

Abstract
Delayed HIV-1 disease progression is associated with a single nucleotide polymorphism upstream of the HLA-C gene that correlates with differential expression of the HLA-C antigen. This polymorphism was recently shown to be a marker for a protective variant in the 3′UTR of HLA-C that disrupts a microRNA binding site, resulting in enhanced HLA-C expression at the cell surface. Whether individuals with ‘high’ HLA-C expression show a stronger HLA-C-restricted immune response exerting better viral control than that of their counterparts has not been established. We hypothesised that the magnitude of the HLA-C-restricted immune pressure on HIV would be greater in subjects with highly expressed HLA-C alleles. Using a cohort derived from a unique narrow source epidemic in China, we identified mutations in HIV proviral DNA exclusively associated with HLA-C which were used as markers for the intensity of the immune pressure exerted on the virus. We found an increased frequency of mutations in individuals with highly expressed HLA-C alleles which also correlated with IFN-γ production by HLA-C-restricted CD8+ T-cells. These findings show that immune pressure on HIV is stronger in subjects with the protective genotype and highlights the potential role of HLA-C-restricted responses in HIV control. This is the first in vivo evidence supporting the protective role of HLA-C-restricted responses in non-Caucasians during HIV infection.
INTRODUCTION
Several host genetic determinants can influence HIV replication and immune responsiveness, thereby having an impact on disease progression. Three genome wide association studies have confirmed the central role of the HLA locus on HIV disease progression by identifying two single nucleotide polymorphisms (SNP) associated with steady-state plasma HIV RNA levels (1-3). One is in strong linkage disequilibrium (LD) with HLA-B*5701, an allele strongly associated with good clinical outcome of HIV infection. The second SNP lies 35 kb upstream of the HLA-C locus and is associated with increased expression of both HLA-C mRNA and protein at the cell surface in subjects homozygous for the protective ‘C’ allele (1, 4). Although previous studies have reported that -35 SNP is in LD with HLA-B*5701, several groups have shown that -35 SNP independently associates with HIV control (1, 4, 5). The -35 SNP is also in strong LD with a variant found in the 3′UTR of the HLA-C gene, which was recently shown to regulate binding of the microRNA miR-148a to its target site, thereby altering HLA-C expression (6). This strongly suggests that HLA-C expression levels play a direct role in HIV-1 control, most probably through the mobilisation of an HLA-C-restricted immune response.
HLA-C is interesting in the context of HIV infection as HIV nef selectively downregulates HLA-A and -B while maintaining HLA-C and -E expression at the cell surface (7). Therefore, highly expressed HLA-C alleles could trigger HIV-specific responses through either increased antigen presentation to cytotoxic CD8 T cells (CTLs), binding to Killing Inhibitory Receptors (KIR) on NK cells or a combination of these mechanisms. The fact that HLA-C-restricted CTLs exert pressure on HIV is an indication of their effectiveness in vivo, and there is a growing body of evidence for the role of HLA-C-mediated responses in other viral infections (4, 6, 8-10). This implies that HLA-C-restricted responses play an under-appreciated role in HIV control. The protective effect of the -35 SNP was clearly shown in Caucasians but, with the exception of one study that showed no protection in African-Americans, there are no data available from other ethnic groups (11). As genetic determinants vary amongst genetic backgrounds, it is crucial to assess the impact of this variant in additional populations to understand how HLA-C-restricted immune responses might contribute to HIV control.
We studied the protective effect of -35 SNP in a unique cohort of former plasma donors in rural China where subjects remained free of anti-retroviral treatment (ART) in the first 9-10 years of infection with an unusually narrow source founder clade B’ HIV-1 strain (12). These subjects are likely to have been infected through the same route and in a short time-frame, thereby providing an exceptional setting in which most usual cohort variables are controlled and where the prevalence of known protective HLA alleles is very low. We investigated whether slow disease progression associated with -35 SNP correlates with a stronger HLA-C-restricted immune response in this cohort as a result of increased HLA-C expression. As the generation of CTL escape variants follows stereotypic mutational pathways based on the HLA-restriction of CTL epitopes, we were able to identify HLA-HIV polymorphism associations that reveal how much cellular immune pressure is inflicted on the virus (13). We used this method to identify HIV mutations that were associated with immune pressure from HLA-C alleles and used these as markers for HLA-C-mediated responses. We report that -35 SNP correlates with delayed disease progression in the Han Chinese population. Moreover, we observe a significant increase in the frequency of HLA-C-associated mutations in the proviral DNA of patients with the protective variant which could indicate stronger HLA-C-restricted immune responses in vivo. To our knowledge, these are the first functional data about the in vivo effect of -35 SNP on HIV evolution. This study provides a new perspective to understanding the mechanisms of immune protection during HIV infection and could potentially lead to the identification of HLA-C-restricted immune responses associated with viral control in HIV-infected individuals.
MATERIALS AND METHODS
Patients
In the mid-1990’s, many Han Chinese residents of an isolated rural community in China were involved in an illegal plasma donation scheme. Ten years later, an HIV screening program revealed that 324 villagers were HIV-1 infected. We estimate that 473 former plasma donors in the village acquired HIV-1 infection based on reports of 149 premature adult deaths with symptoms compatible with AIDS. HIV-1 transmission probably occurred through contaminated blood collection equipment or following the return of pooled red cells to donors. Epidemiological and phylogenetic analyses strongly suggest that all cohort members were infected with an unusually narrow source virus by the same route during the same time-period (12). Members of the cohort were not aware that they had been infected for the first 10 years of infection, therefore information on the infected subjects and uninfected former plasma donors during that period is limited. In 2005, samples from 288 former plasma donors with chronic clade B’ HIV-1 infection were collected and subsequent patient recruitments were organised on a yearly basis. Due to the very narrow source virus in the cohort, genetic divergence in the subjects reveals how much viral diversity at the population level is attributable to host factors. Preliminary analyses showed that HLA-B*57 and −B*27 are not enriched in this cohort of long-term non-progressors (Rai, M.A., et al, manuscript in preparation). Most viral sequences, CD4 count and HLA-typing data were derived from samples collected in 2005, before the onset of anti-retroviral therapy. For some patients, viral load data were not collected until 2007, but are only presented from ART naïve subjects. Clinical data were collected from randomly selected samples. CD3+/CD4+/CD8+ T lymphocyte percentages and true counts were determined by Flow Cytometry as follows: 200 μl of heparin-anticoagulated fresh whole blood was analysed using the BD MultiTest IMK kit (BD, USA) and MultiSetTM software (BD, USA) according to the manufacturer’s instructions. Briefly, multitest antibodies were mixed with the sample and incubated for 15 minutes in the dark at room temperature (RT) before being transferred to BD True count tubes. 450 μl of 1× BD Multitest lysing solution was added to the tube and incubated for another 15 minutes in the dark at RT. Finally, CD3+ /CD4+ /CD8+ T lymphocyte percentages and true counts were calculated using MultiSetTM software. HIV-1 plasma viral load was quantified by Nucleic Acid Sequence Based Amplification (NASBA) in Beijing Youan Hospital. An initial enzymatic amplification of the nucleic acid targets was followed by amplicon detection. Assay lower limit of detection was 50 RNA copies/ml and levels below detection were assigned an arbitrary value of 25 copies/ml. Ethical approval was obtained from Beijing Youan Hospital and the University of Oxford Tropical Ethics Committee (OXTREC). HIV-1 gag, pol and nef sequencing and HLA genotyping was performed on 288 samples as previously described (14, 15). For the analysis described in Fig. 1C, 256 samples from HIV-negative Han Chinese residents of a nearby village were randomly collected and used as a control cohort. A total of 324 HIV-infected subjects were included in this analysis. This population is in Hardy-Weinberg equilibrium (p=0.65).
