | 0.90 | 0.42 | 0.02 | 0 |
| 7 | WT | 4.50 | 0.39 | 0.20 | 0 |
| 8 | WT | 4.50 | 0.39 | 0.06 | 0 |
| 9 | WT | 4.50 | 0.39 | 0.03 | 0 |
| 10 | N-ter | 3.35 | 22.8 | 0.02 | 0 |
| 11 | N-ter | 3.35 | 22.8 | 0 | |
| 12 | NC-ter | 5.00 | 23.6 | 0 | |
| 13 | NC-ter | 5.00 | 23.6 | 11.70 | 55.20 |
| 14 | NC-ter | 5.00 | 23.6 | 0.12 | 17.10 |
| 15 | NC-ter | 1.00 | 4.36 | 0.63 | 2.06 |
At 8 weeks, we further analysis of the type of myeloid cell present and identified granulocytes (hCD33low/hCD15+), granulocytic precursors (hCD33+/hCD15+) and monocytes (hCD33+/hCD14+) (Fig 6C). All the constructs except C-ter behave similarly to control (N-ter/C-ter was not studied as no transduced cells were present at 8 weeks). For the C-ter overexpression, we observed contrary to the in vitro differentiation assay, an increase in granulocytes at the expense of monocytes and granulocytic precursors.
To test whether we could induce leukemia, and to insure that we were able to transduce long-term repopulating cells with our lentivector constructs, we performed secondary transplants from some of these first recipient mice. Purified human cells from transplanted NOD/SCID/β2 mice were injected (intravenously or intrabone) in new irradiated NOD/SCID/β2 mice and human engraftment analyzed 5 - 6 weeks later. Human transduced cells were only detected in the control and NC-ter group (Table 1) with a predominance of lymphoid cells (hCD19+) (data not shown). Thus, comparable to control, the NC-ter transduced cells can retain long-term repopulating activity in vivo, the rest of C/EBPα mutated forms lose their repopulating capacities, probably via myeloid differentiation and/or cell cycle arrest and thus on their own are unable to transform normal HSPC.
Discussion
In our model, over-expression of WT C/EBPα reduced HSC and progenitors in vitro and in vivo, by inducing terminal myeloid differentiation, preferentially of granulocytic lineage, and arresting the cells in G0, consistent with the block in both granulocytic and monocytic differentiation imposed by C/EBPα silencing (54). These data further confirmed the tumor suppressor function of WT CEBPα. These data also indicated that the level of WT C/EBPα is important for the normal hematopoiesis and is similar to other CEBP transcription factor family members whose upregulation accompanying IGH translocations, give rise to B-cell precursor ALL (55, 56). The balance between normal and mutated WT C/EBPα may well be a feature of AML development. Indeed, in addition to the mutations in CEBPA, C/EBPα expression can be controlled in a number of ways, through the effects of specific fusion proteins (35), hypermethylation (57) or post-translation modifications (58).
Although it was initially assumed that all CEBPA mutations are associated with a good prognosis favorable outcome now appears to be restricted to patients with biallelic mutation. We therefore chose to evaluate not only N and C-ter alone but also the different combination of double mutants. Mutations had different effects on normal hematopoiesis which was not wholly surprising given the complex role of C/EBPα in both cell cycle regulation via p21WAF, CDK2-CDK4 interaction and E2F repression (14, 16-18), and on myeloid differentiation (7-9, 11, 21). The N-ter CEBPA mutations (N-ter) failed to induce an arrest in G0 but seems to block the cell in G1 most likely reflecting the critical role for the N-ter domain in E2F repression (11). The N-ter alone showed a slight increase in immature myeloid progenitors (as shown by an increase in LTC-IC), a favorable granulocytic differentiation versus monocytic which is in agreement with recent studies by the Stocking group(53). However, despite this slight increase in immature myeloid progenitors, only a low long-term engraftment was observed in vivo, indicating that the proposed dominant-negative effect of the N-ter mutant is weak, and may merely enrich the pool of myeloid progenitors in which secondary mutations arise and from which the leukemic-initiating cell can emerge. Consistent with this interpretation it is noteworthy that carriers of the germline N-ter mutation develop leukemia after a relatively long latency period (43). In our case, we were unable to look at long term effect as after 8 weeks, we could not detected any engraftment of the N-ter overexpressing cells.
