

Abstract
The rotavirus RNA-dependent RNA polymerase (RdRp), VP1, contains canonical RdRp motifs and a priming loop that is hypothesized to undergo conformational rearrangements during RNA synthesis. In the absence of viral core shell protein VP2, VP1 fails to interact stably with divalent cations or nucleotides and has a retracted priming loop. To identify residues of potential import to nucleotide and divalent cation stabilization, we aligned VP1 of divergent rotaviruses and the structural homolog reovirus λ3. VP1 mutants were engineered and characterized for RNA synthetic capacity in vitro. Conserved aspartic acids in RdRp motifs A and C and arginines in motif F that likely stabilize divalent cations and nucleotides were required for efficient RNA synthesis. Mutation of individual priming loop residues diminished or enhanced RNA synthesis efficiency without obviating the need for VP2, which suggests that the this structure serves as a dynamic regulatory element that links RdRp activity to particle assembly.
Keywords: RNA-dependent RNA polymerase, rotavirus, double-stranded RNA virus, replication, initiation, RNA catalysis, priming loop
INTRODUCTION
RNA-dependent RNA polymerases (RdRps) mediate the highly dynamic process of RNA polymerization. RdRp polymerase domains share a ‘right-handed’ architecture, with fingers, palm, and thumb subdomains, and contain canonical motifs A through F (Fig. 1) (Bruenn, 2003; Ng et al., 2008; O’Reilly and Kao, 1998; Steitz, 1998; Yang et al., 2006). Residues in these motifs interact with templates, incoming nucleotide triphosphates (NTPs), and the divalent metal ions that catalyze phosphodiester bond formation. Due to highly conserved positional identity, these residues likely perform similar functions in divergent RdRps. Initiation mechanisms differ among RdRps, but primer-independent RdRps that initiate at the 3′ end of the template utilize specialized structural elements to stabilize a priming NTP in the catalytic site (Ferrer-Orta et al., 2006; Kao et al., 2001; Ng et al., 2008; van Dijk et al., 2004). The structures of several viral RdRps have provided a wealth of information, but in many cases these static models have failed to fully explain mechanisms of initiation and elongation. Such is the case for VP1, the RdRp of rotavirus (RV).
FIGURE 1. Catalytic centers of rotavirus VP1 and reovirus λ3.

Ribbon drawings of RV VP1 (A–C) and the reovirus λ3 initiation complex (D–H). Conserved RdRp motifs are colored pink (motif A), purple (motif B), green (motif C), cyan (motif D), yellow (motif E), and cornflower blue (motif F). The PL is colored gold, and a bound RNA oligo representing the template is depicted as yellow sticks. In the λ3 structure, divalent cations are colored dark olive, and NTPs occupying the P and N sites are colored orange. The side chains of conserved aspartic acids in motifs A and C, arginines in motif F, and selected residues of the PL are shown. Regions of λ3 and VP1 have been removed for clarity. The RdRp is oriented similarly in (B–C) and (F–H) to the orientation in (A) and (D), respectively. (H) λ3 is rotated approximately 45° to the left in comparison to (D). PLs from the reovirus λ3 initiation complex (gold) or elongation complex (sea green) or the rotavirus VP1 structure (royal blue), and adjacent α-helices, have been overlaid on the λ3 initiation complex structure.
RVs, of which there are eight proposed species (RVA-RVH), are important agents of diarrheal disease (Attoui et al., 2012; Parashar et al., 2009). RV RdRp VP1 synthesizes positive-sense RNAs (+RNAs) from the minus strand of genomic dsRNA (transcription) and genomic dsRNA from viral +RNAs (replication) (Estes and Kapikian, 2007). In each case, VP1 initiates de novo from the 3′ end of the template. VP1 intrinsically and specifically recognizes a consensus sequence at the 3′ terminus of RV +RNAs, but it is catalytically active only in the presence of viral cofactor VP2, which forms, as 60 asymmetric dimers, the innermost layer of RV virions (Lu et al., 2008; McClain et al., 2010; Patton, 1996; Tortorici et al., 2003). Accordingly, crystallized VP1 binds the 3′-consensus sequence, but it does so such that the template RNA has overshot the initiation register by a single nucleotide, and it fails to associate stably with divalent metal ions or NTPs (Fig. 1A) (Lu et al., 2008). Overshot template alignment, which appears to be ‘corrected’ in the presence of bound P-site and N-site NTPs, has been observed for other de novo initiating RdRps (Bressanelli et al., 2002; Butcher et al., 2001; O’Farrell et al., 2003).
In the absence of a structure for a catalytically active form of VP1, comparison with other RdRps may provide insight into mechanisms of VP1 activation, initiation, and polymerization. The closest known homolog of VP1 is λ3, the RdRp of fellow Reoviridae member mammalian orthoreovirus (reovirus) (Lu et al., 2008; McDonald et al., 2009b; Tao et al., 2002). VP1 and λ3 have strikingly similar overall structures, including four-tunneled architecture, a large N-terminal domain and C-terminal bracelet domain that sandwiches the polymerase domain, and an insertion called the priming loop (PL) between the fingers and palm subdomains. Unlike VP1, crystallized λ3 is active and associates stably with divalent cations and NTPs (Tao et al., 2002). In the λ3 initiation complex, templated NTPs occupy the incoming nucleotide site (N site) and the priming site (P site) of the catalytic pocket (Fig. 1D–H). Two consecutive aspartic acids in motif C (Asp734 and Asp735) and an invariant aspartic acid in motif A (Asp585) are involved in coordinating the divalent metal ions that catalyze phosphodiester bond formation and help to orient the N-site NTP through interactions with its triphosphates. A second conserved aspartic acid in motif A (Asp590) interacts with the ribose of the N-site NTP, and basic amino acids in motif F (Args 523, 524, and 526) interact with its triphosphates, likely stabilizing the NTP and assisting PPi leaving. Due to structural similarity with λ3 and conservation of motifs among RdRps, we hypothesize that activated VP1 forms similar interactions with divalent metal ions and N-site NTPs during distinct steps of RNA synthesis.