Figure 1. Protective effect of the -35 SNP in the Han Chinese population.
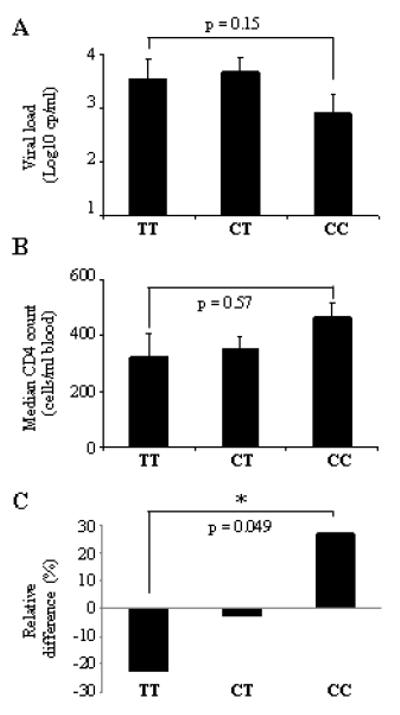
(A) Mean viral load (log10 cp/ml) and (B) median CD4 counts (cells/ml blood) in HIV-infected individuals. Patient samples were grouped based on their genotype at -35. Statistical analyses was performed using the Kruskal-Wallis rank sum test and a two-tailed t-test (C) Enrichment of the -35CC genotype in the HIV-positive cohort. The HIV-negative cohort was used as baseline and relative frequencies of each genotype were compared with those of the HIV-positive cohort. Statistical analysis was performed using a chi-square test. The p-value applies to a comparison of CC and TT rates in cases (HIV+) versus controls (HIV-). Numbers are listed in Table I.
-35 SNP genotyping
The -35 SNP (rs9264942) genotyping was performed as previously described (4). Two PCR reactions (‘C’ and ‘T’ reactions) were set up for each sample, using standard amplification conditions were used. In addition to allele-specific primers, each reaction contained control DRB1-specific primers which allowed failed reactions to be identified. Nucleotides were purchased from GE Healthcare (Illustra range) and Taq from Bioline. Outliers were re-typed by sequencing. Primer sequences are available on request. Genotyping data were generated for 324 members of the cohort; 64 patients showed C-mut (7 subjects had two or more C-mut). Subjects for which viral sequences or genotyping data were not available were excluded from the study with the exception of the case-control analysis shown in Fig. 1C.
Phylogenetic stratification of HLA allele-HIV-1 polymorphism associations
HLA associations were determined as previously described (13). We previously showed that over 50% of amino acid substitutions in HIV gag, pol and nef were strongly associated with HLA class I molecules and are thus attributable to T-cell pressure (12). In this study, we selected nine substitutions in pol, gag and nef exclusively associated with HLA-C. Three mutations were excluded from the analysis as they were less than three positions away from substitutions also associated with HLA-A or −B alleles or found in fewer than three subjects. The six remaining mutations were found in individuals expressing the corresponding HLA-C allele. Only strong associations (0.100 <q-value > 0.250) were included in the analysis (Fig. 2).
Figure 2. -35CC subjects have lower viremia and higher CD4 counts than TT individuals.
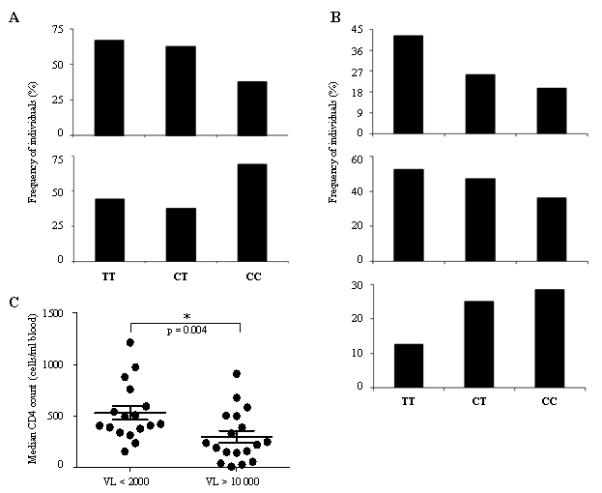
Groups were defined according to the frequencies of subjects with (A) HIV-1 viral load <2000 or >10 000 viral RNA copies per ml plasma and (B) CD4 counts <200, <350 and >500 cells/ml blood which represent different clinical stage of HIV-1 disease progression. (C) Subjects with mVL <2000 exhibit higher CD4 counts than individuals with mVL >10 000. Each dot represents a patient. Statistical analysis was performed using a two-tailed t-test. P = 0.004.
T-cell assays
CTL response specific for the VL8 (VIPMSFAL) epitope was indentified in patient 043 (expressing HLA-Cw*01:02) by IFN-γ ELISpot using 18-mer consensus peptides. Plates were read on an AID plate reader and background was subtracted from wells. A VL8-specific short-term CTL line was generated as described previously and subsequently used for intracellular cytokine staining (ICS) (14). ICS was performed using antibodies for IFN-γ and TNF-α and the Cytofix/Cytoperm kit (Beckton-Dickinson Europe) according to the manufacturer’s instructions. 721.221-HLACw*01:02 transfectants (obtained from Peter Parham) were pulsed with VL8 peptide to re-stimulate the CTL line when performing ICS as well as for maintenance. Untransfected 721.221 cells were used as a negative control.
Statistics
Data were analysed using the R software (R Development Core Team, 2008). The following tests were used when appropriate: t-test, chi-square test, Kruskal-Wallis rank sum test, Fisher’s Exact Test and test for Hardy-Weinberg equilibrium. P values < 0.05 were considered significant.
RESULTS
Study population
A total of 324 HIV-1–infected individuals from a geographically isolated rural community in central China who were exposed to HIV-1 through contaminated blood in the early 1990s were recruited from 2005 to 2010 (12). Members of this cohort are former plasma donors who are likely to have been infected with a single or very narrow range of HIV-1 strains by the same infection route in a two-year period (12). All patients enrolled in the present study made frequent plasma donations in the early 1990’s and have survived without antiretroviral treatment during the first 10 years of infection, but have since progressed to diverse disease outcomes. Very little information is available about individuals who may also have acquired HIV through plasma donation but who subsequently progressed to AIDS and died before recruitment started. Blood samples were collected in 2005 and -35 SNP genotyping data were generated for all cohort members and were analysed in relation to clinical details, proviral sequences and HLA typing. This ethnically homogeneous population infected with a narrow range of HIV-1 clade B’ strains provides an unparalleled opportunity to discern how immune factors drive viral evolution without the confounding effect of ART.