It has previously been shown that mice with a disruption of the C-ter domain fail to promote myeloid differentiation (11). In accordance with these studies we show that the C-ter mutation arrests cells in G0 and increase the number of myeloid precursors mostly by blocking monocytic differentiation. In vivo the C-ter mutation showed a decrease in repopulating capacity compared to control implying that additional lesions are necessary to offer the proliferative advantage needed for these myeloid progenitors in order to acquire leukemic-initiating cell potential.
It appears therefore that N-ter and C-ter mutations alone might not be capable of inducing a full leukemia, which is consistent with the long latency period necessary to obtain overt leukemia described in both the knock-in mouse models (46, 47) or in the overexpression experiment (48). These data are also consistent with clinical findings whereby mutations in FLT3/ITD, NRAS and WT1 are coincident with CEBPA mutations in AML patients (34, 59, 60) suggesting that these or other oncogenic events (like TET2, 61) are required for the development of the leukemic-initiating cell in a CEBPA mutant background.
Despite the capacity of N-ter to maintain more immature progenitors (i.e: LTC-ICs) in vitro and C-ter to induce a block in terminal differentiation, the combination of the two has no additive effect. The monoallelic NC-ter mutation induce a mild in vitro phenotype. Indeed there was an increase in immature progenitors due to the N-ter mutation, a cell cycle arrest and a modest decrease in monocytic differentiation recurrent of the C-ter mutation. Nevertheless in vivo, the cells behave similarly to control. For the biallelic N-ter/C-ter mutation, myeloid differentiation was promoted without reaching terminal differentiation with the C-ter mutation imposing an arrest in cell cycle. This might provide an explanation for the favourable outcome associated with biallelic mutation pattern. Nevertheless, in vivo, these cells fail to give rise to any short or long term repopulation. These data suggest thus additional mutations might be present in patients with biallelic N-ter/C-ter mutation observed in familial cases of mutated C/EBPα contrary to the notion that in these patients, the C-ter mutation acts as a second hit model sufficient to promote the full blow leukemia(12, 36).
Although forced over-expression of mutated forms demonstrated distinct phenotype, we could not exclude that residual wild-type copy present in the umbilical CB had modulated the effects of our over-expression mutant forms. The down-regulation of the wild-type C/EBPα in the original CB by itself is difficult to achieve without interfering with the N-ter and C-ter mutations. However, in our experiments forced expression originated from a viral promoter and expression was considerably higher than endogenous C/EBPα. Nevertheless, it appears that with the biallelic N-ter / C-ter form, the endogenous expression of C/EBPα was increased substantially after induction of myleloid differentiation in vitro suggesting a potential compensatory feedback loop. This might explain at least in part the data obtained here. Nevertheless, it is clear that our results demonstrated some clear differences between mouse and human hematopoietic cells related to the effect of mutated C/EBPα. Indeed we show that by using our constructs, we were able to confirm the results obtained by Kato et al. (48), using the same over-expression strategy demonstrating that the nature of the cells and not the difference in constructs used was responsible for the difference of action.
In conclusion, our data underline the critical role of C/EBPα in human hematopoiesis and demonstrate that C/EBPα mutations (alone or in combination) are insufficient to convert normal human HSPCs into leukemic initiating cells, although individually each altered normal hematopoiesis in a manner characteristic of leukemic transformation. It provides the first insights into the effects of N- and C-ter mutations acting alone but also of the combined effects of N-ter/C-ter double mutants. Contrary to the results obtained recently where the MLL-ENL fusion protein was able to transform primary human hematopoietic cells (49, 50) after a 15 weeks latency, we were not able to propagate the transduced cells in vivo long enough for a potential secondary mutation to occur. Indeed after 6 to 8 weeks, no human repopulating ability could be detected indicating the potential limitations of the xenotransplantation model to recapitulate the multistep transformation process of a leukemia to arise from human normal primary HSC/HPCs (62-64). One question, which is still open is how many mutations do exist in these C/EBPα mutated AML patients and how many are needed to trigger leukemia. Based on how the N and C-ter mutations behave together, the greater challenge in the future is not merely to address the nature of the individual contribution but how mutations compensate and complement each other’s function when partnered together.