In the λ3 initiation complex, two residues (Ser561 and Lys566) in the flexible PL bind the ribose triphosphate of the P-site NTP, which is aligned in-register with the 3′ terminus of the RNA template (Fig. 1F–G) (Tao et al., 2002). While the reovirus PL supports the priming NTP during initiation, it changes conformation to permit RNA product passage during elongation (Fig. 1H). A noteworthy difference between the catalytic regions of λ3 and VP1 is PL conformation (Fig. 1) (Lu et al., 2008; McDonald et al., 2009b). In contrast to the extended λ3 PL, the VP1 PL is retracted and bent away from the catalytic pocket such that it is incapable of supporting a P-site NTP. Nonetheless, the remarkable similarity between VP1 and λ3 suggests the PL serves the same function in each RdRp. Based on this hypothesis, conformational rearrangements of the VP1 PL, triggered by activating interactions with VP2, might permit stabilization of the P-site NTP and, in turn, stabilize the N-site NTP and divalent metal ions, which would result in appropriate positioning of the template for initiation. During elongation, additional changes in PL conformation likely permit passage of nascent RNA products.
The goal of the current study was, in the absence of a structure for the catalytically active RdRp, to dissect the contributions to RNA synthesis of individual VP1 residues that are predicted to help stabilize divalent cations or NTPs in the catalytic pocket. Using alignments and in vitro dsRNA synthesis assays with mutant forms of VP1, we provide evidence that conserved aspartic acids in motifs A and C make critical contributions to RNA synthesis by coordinating divalent metal ions and NTPs. Conserved arginines in motif F are important for dsRNA synthesis, likely because they interact with N-site NTP triphosphates and maintain local RdRp structure. Consistent with a dynamic regulatory element, many individual residues in the PL contribute to RV replication, and mutations at these positions resulted in either enhanced or diminished levels of dsRNA synthesis. None of the PL mutants, or other active-site mutants, was capable of RNA synthesis in the absence of VP2, confirming the essential link between replication and rotavirus particle assembly. Taken together, these findings enhance our understanding of contributions of RdRp motifs and the PL to RV RNA synthesis. Due to conservation of RdRp motifs, these findings may also apply to other viral RdRps.
RESULTS
Conserved aspartic acids in motifs A and C are required for RNA synthesis
To identify conserved aspartic acids in motifs A and C, sequence-based alignments were performed for RVs from divergent groups (Fig. 2A–B). Structure-based alignments were performed between RVA VP1 and reovirus λ3. The length and composition of the two motifs were well conserved among RVs, with the exception of an apparent deletion in motif C for RVH. Aspartic acids at four positions in RVA VP1 were conserved among all RVs and aligned structurally with those in reovirus λ3 that interact with the N-site NTP ribose (λ3, Asp590; VP1, Asp525) and coordinate catalytic divalent metal ions and NTP triphosphates (λ3, Asps 585, 734 735; VP1, Asps 520, 631, 632) in the initiation complex (Fig. 1B,G and 2A B) (Lu et al., 2008; Tao et al., 2002).
FIGURE 2. Contributions of motif A and C aspartic acids to RNA synthesis.

Sequence based alignment of motifs A (A) and C (B) from VP1 of representative strains of RVA-RVD and RVF-RVH, and structure-based alignment of RVA VP1 with reovirus λ3 (Reo). Residues that are identical for all species of RV are denoted with an asterisk. Residues identical for all RdRps are denoted with grey text. (C) Representative autoradiograph of radiolabeled dsRNA synthesis assay products (upper) and Coomassie-stained polyacrylamide gel showing relative amounts of input VP1 protein (lower). Mutations in VP1 are indicated. Mock samples were purified from insect cells infected with baculoviruses not expressing exogenous protein. (D) Graph of relative amounts of products of dsRNA synthesis assays. Error bars represent SD for results of three experiments. Means are indicated above bars. *, P < 0.01.
To determine whether conserved aspartic acids in motifs A and C are required for RNA synthesis, we expressed and purified VP1 proteins with individual alanine substitutions at positions 520, 525, 631, and 632. WT and mutant proteins were recovered with equivalent efficiency (Fig. 2C). The RNA synthetic capacity of the proteins was determined using in vitro dsRNA synthesis assays, in which VP1 was incubated with VP2, Mg2+, NTPs, radiolabeled UTP, and in vitro transcribed RV +RNAs. Products were resolved by polyacrylamide gel electrophoresis and visualized by autoradiography. While products were readily detected for WT VP1, they were not detected for any of the four mutants (Fig. 2C–D), which indicates that each of these highly conserved aspartic acids in VP1 motifs A and C makes a critical contribution to dsRNA synthesis.