-35CC associates with better clinical outcome than -35TT in the Han Chinese
Previous reports showed that the -35 SNP associates with low viral set point and maintenance of peripheral CD4 cells, resulting in slow progression to AIDS and death (1, 4). To determine whether -35CC was also protective in the Han Chinese, each individual was categorised based on the genotype at -35 : CC, CT or TT (Table I). In a comparison of the three genotypes in relation to mean plasma HIV load (mVL), we observed a trend towards lower mVLs for -35CC subjects than for -35TT individuals (Fig. 1A). The effect of -35 SNP on viremia was also observed when subjects were grouped according to their mVLs, where 67% of TT individuals had mVL >10,000, whereas only 38% of CC subjects had uncontrolled viremia, which is in agreement with previous studies (Fig. 2A) (4). We next determined whether the variant also had an effect on peripheral CD4+ T cell numbers. We found that -35 SNP had an effect on progression, as CC subjects had a median CD4 count of 430 compared to only 267 for TT individuals (Fig. 1B). This difference was not statistically significant, nevertheless the effect of -35CC on CD4 counts was considerable as the majority of individuals with normal CD4 counts (>500) had the -35CC genotype whereas most non-controllers with counts <200 were -35TT (Fig. 2B). This was also observed when a cut off of <350 (the current threshold for ART initiation) was used (Fig. 2B). Of note, individuals with low steady-state viremia exhibited significantly higher CD4 counts than subjects with high mVL, suggesting that AIDS progression was delayed in CC subjects (Fig. 2C). In order to test further the inference that -35 SNP is protective, we assessed whether the CC genotype associated with slow disease progression in this population by comparing the -35 genotype frequencies in the cohort to that of an HIV-negative control cohort from a nearby village (Table I). We hypothesised that many HIV-infected TT subjects would have succumbed to HIV disease when ART was not available, resulting in an enrichment of the protective -35CC genotype. Indeed, we found a statistically significant increase (p = 0.049) in the relative frequency of CC subjects in the HIV-positive cohort (Fig. 1C and Table 1). These results suggest that CC individuals progress more slowly to AIDS than their TT counterparts. As no samples were collected for the fast progressors who died during the first 10 years, it is impossible to confirm that those individuals were -35TT. This might also explain why clinical data analyses did not reach statistical significance (Fig. 1A-B). Nevertheless our results are in agreement with two previous studies and show that the -35CC genotype associates with better clinical outcome than -35TT in the Han Chinese population.
Table 1.
Patients genotype at −35.
| Study Group | % TT (n) | % CT (n) | % CC (n) | Total (n) |
|---|---|---|---|---|
| HIV − | 25.3 (65) | 48.8 (125) | 25.8 (66) | 256 |
| HIV+ | 19.3 (63) | 49.8 (162) | 30.8 (99) | 324 |
| A-mut | 17.9 (25) | 52.1 (73) | 30.0 (42) | 140 |
| B-mut | 21.6 (27) | 48.0 (60) | 30.4 (38) | 125 |
| C-mut | 2.1 (3) | 25.0 (35) | 18.6 (26) | 64 |
Individuals with the protective -35CC genotype are more likely than -35TT subjects to show mutations associated with HLA-C in HIV proviral DNA
As -35 SNP correlates with increased HLA-C expression at the cell surface, we assessed whether the variant was also associated with a stronger HLA-C-restricted immune response that could potentially lead to viral control. We hypothesised that, as HLA-C-restricted responses contribute to HIV evolution, the frequency of HLA-C-associated mutations (C-mut) in proviral DNA should correlate with the magnitude of the immune pressure exerted on the virus in vivo (13, 16). Therefore a potent HLA-C-restricted immune response should result in more C-mut in HIV DNA in CC than in TT subjects. We determined which C-mut were exclusively associated with HLA-C alleles and used those C-mut as markers for HLA-C-restricted immune pressure (12). A total of six mutations in HIV gag, pol and nef were selected according to the following criteria: 1) significant association with an HLA-C allele, 2) no association with any HLA-A or –B alleles, and 3) found in at least three individuals expressing the corresponding HLA-C allele (Fig. 3). Interestingly, three mutations lay within previously defined HLA-C-restricted CTL epitopes. To verify that -35CC associated with C-mut, each individual was categorised by -35 genotype and the frequencies of the six mutations were compared. Taking into account that some individuals showed more than one mutation in their proviral DNA, we found that the frequency of C-mut was higher in the presence of the CC variant (29%) than with the TT genotype (4%). Only 3 C-mut occurred in 63 TT subjects as opposed to 29 in 99 CC patients (29%). These results suggest that HLA-C-restricted immune pressure is stronger in the presence of -35CC, the genotype associated with high HLA-C levels (Fig. 4A). Moreover, when patients showing C-mut were compared with the rest of the cohort, CC individuals were five times more likely to exhibit C-mut than TT subjects (Fig. 4B). This finding could be a direct result of enhanced HLA-C expression or reflect LD with protective HLA-B or -A alleles. As previously reported by Thomas and collaborators, we found that -35 SNP is in LD with certain HLA-C alleles (Fig. 5A) (4). However we found no correlation between the frequencies of mutations associated with HLA-A (A-mut) or -B alleles (B-mut) and -35CC (Fig. 4C and Table 1). All HLA Class I associations were more frequent in CC than in TT subjects, however the relationship only achieved significance for C-mut (p = 0.01). Of note, the total numbers of A-mut and B-mut were distinctly higher than the number of C-mut (Table 1). This is consistent with previously published data that showed that CTL responses restricted by HLA-A and –B induce stronger immune pressure than HLA-C-restricted responses (17). Moreover, HLA-C alleles and subtypes are still poorly characterised which could have resulted in some C-mut being overlooked. Nevertheless these figures indicate that -35 SNP specifically correlates with C-mut, an association that could not be indirectly attributed to the presence of protective HLA alleles such as HLA-B*51, which is enriched in subjects showing C-mut (Fig. 5B). When HLA-B*51 individuals were excluded from the analysis, we found that the C-mut frequency was still significantly higher in patients with -35CC, indicating that LD with this allele, known to be protective in Asian populations, is unlikely to account for the observed association (Fig. 5C) (16). Taken together, these results show that C-mut are more frequent when HLA-C expression is enhanced (-35CC), suggesting that these mutations were induced by a potent HLA-C-restricted response in vivo. This is consistent with a previous report which showed that HLA-C-mediated immune responses may restrain the virus to some extent, as Nef variants isolated from -35CC subjects interfere with CD4+ T helper cell activation and MHC-II antigen presentation, possibly to thwart HLA-C mediated immune control (18).