Supplementary Material
Acknowledgements
We would like to thanks the members of FACS service and animal facilities for their help. We are grateful to Dr. Oscar Escribano from Department of Biochemistry and Molecular Biology, Faculty of Pharmacy, Complutense University of Madrid, Madrid, Spain for his technical advice in performing the Western Blot analysis. This project was supported by Cancer Research UK and by European grant (contract No:037632) to DB. O.B-Q was supported by CRUK, LRI fellowship.
Footnotes
Conflict of interest statement: the authors declare no competing financial interests.
Authorship statement: O.B.Q designed and performed research, analyzed and interpreted data, performed statistical analysis and wrote manuscript. S.LL.S, E.G, Y.R and J.V performed part of the research experiments, T.A.L provided vital materials and J.F. interpreted data and wrote manuscript, D.B. designed research and analyzed and interpreted data and wrote manuscript.
Supplementary information is available at Leukemia’s website.
References
- 1.Passegue E, Jamieson CH, Ailles LE, Weissman IL. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11842–11849. doi: 10.1073/pnas.2034201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jordan CT, Guzman ML. Mechanisms controlling pathogenesis and survival of leukemic stem cells. Oncogene. 2004;23:7178–7187. doi: 10.1038/sj.onc.1207935. [DOI] [PubMed] [Google Scholar]
- 3.Warner JK, Wang JC, Hope KJ, Jin L, Dick JE. Concepts of human leukemic development. Oncogene. 2004;23:7164–7177. doi: 10.1038/sj.onc.1207933. [DOI] [PubMed] [Google Scholar]
- 4.Gililand G, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002;1:417–420. doi: 10.1016/s1535-6108(02)00081-8. [DOI] [PubMed] [Google Scholar]
- 5.Pabst T, Mueller BU. Transcriptional dysregulation during myeloid transformation in AML. Oncogene. 2007;26:6829–6837. doi: 10.1038/sj.onc.1210765. [DOI] [PubMed] [Google Scholar]
- 6.Friedman AD. Runx1, c-Myb, and C/EBPalpha couple differentiation to proliferation or growth arrest during hematopoiesis. J Cell Biochem. 2002;86:624–629. doi: 10.1002/jcb.10271. [DOI] [PubMed] [Google Scholar]
- 7.Friedman AD. C/EBPalpha induces PU.1 and interacts with AP-1 and NF-kappaB to regulate myeloid development. Blood Cells Mol Dis. 2007a;39:340–343. doi: 10.1016/j.bcmd.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene. 2007b;26:6816–6828. doi: 10.1038/sj.onc.1210764. [DOI] [PubMed] [Google Scholar]
- 9.Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21:853–863. doi: 10.1016/j.immuni.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 10.Nerlov C. The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007;17:318–324. doi: 10.1016/j.tcb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Nerlov C. C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer. 2004;4:394–400. doi: 10.1038/nrc1363. [DOI] [PubMed] [Google Scholar]
- 12.Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol. 2009;27:619–628. doi: 10.1200/JCO.2008.17.9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calkhoven CF, Muller C, Leutz A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 2000;14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- 14.D’Alo F, Johansen LM, Nelson EA, Radomska HS, Evans EK, Zhang, et al. The amino terminal and E2F interaction domains are critical for C/EBP alpha-mediated induction of granulopoietic development of hematopoietic cells. Blood. 2003;102:3163–3171. doi: 10.1182/blood-2003-02-0479. [DOI] [PubMed] [Google Scholar]