Motif F arginines are required for efficient RNA synthesis
To assess conservation of basic residues in motif F, RV sequences and the reovirus λ3 structure were aligned with RVA VP1 (Fig. 3A). Basic residues were conserved among all the RdRps at RVA VP1 positions 452, 457, and 460. Arg451 and Arg458 were conserved among RVA, RVC, RVD, and RVF, which are phylogenetically closer to one another than to RVB, RVG, and RVH (Attoui et al., 2012; Johne et al., 2011; Ogden et al., in review). These arginines are located adjacent to the predicted N site in the RVA VP1 catalytic pocket (Fig. 1B). Args 452, 457, 458, and 460 in VP1 occupy positions homologous to 518, 523, 524, and 526 in λ3, respectively, and point towards the catalytic pocket in the λ3 initiation complex structure (Fig. 1B,E) (Lu et al., 2008; Tao et al., 2002). Args 523, 524, and 526 of λ3 interact directly with the N-site NTP triphosphates.
FIGURE 3. Contributions of motif F arginines to RNA synthesis.
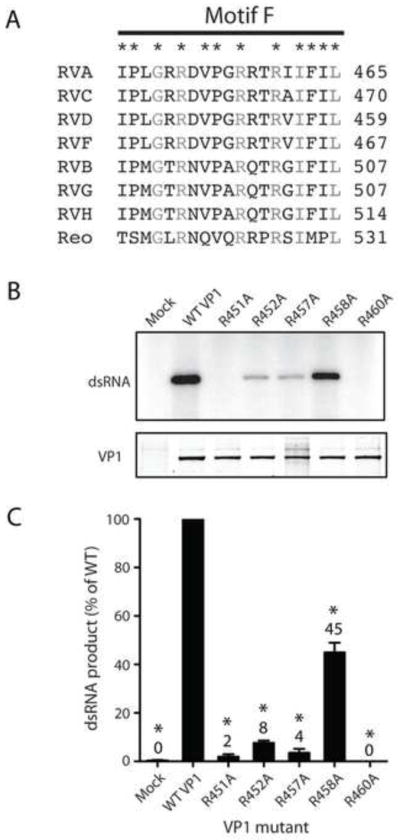
(A) Sequence-based alignment of motif F from VP1 of representative strains of RVA-RVD and RVF-RVH, and structure-based alignment of RVA VP1 with reovirus λ3 (Reo). Residues that are identical for all species of RV are denoted with an asterisk. Residues identical for all RdRps are denoted with grey text. (B) Representative autoradiograph of radiolabeled dsRNA products from an in vitro dsRNA synthesis assay (upper) and Coomassie-stained polyacrylamide gel showing relative amounts of input VP1 protein (lower). Mutations engineered in VP1 are indicated. (C) Graph of relative amounts of products of dsRNA synthesis assays. Error bars represent SEM for results of six experiments. Means are indicated above bars. *, P < 0.01.
To elucidate the functional contributions of conserved motif F arginines to RNA synthesis, VP1 mutants were engineered and purified for biochemical analysis. Each mutant produced fewer full-length dsRNA products than WT VP1 (Fig. 3B–C). Products were undetectable for R460A and no greater than 10% of WT levels for R451A, R452A, and R457A. The R458A mutant was least impaired, exhibiting 45% of WT activity. Together with sequence alignments and comparison with λ3, these results suggest that conserved arginines in VP1 motif F make important contributions to RNA synthesis, with effects likely mediated by direct interactions between Args 457 and 460 with the N-site NTP.
Rotavirus priming loops vary in length and composition
In alignments of VP1 from divergent RVs, the PLs of more closely related RVA, RVC, RVD, and RVF were all the same length and conserved several positions of amino acid identity (Fig. 4A). The more distantly related RVB, RVG, and RVH PLs were three residues longer than those of RVA. All RV PLs conserved identity at positions Glu492 and Tyr494 and similarity at Tyr490. Despite the remarkable overall similarity of the VP1 and λ3 structures, the PLs of these RdRps fail to align well structurally, due to obvious differences in conformation (Figs. 1 and 4B). The PLs, however, do occupy the same topological position between two α-helices in the polymerase domain (Lu et al., 2008; Tao et al., 2002). The RVA VP1 PL is 12 amino acids long. The λ3 PL contains 15 amino acids, which is the apparent length of RVB, RVG, and RVH PLs (Fig. 4A). Sequence alignments of VP1 and λ3 suggest that there is little positional identity or similarity between the PLs (Fig. 4B).
FIGURE 4. Comparison of priming loop sequence and composition.

(A) Sequence-based alignment of PLs from VP1 of representative strains of RVA-RVD and RVF-RVH. Identical residues are denoted in gray text, and similar residues are indicated with an asterisk. (B) Alignment of RVA VP1 and reovirus λ3 (Reo) PL regions based on structure (upper) and sequence (lower). Residues in the PL are indicated by bold oblique text. (C) Representative autoradiograph of radiolabeled dsRNA synthesis assay products (upper) and Coomassie-stained polyacrylamide gel showing relative amounts of input VP1 protein (lower). PL swaps engineered in VP1 are indicated. (D) Graphs of relative amounts of products of dsRNA synthesis assays. Error bars represent SD for results of three experiments. Means are indicated above bars. *, P < 0.01.