Figure 3. Selection of C-mut.
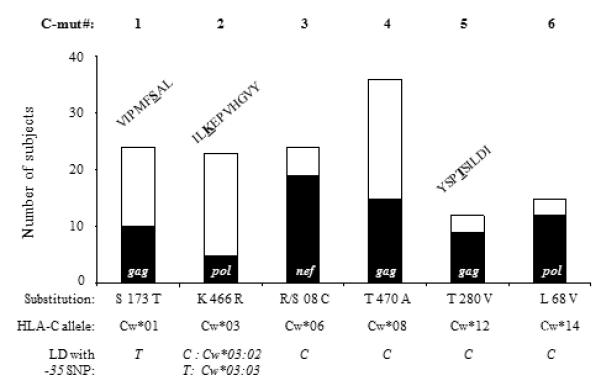
Numbers of patients showing C-mut (black) or not (white). Six mutations associated with HLA-C exclusively were found in individuals expressing the corresponding HLA-C alleles. Substitutions and LD between HLA-C alleles associated and -35 SNP are shown below the graph. When substitutions were found inside known epitopes, the epitope sequences are shown above the corresponding bars. C-mut # 3 (KWSKCSMIGWPRVRERMR), #4 (TAPPEESFRFGEETTTPSQK) and #6 (WQLDCTHVEGKIILVAVH) were found in unpublished epitopes.
Figure 4. Association between C-mut and -35 SNP.
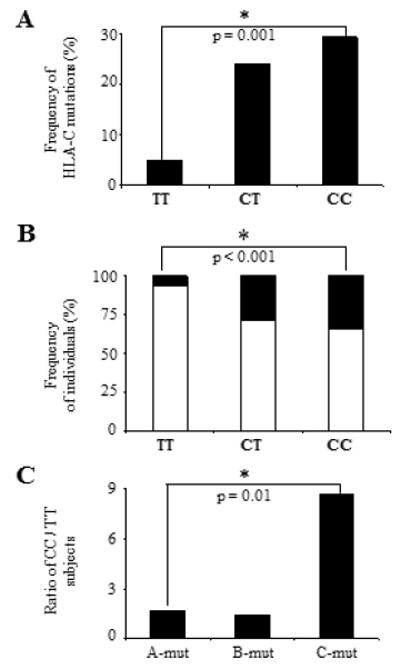
(A) Increased frequency of C-mut in presence of the CC genotype. Subjects were categorised in three groups based on their genotype at -35 (TT, CT and CC). The total numbers of C-mut in each group were counted and the frequencies were calculated based on numbers listed in Table 1 (some subjects showed more than one C-mut). (B) Distribution of -35 SNP in the cohort. Frequencies of subject with C-mut are shown in black and frequencies of individuals not exhibiting C-mut (rest of the cohort) are shown in white (Table 1). The likelihood to show C-mut is five times higher in ‘CC’ than in ‘TT’ subjects (black). (C) CC subjects exhibit an increased frequency of substitutions in HIV pro DNA associated with HLA-C but not with HLA-A or –B alleles. Ratios of subjects with the CC genotype versus individuals with the TT genotype are shown for each HLA Class I association. Statistical analyses were performed using a chi-square test using numbers from Table 1. In C, the p-value applies to a comparison between the numbers of CC versus TT subjects with A-mut, B-mut or C-mut.
Figure 5. LD between -35 SNP and HLA-alleles.
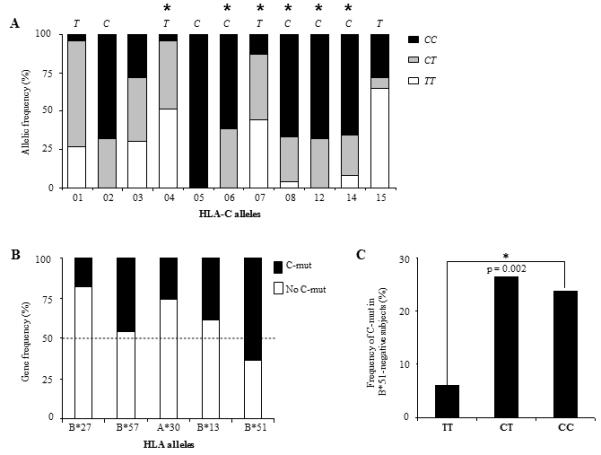
(A) -35 SNP is in LD with some HLA-C alleles. Allelic frequencies of the different HLA-C alleles are shown according to the genotypes at -35: CC (black), CT (grey) and TT (white). Annotations above each bar indicate which of the ‘C’ or ‘T’ allele at -35 is associated with HLA-C alleles. Significant associations are represented by *. HLA-Cw*03 subtypes are differentially associated with -35 SNP: Cw*03:02 associates with ‘C’ whereas Cw*03:03 is in LD with ‘T’. (B) Frequencies of the main protective HLA-alleles in subjects showing C-mut. HLA-B*51 is the only allele enriched in patients with C-mut (black). Frequencies of subjects without C-mut are shown in white. (C) Increased frequency of C-mut in CC subjects in absence of HLA-B*51. Analysis described in Fig. 4A was repeated after exclusion of all HLA-B*51 subjects showing C-mut. Statistical analysis was performed using a Chi-square test. In addition, a logistic regression analysis adjusting for HLA-B*51 showed that its presence did not affect the results.
C-mut give rise to escape variants in subject exhibiting HLA-C-restricted CTL responses
We next determined whether T cells were responsible for inducing C-mut in CC subjects. The fact that three out of six C-mut (C-mut # 1, 2 and 5) were found in regions coding for CTL epitopes suggests that these substitutions have been induced by CTL pressure (Fig. 3) (19, 20). To verify that a T-cell response was present in patients expressing HLA-C alleles associated with each C-mut, we performed ELISpot assays using consensus 18-mer HIV peptides spanning regions where C-mut had been mapped (Fig. 5 and Fig. 6A). Peptide-specific IFN-γ responses were observed for all tested peptides, showing that these non-adapted sequences are immunogenic and presumably contain CTL epitopes. No T-cell epitopes have previously been reported in the region where C-mut #3, 4 and 6 were found but, as only a few HLA-C-restricted CTL epitopes have been mapped, particularly in non-Caucasian populations, those C-mut are likely to mark new HIV epitopes subject to CTL selection (Fig. 4A). We and others have previously confirmed that HLA-associations can be used to predict new T-cell epitopes (12, 21, 22). To confirm the role of HLA-C as a T-cell restriction element and mediator of CTL activation, a short-term CTL line was generated using the consensus VL-8 peptide (VIPMSFAL) and frozen PBMCs from a Cw*01:02 patient exhibiting the S173T substitution (C-mut #1). Using 721.221-HLACw*01:02 transfectants as antigen-presenting cells only able to express HLA-Cw*01:02, we performed intracellular cytokine staining and found that TNF-α and IFN-γ production by CTLs is dependent on Cw*01:02 expression (Fig. 6B). We next verified that this response was peptide-specific and that the S173T substitution was an escape mutation in a peptide titration assay. The peptide containing the S173T substitution was less well recognised than the non-adapted peptide. Notably, a statistically significant difference was found at a peptide concentration of 0.5 ug/ml, indicating that the mutation led to an escape variant (Fig. 6C). In addition, we performed a peptide titration using PBMCs from a HLA-Cw*08 patient responding to a consensus peptide where no CTL epitopes had been previously mapped and confirmed that the peptide containing the T470A substitution (C-mut #4) was more weakly recognised than the WT peptide, indicating that T470A was also an escape variant (Fig. 6D). Escape mutations in highly conserved epitopes can have a beneficial impact for the patient as shown in the context of HLA-B*5701 KW10 epitope (23). On the other hand, escape mutations can result in viral adaptation and lead to disease progression, as in the case of the immunodominant HLA-B*51 CTL response (24). We did not assess whether those escape mutations were beneficial for the patients, but rather used them as markers for the intensity of the immune response.