- 15.Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR, et al. c-Myc is a critical target for c/EBPalpha in granulopoiesis. Mol Cell Biol. 2001;21:3789–3806. doi: 10.1128/MCB.21.11.3789-3806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timchenko NA, Harris TE, Wilde M, Bilyeu TA, Burgess-Beusse BL, Finegold MJ, et al. CCAAT/enhancer binding protein alpha regulates p21 protein and hepatocyte proliferation in newborn mice. Mol Cell Biol. 1997;17:7353–7361. doi: 10.1128/mcb.17.12.7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timchenko NA, Wilde M, Nakanishi M, Smith JR, Darlington GJ. CCAAT/enhancer-binding protein alpha (C/EBP alpha) inhibits cell proliferation through the p21 (WAF-1/CIP-1/SDI-1) protein. Genes Dev. 1996;10:804–815. doi: 10.1101/gad.10.7.804. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ, et al. C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell. 2001;8:817–828. doi: 10.1016/s1097-2765(01)00366-5. [DOI] [PubMed] [Google Scholar]
- 19.Porse BT, Pedersen TA, Xu X, Lindberg B, Wewer UM, Friis-Hansen L, et al. E2F repression by C/EBPalpha is required for adipogenesis and granulopoiesis in vivo. Cell. 2001;107:247–258. doi: 10.1016/s0092-8674(01)00516-5. [DOI] [PubMed] [Google Scholar]
- 20.Porse BT, Bryder D, Theilgaard-Monch K, Hasemann MS, Anderson K, Damgaard I, et al. Loss of C/EBP alpha cell cycle control increases myeloid progenitor proliferation and transforms the neutrophil granulocyte lineage. J Exp Med. 2005;202:85–96. doi: 10.1084/jem.20050067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNagny KM, Sieweke MH, Doderlein G, Graf T, Nerlov C. Regulation of eosinophil-specific gene expression by a C/EBP-Ets complex and GATA-1. Embo J. 1998;17:3669–3680. doi: 10.1093/emboj/17.13.3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kummalue T, Friedman AD. Cross-talk between regulators of myeloid development: C/EBPalpha binds and activates the promoter of the PU.1 gene. J Leukoc Biol. 2003;74:464–470. doi: 10.1189/jlb.1202622. [DOI] [PubMed] [Google Scholar]
- 23.Tenen DG, Hromas R, Licht JD, Zhang DE. Transcription factors, normal myeloid development, and leukemia. Blood. 1997;90:489–519. [PubMed] [Google Scholar]
- 24.Kovacs KA, Steinmann M, Magistretti PJ, Halfon O, Cardinaux JR. CCAAT/enhancer-binding protein family members recruit the coactivator CREB-binding protein and trigger its phosphorylation. J Biol Chem. 2003;278:36959–36965. doi: 10.1074/jbc.M303147200. [DOI] [PubMed] [Google Scholar]
- 25.Nerlov C, Ziff EB. CCAAT/enhancer binding protein-alpha amino acid motifs with dual TBP and TFIIB binding ability co-operate to activate transcription in both yeast and mammalian cells. Embo J. 1995;14:4318–4328. doi: 10.1002/j.1460-2075.1995.tb00106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller C, Calkhoven CF, Sha X, Leutz A. The CCAAT enhancer-binding protein alpha (C/EBPalpha) requires a SWI/SNF complex for proliferation arrest. J Biol Chem. 2004;279:7353–7358. doi: 10.1074/jbc.M312709200. [DOI] [PubMed] [Google Scholar]
- 27.Pedersen TA, Kowenz-Leutz E, Leutz A, Nerlov C. Cooperation between C/EBPalpha TBP/TFIIB and SWI/SNF recruiting domains is required for adipocyte differentiation. Genes Dev. 2001;15:3208–3216. doi: 10.1101/gad.209901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barjesteh van Waalwijk van Doorn-Khosrovani S, Erpelinck C, Meijer J, van Oosterhoud S, van Putten WL, Valk PJ, et al. Biallelic mutations in the C/EBPA gene and low C/EBPA expression levels as prognostic markers in intermediate-risk AML. Hematol J. 2003;4:31–40. doi: 10.1038/sj.thj.6200216. [DOI] [PubMed] [Google Scholar]
- 29.Bienz M, Ludwig M, Leibundgut EO, Mueller BU, Ratschiller D, Solenthaler, et al. Risk assessment in patients with acute myeloid leukemia and a normal karyotype. Clin Cancer Res. 2005;11:1416–1424. doi: 10.1158/1078-0432.CCR-04-1552. [DOI] [PubMed] [Google Scholar]
- 30.Frohling S, Dohner H. Disruption of C/EBPalpha function in acute myeloid leukemia. N Engl J Med. 2004;351:2370–2372. doi: 10.1056/NEJMp048241. [DOI] [PubMed] [Google Scholar]
- 31.Gombart AF, Hofmann WK, Kawano S, Takeuchi S, Krug U, Kwok SH, et al. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood. 2002;99:1332–1340. doi: 10.1182/blood.v99.4.1332. [DOI] [PubMed] [Google Scholar]
- 32.Leroy H, Roumier C, Huyghe P, Biggio V, Fenaux P, Preudhomme C. C/EBPA point mutations in hematological malignancies. Leukemia. 2005;19:329–334. doi: 10.1038/sj.leu.2403614. [DOI] [PubMed] [Google Scholar]