To determine whether a similar or more divergent PL could substitute functionally for the RVA VP1 PL, we engineered RVA VP1 with an RVB or RVC PL. The RVA to RVC PL swap resulted in four amino acid sequence changes: H486R, T487E, A491S, and S497A (Fig. 4A). These residues are close to the ends of the PL and are not highly conserved. The RVA to RVB PL swap increased PL length by three amino acids and dramatically altered its composition. An RVA to λ3 PL swap was also constructed, but we were unable to efficiently recover the mutant protein (data not shown). VP1 proteins with PL swaps were purified, and their in vitro dsRNA synthetic capacity was quantified (Fig. 4C–D). The RVB PL swap resulted in a catalytically inactive polymerase, whereas the RVC PL swap yielded a poorly functional polymerase (5% WT activity). These results suggest that both the length and composition of the PL are important for function, and that His486, Thr487, Ala491, and Ser497 are important, but not strictly required, for VP1 polymerase activity.
Several residues of the VP1 priming loop contribute to RNA synthesis
To dissect contributions of individual PL residues to RNA synthesis, we engineered alanine substitutions across the RVA VP1 PL. Each PL residue that did not align with alanine for at least one RV strain in our alignment was mutated, with the exception of Thr487, which occupies a highly variable position (Fig. 4A). We also included in this group a mutant of Lys679, a conserved residue in motif E that points towards the catalytic pocket and forms a three-way stack with PL residues Phe493 and Glu492 (Fig. 1C) (Lu et al., 2008). Arg488 stacks against the conserved Tyr494 side chain, and Asn498 accepts a hydrogen bond from the Leu500 main chain. Mutant VP1 proteins were purified to levels similar to those of WT VP1 (Fig. 5A). The only exception was the Y490A mutant, which we could not purify to sufficient levels for experimentation (data not shown). The E489A mutant produced dsRNA nearly as efficiently (87%) as WT VP1, and the N498A had nearly half (45%) of WT activity (Fig. 5B). The R488A, E492A, F493A, Y494A, and K679A mutants all exhibited significantly reduced RNA synthetic capacity, with R488A, E492A, and K679A most severely impaired (<10% WT activity). In contrast, the S495A and Q496A mutants reproducibly synthesized dsRNA about twice as efficiently as WT VP1. These results suggest that multiple priming loop residues function in concert, possibly through interactions with a P-site nucleotide or with one another, to regulate dsRNA synthesis.
FIGURE 5. Contributions of VP1 priming loop residues to RNA synthesis.
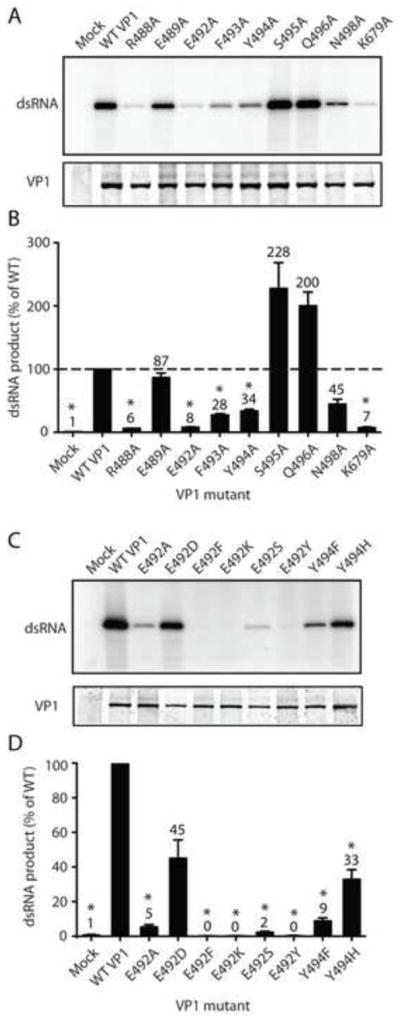
(A,C) Representative autoradiographs of radiolabeled dsRNA synthesis assay products (upper) and Coomassie-stained polyacrylamide gels showing relative amounts of input VP1 protein (lower). Point mutations engineered in VP1 are indicated. (B,D) Graphs of relative amounts of products of dsRNA synthesis assays. Error bars represent SD for results of at least three experiments. Means are indicated above bars. *, P < 0.01. Dashed line represents replication levels for WT VP1 (B).
Amino acid identity was conserved among all RV PLs at Glu492 and Tyr494 (Fig. 4A). To determine whether amino acids other than alanine could function in place of these residues, we replaced Glu492 with aspartic acid, phenylalanine, tyrosine, lysine, or serine and Tyr494 with phenylalanine or histidine. Only aspartic acid was capable of complementing the function of Glu492 with regards to RNA synthesis, suggesting that the acidic nature of this residue contributes to its function (Fig. 5C–D). While Y494H synthesized higher levels of dsRNA than Y494F, neither residue substituted efficiently for tyrosine. These observations suggest that the specific chemistry of tyrosine is important for PL function.
Dinucleotides render initiation resistant to salt
A step in the initiation of dsRNA synthesis by RV subviral particles, open cores, and by recombinant VP1 is sensitive to the presence of NaCl (Chen and Patton, 2000; Tortorici et al., 2003). However, pre-incubation of RV open cores with pGpG permits stable initiation complex formation and renders the elongation step of dsRNA synthesis salt-insensitive. To determine whether dinucleotides or trinucleotides could function in a similar capacity for recombinant VP1, we performed in vitro salt sensitivity assays (Fig. 6A–B). In these assays, VP1 was incubated with VP2, +RNA, Mg2+, and either GTP, dinucleotides, or trinucleotides, to permit stable initiation complex formation. Salt was then added to inhibit additional initiation, followed by the addition of ATP, CTP, GTP, and UTP to promote elongation from pre-formed initiation complexes. The phosphorylated dinucleotide pGpG and the trinucleotide pGpGpU were used as ‘primers’ in these assays because they represent the Watson-Crick base-pairing partners for the 3′-terminal residues of +RNA templates. For samples in which no GTP or ‘primers’ were included in pre-incubation, a low level of dsRNA synthesis was observed (Fig. 6B), likely due to carryover of NTPs with the +RNA templates added to the reaction. Upon pre-incubation with GTP, initiation occurred at levels slightly higher than background, and upon pre-incubation with pGpG, initiation efficiency appeared substantially enhanced. However, pre-incubation with pGpGpU not only failed to enhance initiation, but it reduced full-length dsRNA production to undetectable levels. These findings demonstrate that pGpG can prime dsRNA synthesis for recombinant VP1 and suggest that trinucleotides inhibit stable RV initiation complex formation.