Figure 6. C-mut are escape mutations and markers for immunogenic epitopes.
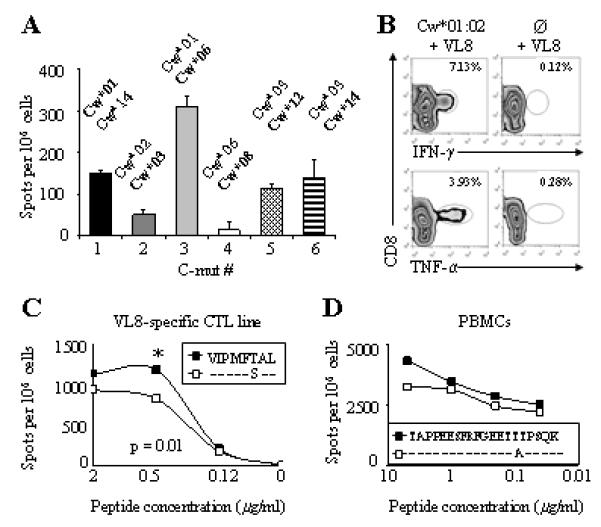
(A) Magnitude of IFN-γ responses to 18-mer HIV-1 consensus peptides. PBMCs from patients expressing HLA-C alleles associated with each C-mut were used in an ELISpot assay. Each bar represents one patient. HLA-C type of each subject is shown above the bars; the HLA-C alleles associated with C-mut are in bold (Fig. 2). Two to five responding patients were identified for each peptide. (B) TNF-α and IFN-γ production is CD8+ T cell mediated, HLA-Cw*01:02-restricted and peptide-specific. A short-term CTL line was re-stimulation with VL8-pulsed .221HLACw*01:02 transfectants (left). Untransfected VL8-pulsed .221 cells were used as control (right). Numbers indicate percentage of positive cells. Gated on CD3+ T cells. (C and D) S173T and T470A substitutions are escape mutations. Peptide titration assay with CTL line or PBMCs in presence of decreasing concentrations of adapted (white) and nonadapted (black) (C) VL-8 or (D) TAPPEESFRFGEETTTPSQK peptides in an ELIspot assay. Data representative of two independent experiments. Statistical analyses were performed using a two-tailed t-test. Significant differences are indicated by the presence of *.
We next assessed whether C-mut were the result of NK cell interactions with peptide-HLA-C complexes. As KIR data are not yet available for the cohort, we categorised each individuals based on the two major KIR-ligand categories: C1 and C2 (similar to the Bw4 and Bw6 determinants). A weak interaction between an inhibitory KIR and the HLA-C molecule, such as the one between KIR2DL3 and C1 alleles, can easily be overcome and lead to NK-cell activation (25). We found no association between the distribution of C1 or C2 alleles and -35 SNP, indicating that neither of the KIR-ligand groups is in LD with -35CC (Supplemental Fig. 1). Although these data do not exclude a role for NK-cells in the control of viral replication in the presence of the variant (for which KIR typing data are required), our results suggest that the C-muts are unlikely to be induced by NK-cell pressure. Taken together these results strongly suggest that C-muts are induced by HLA-C-restricted CTLs, which might therefore play a role in the protective effect of the -35 SNP.
The binding site for miR148a/miR148b is disrupted in -35CC subjects
There has been some controversy about the biological relevance of -35 SNP and its association with differential cell-surface expression of HLA-C alleles. It was reported that HLA-C expression is not always significantly higher in -35CC subjects and does not systematically correlate with lower viremia, suggesting that -35 SNP is just in strong LD with protective HLA alleles and does not directly mediate any protective effect (26). Nonetheless a recent report supports the initial notion that -35 SNP associates with surface HLA-C expression. Another variant located in the 3′UTR of HLA-C promotes high surface expression of HLA-C alleles which subsequently escapes post-transcriptional regulation by microRNAs (6). A deletion in position 263 of the 3′UTR (263del) results in a disrupted binding site for miR148a/miR148b which leads to increased cell-surface HLA-C expression. Therefore HLA-C alleles can be categorised based on the presence of the 263del, which correlates with increased levels at the cell surface and associates with low mVL (<2000). Importantly the 3′UTR variant is in strong LD with -35 SNP as the majority of -35C-associated HLA-C alleles exhibit the 263del whereas most -35T alleles have an insertion at position 263 (263ins). In line with this, we investigated whether the protective effect of -35 SNP also correlated with the 3′UTR variant in the Han Chinese. We found that subjects homozygous for 263del (del/del) had lower mVL and higher CD4 counts than subjects homozygous for ‘low’ HLA-C alleles with the 263ins/ins genotype (Fig. 7A-B). Moreover, 263del/del individuals with the CC genotype were more likely to exhibit C-mut (Fig. 7C-D). We sequenced the 3′UTR of individuals exhibiting C-mut and found that all ‘high’ expressing HLA-C alleles containing 263del exhibit the -35CC genotype, with the exception of Cw*14 which contains an insertion at position 263 of the 3′UTR but associates with -35CC (Supplemental Fig. 2). These results confirm that CC subjects with C-mut also have a disrupted binding site for miR-148a (263del) which is likely to result in increased cell-surface HLA-C expression in these individuals compared to TT subjects exhibiting an intact miRNA binding site (263ins). Therefore, it is likely that C-muts were induced by enhanced HLA-C-restricted immune pressure in CC individuals.
Figure 7. 3′UTR variant associates with protection and C-mut in the Han Chinese population.