- 33.Lin LI, Chen CY, Lin DT, Tsay W, Tang JL, Yeh YC, et al. Characterization of C/EBPA mutations in acute myeloid leukemia: most patients with C/EBPA mutations have biallelic mutations and show a distinct immunophenotype of the leukemic cells. Clin Cancer Res. 2005;11:1372–1379. doi: 10.1158/1078-0432.CCR-04-1816. [DOI] [PubMed] [Google Scholar]
- 34.Mueller BU, Pabst T. C/EBPalpha and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol. 2006;13:7–14. doi: 10.1097/01.moh.0000190110.08156.96. [DOI] [PubMed] [Google Scholar]
- 35.Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of C/EBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–270. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 36.Pabst T, Eyholzer M, Haefliger S, Schardt J, Mueller BU. Somatic C/EBPA mutations are a frequent second event in families with germline C/EBPA mutations and familial acute myeloid leukemia. J Clin Oncol. 2008;26:5088–5093. doi: 10.1200/JCO.2008.16.5563. [DOI] [PubMed] [Google Scholar]
- 37.Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of C/EBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–270. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 38.Preudhomme C, Sagot C, Boissel N, Cayuela JM, Tigaud I, de Botton S, et al. Favorable prognostic significance of C/EBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA) Blood. 2002;100:2717–2723. doi: 10.1182/blood-2002-03-0990. [DOI] [PubMed] [Google Scholar]
- 39.Snaddon J, Smith ML, Neat M, Cambal-Parrales M, Dixon-McIver A, Arch R, et al. Mutations of C/EBPA in acute myeloid leukemia FAB types M1 and M2. Genes Chromosomes Cancer. 2003;37:72–78. doi: 10.1002/gcc.10185. [DOI] [PubMed] [Google Scholar]
- 40.Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double C/EBPA mutations, but not single C/EBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–3091. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fitzgibbon J, Smith LL, Raghavan M, Smith ML, Debernardi S, Skoulakis S, et al. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res. 2005;65:9152–154. doi: 10.1158/0008-5472.CAN-05-2017. [DOI] [PubMed] [Google Scholar]
- 42.Hou HA, Lin LI, Tien HF. Reply to “Heterogeneity within AML with CEBPA mutations; only CEBPA double mutations, but not single CEBPA mutations are associated with favorable prognosis”. Br. J Cancer. 2009;100:1–3. doi: 10.1038/sj.bjc.6605207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pabst T, Eyholzer M, Fos C, Mueller BU. Heterogeneity within AML with CEBPA mutations; only CEBPA double mutations, but not single CEBPA mutations are associated with favourable prognosis. Br. J. Cancer. 2009;100:1343–1346. doi: 10.1038/sj.bjc.6604977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of C/EBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351:2403–2407. doi: 10.1056/NEJMoa041331. [DOI] [PubMed] [Google Scholar]
- 45.Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, et al. for the German–Austrian Acute Myeloid Leukemia Study Group Mutations and Treatment Outcome in Cytogenetically Normal Acute Myeloid Leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 46.Renneville A, Mialou V, Philippe N, Kagialis-Girard S, Biggio V, Zabot MT, et al. Another pedigree with familial acute myeloid leukemia and germline C/EBPA mutation. Leukemia. 2009;23:804–806. doi: 10.1038/leu.2008.294. [DOI] [PubMed] [Google Scholar]
- 47.Kirstetter P, Schuster MB, Bereshchenko O, Moore S, Dvinge H, Kurz E, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13:299–310. doi: 10.1016/j.ccr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 48.Bereshchenko O, Mancini E, Moore S, Bilbao D, Mansson R, Luc S, et al. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell. 2009;16(5):390–400. doi: 10.1016/j.ccr.2009.09.036. [DOI] [PubMed] [Google Scholar]
- 49.Kato N, Kitaura J, Komeno Y, Watanabe-Okochi N, Togami K, Nakahara F, et al. Two types of C/EBPα mutations play distinct but collaborative roles in leukemogenesis: lessons from clinical data and BMT models. Blood. 2011;117:221–233. doi: 10.1182/blood-2010-02-270181. [DOI] [PubMed] [Google Scholar]