FIGURE 6. Salt sensitivity assays of WT and mutant VP1.

(A) Steps of salt sensitivity assays. (B) Autoradiograph showing radiolabeled dsRNA products from a salt sensitivity assay in which the indicated NTP, dinucleotide, or trinucleotide was present at 1.25 mM during pre-incubation. The first lane is a control experiment in which VP1 was not added. (C) Autoradiograph showing radiolabeled dsRNA products from a salt sensitivity assay in which the indicated NTP, dinucleotide, or trinucleotide was present in the pre-incubation at 1.25 mM. The presence (+) or absence (−) of 250 mM NaCl in the pre-incubation is indicated. (D) Graph of quantified dsRNA products from salt sensitivity assays, in which 0.125 mM pGpG was included in the pre-incubation. Error bars represent SD for results of two independent experiments. Means are indicated above bars. Dashed line represents replication levels for WT VP1.
While initiation has been shown to be a salt-sensitive process (Chen and Patton, 2000; Tortorici et al., 2003), it is not known whether pGpG confers salt resistance to the RV initiation complex. To determine whether VP1 can form stable initiation complexes in the presence of salt, assays were performed with GTP, pGpG, or pGpGpU and 250 mM NaCl present during the pre-incubation. Upon pre-incubation in the presence of salt, VP1 was essentially unable to form stable initiation complexes with GTP or pGpGpU, as reflected by the absence of dsRNA products (Fig. 6C). Upon pre-incubation with pGpG in the presence of salt, levels of dsRNA synthesis were equivalent or higher than in its absence, suggesting that stable initiation complexes containing pGpG form efficiently in the presence of salt. These results demonstrate that pGpG permits VP1 to bypass salt-sensitive step(s) of initiation during dsRNA synthesis.
‘Priming’ with pGpG complements dsRNA synthesis for some priming loop mutants
Alanine substitution mutants of the PL exhibited a variety of phenotypes in dsRNA synthesis assays (Fig. 5B). To gain insight into the step(s) of dsRNA synthesis during which PL mutations meditated their phenotypic effects, salt sensitivity assays were performed. In the assays, VP1 PL mutants were pre-incubated with pGpG before the addition of NTPs (Fig. 6D). In general, relative levels of dsRNA synthesis showed trends similar to those observed in dsRNA synthesis assays (compare Fig. 5B to 6D). However, several specific differences also were observed. For example, pre-incubation with pGpG in the absence of salt resulted in at least partial complementation of dsRNA synthetic activity for the R488A, E492A, Y494A, N498A, and K679A mutants, with the most efficient complementation observed for R488A and K679A. These findings suggest that the indicated mutations affect events at or upstream of the salt-sensitive initiation step of dsRNA synthesis. While partial complementation was observed for E492A and Y494A, no complementation of dsRNA synthesis was observed for the F493A mutant, consistent with the idea that these mutated residues contribute to events downstream of the salt-sensitive step of initiation. The S495A and Q496A mutants retained enhanced dsRNA synthetic capacity, similar to results observed during in vitro dsRNA synthesis assays. For WT VP1 and all PL mutants tested, dsRNA synthesis remained VP2 dependent, despite pre-incubation with pGpG (data not shown).
DISCUSSION
For primer-independent RdRps, RNA polymerization proceeds through several steps, including (i) template binding, (ii) stabilization of divalent metal ions, P-site, and N-site NTPs, (iii) formation of the first phosphodiester bond and PPi release, (iv) ratcheting forward of the template and nascent strand, such that N site is unoccupied and N-site NTP has moved into P site, and (v) repetition of the process, with the 3′OH of the most recently added nucleotide serving as a ‘primer’ for the addition of the next N-site NTP. Residues in the RdRp active site must act in concert to engage, translocate, and release substrates and to catalyze phosphodiester bond formation. For rotavirus RdRp VP1, the mechanism of template binding is known, but downstream events are unclear. The studies presented herein provide clues about the specific residues that are involved in orchestrating these polymerization events for VP1.
Aspartic acids at VP1 positions 520, 631, and 632 likely coordinate divalent metal ions in the catalytic site. The absolute conservation of these motif A and C aspartic acid among rotaviruses from divergent groups and the observed coordination of Mn2+ by homologous residues in λ3 structures (Fig. 2A) are consistent with this hypothesis. In accord with the critical role of divalent metal ions in catalysis, alanine substitution of these residues ablated dsRNA synthesis by VP1 (Fig. 2B–C). Enzymes that use a two metal ion mechanism of catalysis commonly demonstrate catalytically relevant ion binding only in the presence of substrate (Yang et al., 2006). Thus, it is likely that crystallized VP1 fails to stably associate with Mg2+ because it is unable to stabilize NTPs in the catalytic pocket (Lu et al., 2008).