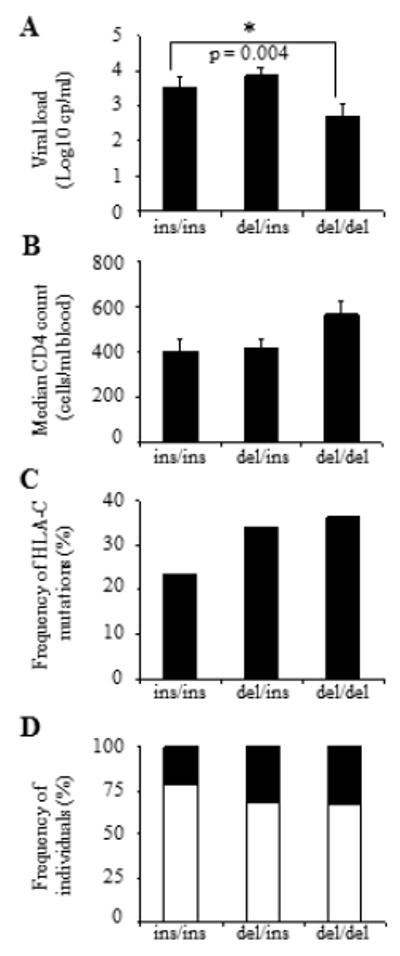
(A) Mean viral load (log10 cp/ml) and (B) median CD4 counts (cells/ml blood) in HIV-infected individuals in relation to 263 del/ins. (C) Increased frequency of C-mut in CC subjects. Frequencies of C-mut in the cohort in relation to 263del/ins. (D) Frequencies of subjects with (black) or without (white) C-mut. Subjects were grouped based on the presence of 263del/ins. Frequencies of individuals are shown. Statistical analyses were performed using Kruskal-Wallis rank sum test and chi-square test.
DISCUSSION
After more than 30 years of active research on HIV/AIDS, many challenging questions remain unanswered. For example, some individuals progress to AIDS within a year after HIV acquisition whilst others never develop the disease. Host genetic factors play a role in disease progression but the precise pathways are still unknown. Studies aimed at deciphering this are hampered by inter-individual variability and HIV’s high propensity to mutate: as a consequence the host-virus interactions can vary significantly between different infected people. How innate and adaptive immune mechanisms slow HIV replication is still ill defined and differs substantially amongst different ethnic groups. As most studies to date have focused on patients of European ancestry, it has become a global health priority to determine how HIV control is achieved in non-Caucasians (27). In the present study, we assessed the protective effect of -35 SNP in Han Chinese subjects who were infected in a short time-frame by a very narrow range of HIV-1 strains. In addition to being the first report on the effect of -35 SNP in an Asian population, this study confirms the protective effect of the variant in a setting where confounders such as viral strain diversity and time after HIV acquisition are largely controlled. We analysed six substitutions in HIV’s proviral DNA which were associated with HLA-C (C-mut) in order to determine whether immune pressure on the virus was stronger in -35CC than in -35TT subjects. C-mut were identified using sequencing data from PBMC proviral DNA rather than plasma RNA, as mutations that had reached fixation in the population were more relevant to the purpose of the analysis than quasispecies from an actively replicating population (28). The key finding of this study is that subjects with the protective CC genotype at -35 show a higher C-mut frequency than their TT counterparts, suggesting that the virus is exposed to a stronger immune pressure in presence of the CC genotype. We suggest that HLA-C-restricted CTLs play a role in the protective effect of the variant, without excluding the possibility that NK-cells or some HLA alleles also contribute to viral control.
Of all SNPs associated with delayed HIV disease progression, -35 SNP is consistently prominent, clearly implicating HLA-C as a key player in HIV control. The mechanisms underlying this have been partially elucidated with the identification of the 3′UTR variant but remain subject to considerable debate. On the other hand, there are associations between HLA-C and several autoimmune diseases (29). Notably, -35 SNP is also associated with increased susceptibility to psoriasis where disease is thought to be mediated by HLA-Cw*06 restricted CTLs specific for self peptides, a response that could potentially be elicited as a result of enhanced HLA-C expression (30, 31). HLA-C is involved in other autoimmune disorders suggesting that it is somehow implicated in T-cell tolerance. HLA-C binds a restricted range of self peptides as a result of limited polymorphism in its α1 domain and prolonged association with chaperones in the endoplasmic reticulum. This could result in HLA-C molecules being more available to bind pathogen-derived peptides, which could result in specific targeting of key viral molecules, thus countering pathogen replication. Supporting this, the molecule H-2Ld corresponding to HLA-C in the mouse, is poorly expressed and defective in binding endogenous self peptides but is the dominant CTL-restriction element for recognition of Vesicular Stomatitis and Lymphocytic Choriomeningitis viruses (32). Therefore, the contribution of HLA-C to pathogen recognition in peripheral and lymphoid organs could be distinct but complementary to that of HLA-A and –B.
HLA-C molecules could also play a role during thymic T-cell selection (33). The efficacy of thymic negative selection is proportional to the expression level of MHC complexes in the thymic medulla, which means that fewer clones with a high avidity for self MHC-peptide complexes are deleted when MHC expression is low. Assuming that HLA-C expression in medullary epithelial and dendritic cells is lower than that of HLA-A and –B (as it is in other tissues), HLA-C-mediated negative selection might be less stringent. Thymic negative selection imposes specificity on the T-cell repertoire, consequently a less efficient negative selection leads to the generation of a highly avid T-cell repertoire (34). Therefore, it is tempting to speculate that the T-cell repertoire restricted by HLA-C comprises several high-avidity clones able to effectively recognise viral peptides in the periphery. Selection of a high avidity T-cell repertoire could be a double-edged sword: it might lead to better pathogen recognition but also result in the generation of auto-immune pathologies. The dual association of -35 SNP linking HLA-C to better HIV control and susceptibility to psoriasis seems to corroborate this hypothesis. Moreover differential HLA-C expression could also influence thymic positive selection by allowing a more diverse range of T-cell clones to be selected in subjects with the CC genotype where HLA-C expression is thought to be higher than in TT individuals. This hypothesis remains to be tested.
We report here that increased expression of HLA-C in the -35 SNP leads to the generation of escape mutations in HIV. This is the first function-based evidence linking -35 SNP to HIV control in vivo. Whether HLA-C expression levels are directly responsible for the protective effect of -35 SNP (or 263del) or strong LD between the variants and other protective genes in the HLA locus exert viral control is still unclear. This is extremely difficult to disentangle, therefore we cannot exclude the possibility that some protective HLA-alleles have an effect on delayed progression to AIDS independently or in conjunction with -35 SNP. On the other hand, as LD between HLA alleles in the Han Chinese substantially differs from that of the Caucasians, we were able to partially separate some associations with -35 SNP to conclude that the protective effect of the variant can be dissociated from the effect of the main protective HLAs (i.e. B*57, B*27). Although polymorphisms in the binding site of miRNA-148a (HLA-C 3′UTR) provide a mechanism explaining how HLA-C expression can be modulated, we cannot exclude that HLA-C alleles in LD with -35CC play a role in protection. However a growing body of evidence support the concept that increased HLA-C expression results in a more effective immune response through mechanisms such as NK-cell licensing and activation, antigen cross presentation to CTLs and increased survival of APCs (35). Further studies are required, nevertheless our data support the concept that HIV-specific CTLs are key components of HIV control in vivo and highlight the relevance of exploring HLA-C-restricted CTLs in the design of a potential HIV vaccine.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Jane Holmes, Ly-Mee Yu, Daniel Lunn, Joe Parker and Louis-Marie Yindom for help with statistical analyses, Peter Parham for providing 721.221-HLACw*01:02 transfectants, Smita Kulkarni and Mary Carrington for help with the 3′UTR PCR for sequencing protocol, and Simon Brackenridge for help with -35 SNP genotyping.