- 50.Barabe F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science. 2007;316:600–604. doi: 10.1126/science.1139851. [DOI] [PubMed] [Google Scholar]
- 51.Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 2008;13:483–95. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Almarza E, Rio P, Meza NW, Aldea M, Agirre X, Guenechea G, et al. Characteristics of lentiviral vectors harboring the proximal promoter of the vav proto-oncogene: a weak and efficient promoter for gene therapy. Mol Ther. 2007;15:1487–1494. doi: 10.1038/sj.mt.6300213. [DOI] [PubMed] [Google Scholar]
- 53.Cammenga J, Mulloy JC, MacGrogan D, Viale A, Nimer SD. Induction of CEBPalpha activity alters gene expression and differentiation of human CD34+ cells. Blood. 2003;101:2206–14. doi: 10.1182/blood-2002-05-1546. [DOI] [PubMed] [Google Scholar]
- 54.Paz-Priel I, Ghosal AK, Kowalski J, Friedman AD. C/EBPalpha or C/EBPalpha oncoproteins regulate the intrinsic and extrinsic apoptotic pathways by direct interaction with NF-kappaB p50 bound to the bcl-2 and FLIP gene promoters. Leukemia. 2009;23(2):365–74. doi: 10.1038/leu.2008.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niebuhr B, Iwanski GB, Schwieger M, Roscher S, Stocking C, Cammenga J. Investigation of C/EBPalpha function in human (versus murine) myelopoiesis provides novel insight into the impact of C/EBPA mutations in acute myelogenous leukemia (AML) Leukemia. 2009;23:978–983. doi: 10.1038/leu.2008.332. [DOI] [PubMed] [Google Scholar]
- 56.Chapiro E, Russell L, Radford-Weiss I, Bastard C, Lessard M, Struski S, et al. Overexpression of CEBPA resulting from the translocation t(14;19)(q32;q13) of human precursor B acute lymphoblastic leukemia. Blood. 2006;108:3560–3. doi: 10.1182/blood-2006-03-010835. [DOI] [PubMed] [Google Scholar]
- 57.Akasaka T, Balasas T, Russell LJ, Sugimoto K, Majid A, Walewska R, et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocation in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) Blood. 2007;109:3451–3461. doi: 10.1182/blood-2006-08-041012. [DOI] [PubMed] [Google Scholar]
- 58.Wouters BJ, Jorda MA, Keeshan K, Louwers I, Erpelinck-Verschueren CA, Tielemans D, et al. Distinct gene expression profiles of acute myeloid/T-lymphoid leukemia with silenced C/EBPA and mutations in NOTCH1. Blood. 2007;110:3706–3714. doi: 10.1182/blood-2007-02-073486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geletu M, Balkhi MY, Peer Zada AA, Christopeit M, Pulikkan JA, Trivedi AK, et al. Target proteins of C/EBPalphap30 in AML: C/EBPalphap30 enhances sumoylation of C/EBPalphap42 via up-regulation of Ubc9. Blood. 2007;110:3301–3309. doi: 10.1182/blood-2007-01-071035. [DOI] [PubMed] [Google Scholar]
- 60.Shih LY, Liang DC, Huang CF, Wu JH, Lin TL, Wang PN, et al. AML patients with C/EBPalpha mutations mostly retain identical mutant patterns but frequently change in allelic distribution at relapse: a comparative analysis on paired diagnosis and relapse samples. Leukemia. 2006;20:604–609. doi: 10.1038/sj.leu.2404124. [DOI] [PubMed] [Google Scholar]
- 61.Sellick GS, Spendlove HE, Catovsky D, Pritchard-Jones K, Houlston RS. Further evidence that germline C/EBPA mutations cause dominant inheritance of acute myeloid leukaemia. Leukemia. 2005;19:1276–1278. doi: 10.1038/sj.leu.2403788. [DOI] [PubMed] [Google Scholar]
- 62.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, et al. Mutations in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 63.Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. 2003;3:639–649. doi: 10.1038/nrc1164. [DOI] [PubMed] [Google Scholar]
- 64.Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008;319:336–339. doi: 10.1126/science.1150648. [DOI] [PubMed] [Google Scholar]
- 65.Rossi DJ, Jamieson CH, Weissman IL. Stems cells and the pathways to aging and cancer. Cell. 2008;132:681–96. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.