Several components of the VP1 catalytic pocket likely interact with the N-site NTP. Based on comparison with reovirus λ3, VP1 motif F Args 452, 457, 458, and 460 are predicted to interact directly with N-site NTP triphosphates, and motif A Asp525 is predicted to interact with its ribose (Figs. 1B,E, 2A, and 3A). These hypotheses are supported by the findings that most of these residues are conserved among RVs and that mutation of Arg452, Arg457, Arg460, and Asp525 severely impaired or ablated dsRNA synthesis (Figs. 2 and 3). In published structures of VP1, Arg458 points away from the catalytic pocket (Fig. 1B) (Lu et al., 2008). While Arg458 is not critical for dsRNA synthesis (Fig. 3B–C), upon transit of an NTP through the entry tunnel, the Arg458 side chain might shift to permit direct interaction with the nucleotide triphosphates. In VP1, the Arg451 side chain forms a salt bridge with Asp453 and donates H to the Gln337 main chain oxygen (Lu et al., 2008). These interactions might help stabilize motif F conformation, which would explain the loss of activity for the R451A mutant. In addition to motif F residues, divalent metal ions also are likely involved in interactions with the P-site NTP triphosphates. Upon assembly of a stable initiation complex, Watson-Crick pairing with the template RNA and stacking with the P-site NTP base also likely contribute to N-site NTP stabilization.
The mechanism by which a P-site NTP is stabilized in the catalytic pocket during initiation is still unknown. However, stabilization of this NTP mediated by the PL in an alternate conformation remains the most plausible mechanism. Polymerization is a highly dynamic process, and loops are commonly flexible elements within protein structures. In addition to striking structural similarities between their RdRps, reovirus and RV are members of the same virus family, which likely share a common ancestor and, therefore, common structural and functional properties. In a practical sense, it seems quite possible that the VP1 PL can function as an initiation platform. Few contacts external to the loop itself anchor the VP1 PL in its observed conformation, and ample space is available in the hollow center of VP1 to accommodate PL conformational rearrangements (Lu et al., 2008). Our alignments suggest that RVA, RVC, RVD, and RVF PLs are three amino acids shorter, and the RVB, RVG, and RVH PLs are the same length as the reovirus λ3 PL (Fig. 4A) (Tao et al., 2002). Thus, the PL could potentially extend to a similar length and reach the P-site NTP that needs stabilization in the catalytic pocket during initiation.
In combination with dsRNA synthesis assays, the results of salt sensitivity assays suggest a role for several PL residues in stable initiation complex formation. Pre-incubation of WT VP1 with pGpG permitted bypass of a salt-sensitive step of initiation (Fig. 6B). Although the precise step in initiation that is sensitive to salt is not known, it is likely a rate limiting step that occurs after template binding and at or prior to formation of the first phosphodiester bond of the nascent strand {Chen, 2000 #21}. VP1 mutants R488A, E492A, F493A, Y494A, and K679A all exhibited significantly diminished dsRNA synthetic capacity compared to WT VP1 (Fig. 5A–B). Pre-incubation with pGpG at least partially complemented defects in dsRNA synthesis for the R488A, E492A, Y494A, and K679A mutants, with product levels increasing approximately tenfold for the R488A and K679A mutants (Fig. 6D). The observed pGpG complementation suggests that these residues are involved in events at or upstream of the salt-sensitive step of initiation. The λ3 homolog (Lys783) of VP1 motif E residue Lys679 is oriented towards the divalent metal ions and N-site nucleotide triphosphates (Fig. 1F–G) (Tao et al., 2002). However, VP1 Lys679 forms the end of a three way stack with PL residues Glu492 and Phe493, and it is positioned to most likely interact with the triphosphates of a P-site NTP (Fig. 1C) (Lu et al., 2008). These results suggest that Lys679 of VP1 is likely to play a role in P-site NTP stabilization, rather than in divalent cation coordination. Arg488 is located close to the end of the PL (Fig. 1C). Although it is somewhat unlikely, based on position, that Arg488 interacts directly with a P-site NTP, this residue may have important effects on PL dynamics. The high level of conservation of residues Glu492 and Tyr494, their importance for dsRNA synthesis, and their location near the middle of the PL suggest that these residues might help to directly support a P-site NTP during initiation, were the PL to occupy an extended active conformation (Figs. 1C, 4A, and 5C–D). Consistent with this hypothesis is the observation that pre-incubation with pGpG partially complemented dsRNA synthesis defects for the E492A and Y494A mutants (Fig. 6D).