1. This work is funded by Medical Research Council UK, Li Ka Shing Foundation, Royal Society UK, Beijing Natural Science Foundation (7111005), Beijing Talents Building Projects (PYZZ091016001765), Beijing Fengtai Health Bureau and Beijing Municipal Science & Technology Commission (D09050703590904, D09050703560903, D09050703590901), China National Science & Technology Key Program (2012ZX10001-006). MEB holds a postdoctoral fellowship from Fond de la Recherche en Santé du Québec and YHZ was funded by the Drs Richard Charles and Esther Yewpick Lee Charitable Foundation.
3. Abbreviations used in this article
- C-mut
HLA-C-associated mutations
- HIV nef
HIV negative replication factor protein
Footnotes
The authors declare no competing financial interests.
REFERENCES
- 1.Fellay J, Ge D, Shianna KV, Colombo S, Ledergerber B, Cirulli ET, Urban TJ, Zhang K, Gumbs CE, Smith JP, Castagna A, Cozzi-Lepri A, De Luca A, Easterbrook P, Gunthard HF, Mallal S, Mussini C, Dalmau J, Martinez-Picado J, Miro JM, Obel N, Wolinsky SM, Martinson JJ, Detels R, Margolick JB, Jacobson LP, Descombes P, Antonarakis SE, Beckmann JS, O’Brien SJ, Letvin NL, McMichael AJ, Haynes BF, Carrington M, Feng S, Telenti A, Goldstein DB. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5:e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, Cozzi-Lepri A, De Luca A, Easterbrook P, Francioli P, Mallal S, Martinez-Picado J, Miro JM, Obel N, Smith JP, Wyniger J, Descombes P, Antonarakis SE, Letvin NL, McMichael AJ, Haynes BF, Telenti A, Goldstein DB. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.IntHIVContStud The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas R, Apps R, Qi Y, Gao X, Male V, O’HUigin C, O’Connor G, Ge D, Fellay J, Martin JN, Margolick J, Goedert JJ, Buchbinder S, Kirk GD, Martin MP, Telenti A, Deeks SG, Walker BD, Goldstein D, McVicar DW, Moffett A, Carrington M. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat Genet. 2009;41:1290–1294. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catano G, Kulkarni H, He W, Marconi VC, Agan BK, Landrum M, Anderson S, Delmar J, Telles V, Song L, Castiblanco J, Clark RA, Dolan MJ, Ahuja SK. HIV-1 disease-influencing effects associated with ZNRD1, HCP5 and HLA-C alleles are attributable mainly to either HLA-A10 or HLA-B*57 alleles. PLoS One. 2008;3:e3636. doi: 10.1371/journal.pone.0003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulkarni S, Savan R, Qi Y, Gao X, Yuki Y, Bass SE, Martin MP, Hunt P, Deeks SG, Telenti A, Pereyra F, Goldstein D, Wolinsky S, Walker B, Young HA, Carrington M. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472:495–498. doi: 10.1038/nature09914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10:661–671. doi: 10.1016/s1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 8.Adnan S, Balamurugan A, Trocha A, Bennett MS, Ng HL, Ali A, Brander C, Yang OO. Nef interference with HIV-1-specific CTL antiviral activity is epitope specific. Blood. 2006;108:3414–3419. doi: 10.1182/blood-2006-06-030668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makadzange AT, Gillespie G, Dong T, Kiama P, Bwayo J, Kimani J, Plummer F, Easterbrook P, Rowland-Jones SL. Characterization of an HLA-C-restricted CTL response in chronic HIV infection. Eur J Immunol. 2010;40:1036–1041. doi: 10.1002/eji.200939634. [DOI] [PubMed] [Google Scholar]
- 10.Mkhwanazi N, Thobakgale CF, van der Stok M, Reddy S, Mncube Z, Chonco F, Walker BD, Altfeld M, Goulder PJ, Ndung’u T. Immunodominant HIV-1-specific HLA-B- and HLA-C-restricted CD8+ T cells do not differ in polyfunctionality. Virology. 2010;405:483–491. doi: 10.1016/j.virol.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shrestha S, Aissani B, Song W, Wilson CM, Kaslow RA, Tang J. Host genetics and HIV-1 viral load set-point in African-Americans. AIDS. 2009;23:673–677. doi: 10.1097/QAD.0b013e328325d414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong T, Zhang Y, Xu KY, Yan H, James I, Peng Y, Blais ME, Gaudieri S, Chen X, Lun W, Wu H, Qu WY, Rostron T, Li N, Mao Y, Mallal S, Xu X, McMichael A, John M, Rowland-Jones SL. Extensive HLA-driven viral diversity following a narrow-source HIV-1 outbreak in rural China. Blood. 2011;118:98–106. doi: 10.1182/blood-2010-06-291963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science. 2002;296:1439–1443. doi: 10.1126/science.1069660. [DOI] [PubMed] [Google Scholar]
- 14.Dong T, Stewart-Jones G, Chen N, Easterbrook P, Xu X, Papagno L, Appay V, Weekes M, Conlon C, Spina C, Little S, Screaton G, van der Merwe A, Richman DD, McMichael AJ, Jones EY, Rowland-Jones SL. HIV-specific cytotoxic T cells from long-term survivors select a unique T cell receptor. J Exp Med. 2004;200:1547–1557. doi: 10.1084/jem.20032044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rauch A, James I, Pfafferott K, Nolan D, Klenerman P, Cheng W, Mollison L, McCaughan G, Shackel N, Jeffrey GP, Baker R, Freitas E, Humphreys I, Furrer H, Gunthard HF, Hirschel B, Mallal S, John M, Lucas M, Barnes E, Gaudieri S. Divergent adaptation of hepatitis C virus genotypes 1 and 3 to human leukocyte antigen-restricted immune pressure. Hepatology. 2009;50:1017–1029. doi: 10.1002/hep.23101. [DOI] [PubMed] [Google Scholar]