Available evidence suggests the PL is a dynamic structure involved in the regulation of multiple steps of RNA synthesis. In this study, nearly every residue of the VP1 PL was altered (Fig. 5). While none of the PL residues tested was absolutely required for polymerase activity, mutation of nearly any individual PL residue affected the efficiency of RNA synthesis in vitro, with S495A and Q496A actually enhancing product formation. These results are consistent with the hypothesis that multiple PL residues help to maintain different conformations of the loop during pre-initiation, initiation, and elongation. Van der Waals interactions between the stacked side chains of Arg488 and Tyr494 or between Glu492, Phe493, and Lys679 likely contribute to the observed orientation of the PL in VP1 crystal structures (Fig. 1C) (Lu et al., 2008). Alanine substitution at any of these positions (488, 492, 493, 494, or 679) resulted in diminished levels of dsRNA synthesis (Fig. 5A–B). Furthermore, only partial complementation of dsRNA synthesis was observed for the E492A and Y494A mutants during salt sensitivity assays, and this treatment failed to complement defects in dsRNA synthesis for the F493A mutant (Fig. 6D), which suggests that these mutations exert effects on events downstream of the salt-sensitive step of initiation. Due to disruption of stacking interactions within the PL, any of these mutations could result in local PL destabilization. Such destabilization could permit the PL to move more easily into an extended conformation, but it might also hinder movement into a position that permits RNA product passage during elongation. It is currently unclear how alanine substitutions at positions 495 and 496 resulted in VP1 proteins with enhanced dsRNA synthetic activity (Fig. 5A–B). That the S495A and Q496A mutants were little affected by pre-incubation with pGpG during salt sensitivity assays (Fig. 6D) might suggest that these mutations enhance elongation efficiency for VP1, but further studies would be necessary to support such a claim. In VP1 structures, the only contact observed for the Ser495 side chain is donation of H to the Gln496 main chain (Lu et al., 2008). Nonetheless, alanine substitutions at these positions might promote favorable conformational changes or enhance PL mobility.
Pre-incubation with pGpGpU completely inhibited dsRNA synthesis during salt sensitivity assays (Fig. 6A–B). We envision three possible explanations for this inhibition. (i) pGpGpU was bound within the hollow catalytic center of VP1, occupying the P and N sites, as well as the site opposite the overshot RNA residue (P–1). In this situation, the primer could pair with the template RNA and hold it in an overshot conformation, possibly preventing dynamic conformational changes necessary for elongation. (ii) pGpGpU paired with template RNA outside of the polymerase, preventing the template from accessing the VP1 entry tunnel, which is too narrow to accommodate dsRNA (Lu et al., 2008). (iii) pGpGpU occupied the template entry tunnel, NTP entry tunnel, or another region of VP1, directly blocking RNA or NTP/ PPi entry or exit.
Chen and Patton previously showed that pGpG dinucleotides function as specific primers for dsRNA synthesis, rendering open cores insensitive to salt after pre-incubation with pGpG (Chen and Patton, 2000). Here, we confirmed this finding for recombinant VP1 and further demonstrated that incubation with pGpG renders a salt-sensitive step of initiation salt-resistant (Fig. 6A–B). The most likely explanation for the observed result is that pGpG entered the VP1 catalytic pocket, occupied the P and N sites, and paired with the terminal CC nucleotides of the template, permitting VP1 to bypass formation of the first phosphodiester bond. However, it is also possible that pGpG acted as an allosteric enhancer. If pGpG did occupy the P and N sites of VP1, it is likely that the same conformational changes in the catalytic pocket required to support the P-site NTP during initiation were also required to support the dinucleotide, at least temporarily. This hypothesis is supported by the observation that pre-incubation of VP1 with pGpG in the presence or absence of salt during failed to render dsRNA synthesis VP2-independent (data not shown).
The 120 copies of VP2 that form the inner shell of RV virions are arranged as twelve decamers, each of which forms an icosahedral vertex thought to associate with a single VP1 molecule (Lawton et al., 1997; Li et al., 2009; McClain et al., 2010; Pesavento et al., 2006). A region on the interior surface of the VP2 shell provides RV species-specific VP1 activation signals (McDonald and Patton, 2011). Fitting of the VP1 structure into cryo-EM image reconstructions has reinforced the idea that VP1 interacts directly with the VP2 shell interior (Estrozi and Navaza, 2010). VP1 is oriented such that the surface of the molecule containing the +RNA exit tunnel abuts the inner VP2 shell, proximal to the five-fold icosahedral vertex (S. Harrison and E. Settembre, unpublished observations). The retracted PL resides very near the surface of VP1, adjacent to the +RNA exit tunnel. Thus, VP2 may trigger conformational rearrangements in VP1 through direct interaction with residues in or adjacent to the PL. One of our initial goals in this study was to purify a VP1 mutant that was capable of synthesizing RNA in the absence of VP2. Although none of the mutants we engineered was capable of such activity, future studies to identify specific interacting residues of VP1 and VP2 may permit design of a constitutively active form of the RdRp, which could be used for molecular biological and structural studies. Such a molecule could be used to clarify mechanisms of VP1 initiation and elongation and determine the precise role of the PL in these processes.
MATERIALS AND METHODS
Alignments
GenBank accession numbers for VP1 amino acid sequences are: RVA (SA11), ABG75815; RVB (Cal-1), ACD39820; RVC (Bristol), Q91E95; RVD (05V0049), ADN06423; RVF (03V0568), JN596591; RVG (03V0567), JN596592; and RVH (J19), AAZ03485 (Chen et al., 2002; Jiang et al., 2008; Small et al., 2007; Trojnar et al., 2010). Sequences of representative RV strains were aligned using the ClustalW function, with default settings, of MacVector, version 12.0.3. Reovirus λ3 (PDB 1N1H and 1N35) was aligned to RVA SA11 VP1 (PDB 2R7R) using the MatchMaker function in the UCSF Chimera package (Lu et al., 2008; Tao et al., 2002). Sequence alignments were generated from aligned structures using the Match→Align function in Chimera, with circular permutations permitted and alignments iterated until convergence. For PL alignments, the residue-residue distance cutoff was raised from the default value of 5Å to 10Å, to permit alignment despite significant differences in the conformation of this structure.