- 16.Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, Kuse N, Oka S, Duda A, Prendergast A, Crawford H, Leslie A, Brumme Z, Brumme C, Allen T, Brander C, Kaslow R, Tang J, Hunter E, Allen S, Mulenga J, Branch S, Roach T, John M, Mallal S, Ogwu A, Shapiro R, Prado JG, Fidler S, Weber J, Pybus OG, Klenerman P, Ndung’u T, Phillips R, Heckerman D, Harrigan PR, Walker BD, Takiguchi M, Goulder P. Adaptation of HIV-1 to human leukocyte antigen class I. Nature. 2009;458:641–645. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L, Zimbwa P, Moore S, Allen T, Brander C, Addo MM, Altfeld M, James I, Mallal S, Bunce M, Barber LD, Szinger J, Day C, Klenerman P, Mullins J, Korber B, Coovadia HM, Walker BD, Goulder PJ. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–775. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 18.Specht A, Telenti A, Martinez R, Fellay J, Bailes E, Evans DT, Carrington M, Hahn BH, Goldstein DB, Kirchhoff F. Counteraction of HLA-C-mediated immune control of HIV-1 by Nef. J Virol. 2010;84:7300–7311. doi: 10.1128/JVI.00619-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buranapraditkun S, Hempel U, Pitakpolrat P, Allgaier RL, Thantivorasit P, Lorenzen SI, Sirivichayakul S, Hildebrand WH, Altfeld M, Brander C, Walker BD, Phanuphak P, Hansasuta P, Rowland-Jones SL, Allen TM, Ruxrungtham K. A Novel Immunodominant CD8+ T Cell Response Restricted by a Common HLA-C Allele Targets a Conserved Region of Gag HIV-1 Clade CRF01_AE Infected Thais. PLoS One. 2011;6:e23603. doi: 10.1371/journal.pone.0023603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honda K, Zheng N, Murakoshi H, Hashimoto M, Sakai K, Borghan MA, Chikata T, Koyanagi M, Tamura Y, Gatanaga H, Oka S, Takiguchi M. Selection of escape mutant by HLA-C-restricted HIV-1 Pol-specific cytotoxic T lymphocytes carrying strong ability to suppress HIV-1 replication. Eur J Immunol. 2011;41:97–106. doi: 10.1002/eji.201040841. [DOI] [PubMed] [Google Scholar]
- 21.Almeida CA, Bronke C, Roberts SG, McKinnon E, Keane NM, Chopra A, Kadie C, Carlson J, Haas DW, Riddler SA, Haubrich R, Heckerman D, Mallal S, John M. Translation of HLA-HIV Associations to the Cellular Level: HIV Adapts To Inflate CD8 T Cell Responses against Nef and HLA-Adapted Variant Epitopes. J Immunol. 2011;187:2502–2513. doi: 10.4049/jimmunol.1100691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, Weinhold KJ, Moore S, Letvin N, Haynes BF, Cohen MS, Hraber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Peng Y, Yan H, Xu K, Saito M, Wu H, Chen X, Ranasinghe S, Kuse N, Powell T, Zhao Y, Li W, Zhang X, Feng X, Li N, Leligdowicz A, Xu X, John M, Takiguchi M, McMichael A, Rowland-Jones S, Dong T. Multilayered Defense in HLA-B51-Associated HIV Viral Control. J Immunol. 2011;187:684–691. doi: 10.4049/jimmunol.1100316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahlenstiel G, Martin MP, Gao X, Carrington M, Rehermann B. Distinct KIR/HLA compound genotypes affect the kinetics of human antiviral natural killer cell responses. J Clin Invest. 2008;118:1017–1026. doi: 10.1172/JCI32400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corrah TW, Goonetilleke N, Kopycinski J, Deeks SG, Cohen MS, Borrow P, McMichael A, Brackenridge S. Reappraisal of the relationship between the HIV-1-protective single-nucleotide polymorphism 35 kilobases upstream of the HLA-C gene and surface HLA-C expression. J Virol. 2011;85:3367–3374. doi: 10.1128/JVI.02276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fellay J, Shianna KV, Telenti A, Goldstein DB. Host genetics and HIV-1: the final phase? PLoS Pathog. 2010;6:e1001033. doi: 10.1371/journal.ppat.1001033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaye S, Comber E, Tenant-Flowers M, Loveday C. The appearance of drug resistance-associated point mutations in HIV type 1 plasma RNA precedes their appearance in proviral DNA. AIDS Res Hum Retroviruses. 1995;11:1221–1225. doi: 10.1089/aid.1995.11.1221. [DOI] [PubMed] [Google Scholar]
- 29.Blais ME, Dong T, Rowland-Jones S. HLA-C as a mediator of natural killer and T-cell activation: spectator or key player? Immunology. 2011;133:1–7. doi: 10.1111/j.1365-2567.2011.03422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, Ruether A, Schreiber S, Weichenthal M, Gladman D, Rahman P, Schrodi SJ, Prahalad S, Guthery SL, Fischer J, Liao W, Kwok PY, Menter A, Lathrop GM, Wise CA, Begovich AB, Voorhees JJ, Elder JT, Krueger GG, Bowcock AM, Abecasis GR. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, Barton A, Band G, Bellenguez C, Bergboer JG, Blackwell JM, Bramon E, Bumpstead SJ, Casas JP, Cork MJ, Corvin A, Deloukas P, Dilthey A, Duncanson A, Edkins S, Estivill X, Fitzgerald O, Freeman C, Giardina E, Gray E, Hofer A, Huffmeier U, Hunt SE, Irvine AD, Jankowski J, Kirby B, Langford C, Lascorz J, Leman J, Leslie S, Mallbris L, Markus HS, Mathew CG, McLean WH, McManus R, Mossner R, Moutsianas L, Naluai AT, Nestle FO, Novelli G, Onoufriadis A, Palmer CN, Perricone C, Pirinen M, Plomin R, Potter SC, Pujol RM, Rautanen A, Riveira-Munoz E, Ryan AW, Salmhofer W, Samuelsson L, Sawcer SJ, Schalkwijk J, Smith CH, Stahle M, Su Z, Tazi-Ahnini R, Traupe H, Viswanathan AC, Warren RB, Weger W, Wolk K, Wood N, Worthington J, Young HS, Zeeuwen PL, Hayday A, Burden AD, Griffiths CE, Kere J, Reis A, McVean G, Evans DM, Brown MA, Barker JN, Peltonen L, Donnelly P, Trembath RC. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lie WR, Myers NB, Gorka J, Rubocki RJ, Connolly JM, Hansen TH. Peptide ligand-induced conformation and surface expression of the Ld class I MHC molecule. Nature. 1990;344:439–441. doi: 10.1038/344439a0. [DOI] [PubMed] [Google Scholar]
- 33.Zemmour J, Parham P. Distinctive polymorphism at the HLA-C locus: implications for the expression of HLA-C. J Exp Med. 1992;176:937–950. doi: 10.1084/jem.176.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stojakovic M, Salazar-Fontana LI, Tatari-Calderone Z, Badovinac VP, Santori FR, Kovalovsky D, Sant’Angelo D, Harty JT, Vukmanovic S. Adaptable TCR avidity thresholds for negative selection. J Immunol. 2008;181:6770–6778. doi: 10.4049/jimmunol.181.10.6770. [DOI] [PubMed] [Google Scholar]
- 35.Kulpa DA, Collins KL. The emerging role of HLA-C in HIV-1 infection. Immunology. 2011;134:116–122. doi: 10.1111/j.1365-2567.2011.03474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.