Recombinant baculovirus generation
Recombinant baculoviruses expressing mutant VP1 proteins were made using the BaculoDirect Expression System (Invitrogen). The parental pENTR-A-VP1 plasmid and baculoviruses expressing WT RVA (SA11) VP1 or VP2 were generated as previously described (McDonald et al., 2009a). Point mutations (R451A, R452A, R457A, R458A, R460A, R488A, E489A, E492A, E492D, E492F, E492K, E492S, E492Y, F493A, Y494A, Y494F, Y494H, S495A, Q496A, N498A, D632A, and K679A) were engineered in the VP1 open reading frame in pENTR-A-VP1 by outward PCR using Accuprime Pfx Supermix (Invitrogen) and 5′-phosphorylated primers containing specific nucleotide changes. Substitution of nucleic acid changes encoding the RVC (Bristol) RV PL for that of RVA were engineered in pENTR-A-VP1 by outward PCR using primers encoding the altered amino acid sequences (HTREYAEFYSQSNQ > REREYSEFYSQANQ). Mutagenic primer sequences are available upon request. cDNA cassettes encoding a fragment of VP1 that contained a D520A, D525A, or D631A mutation were synthesized de novo by Blue Heron Biotechnology (Bothell, WA) and cloned into pENTR-A-VP1 by restriction digestion and ligation. A cDNA cassette encoding a fragment of RVA VP1 with the PL sequence from RVB VP1 (Cal-1; HTREYAEFYSQSNQL > QGSFAEAYTAKTASLTYG) was synthesized de novo by GeneArt AG (Regensburg, Germany) and cloned into pENTR-A-VP1 by restriction digestion and ligation. Following sequence confirmation, VP1-encoding sequences were inserted into linearized baculovirus DNA, using the BaculoDirect C-Term Transfection Kit (Invitrogen). Baculoviruses were selected using ganciclovir and harvested from the medium.
Recombinant VP1 and VP2 purification
Recombinant VP2 and His-tagged VP1 proteins were prepared as described (Ogden et al., 2011).
In vitro transcription of RNA templates
RNA templates identical to RV SA11 gene 8 +RNAs were synthesized, purified, and quantified as described (Ogden et al., 2011), with the addition of phenol-chloroform extraction and ethanol precipitation immediately following transcript generation.
Double-stranded RNA synthesis assays
Reaction mixtures (20 μl) contained 50 mM Tris-HCl, pH 7.1, 1.5% PEG 8000, 2.5 mM dithiothreitol, 20 mM magnesium chloride, 1.25 mM each of ATP, CTP, GTP, and UTP, 2 U RNase inhibitor (New England Biolabs), 10 μCi [α32P]-UTP (3000 Ci/mmol), 8 pmol of SA11 gene 8 RV +RNA, ~2 pmol of VP1 and ~20 pmol of VP2 (Tortorici et al., 2003). After incubation for 3 h at 37°C, 32P-labeled gene 8 dsRNA products were resolved by electrophoresis in 12% polyacrylamide gels, detected by autoradiography, and quantified by phosphorimage analysis using a Typhoon 8600 Variable Mode Imager (GE Healthcare). At least three experiments were quantified for statistical analysis. One-sample t-tests of the mean were performed for each data set using Smith’s Statistical Package, version 2.80. P values of <0.01, in comparison to the set value of 100% for WT VP1, were considered statistically significant.
Salt sensitivity assays
Reaction mixtures (20 μl) containing 50 mM Tris-HCl, pH 7.1, 1.5% PEG 8000, 2.5 mM dithiothreitol, 20 mM magnesium chloride, 2 U RNase inhibitor, the indicated amount of GTP, pGpG or pGpGpU (Dharmacon), 8 pmol of SA11 gene 8 RV +RNA, ~2 pmol of VP1 and ~20 pmol of VP2 were incubated for 1 h at 37°C to promote initiation complex formation. NaCl was added to 250 mM to prevent further initiation. Then, 1.25 mM ATP, CTP, GTP, and UTP, and 10 μCi [α32P] UTP (3000 Ci/mmol) were added. Reaction mixtures were incubated for an additional 3 h at 37°C to permit elongation, prior to addition of SDS sample buffer. The 32P-labeled dsRNA products were analyzed identically to those of the dsRNA synthesis assays.
Images
Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081) and the aforementioned Protein Data Bank files (Lu et al., 2008; Petterson, 2004; Tao et al., 2002). Images showing 32P-labeled products of dsRNA synthesis assays or amounts of input VP1 protein were trimmed and processed for clarity using Adobe Photoshop CS5, version 12.0. Adjustments to levels and contrast were applied to entire images.
Made alignments and mutagenized rotavirus RNA polymerase (VP1) active site motifs
Aspartic acids thought to coordinate Mg2+ were critical for RNA synthesis
Motif F arginines that likely interact with NTPs were important for RNA synthesis
Individual priming loop mutations enhanced or diminished RNA synthesis
VP1 priming loop appears to be a dynamic regulatory element involved in initiation
Acknowledgments
The authors would like to thank Michelle Arnold, Natalie Leach, Marco Morelli, Aitor Navarro, and Shane Trask for helpful discussions and critical reading of the manuscript and Tamara Bar-Magen for technical assistance. We also thank Stephen Harrison and Ethan Settembre for sharing findings regarding the location and orientation of VP1 within rotavirus particles. This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases at the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Attoui H, Becnel J, Belaganahalli S, Bergoin M, Brussaard CP, Chappell JD, Ciarlet M, del Vas M, Dermody TS, Dormitzer PR, Duncan R, Fang Q, Graham R, Guglielmi KM, Harding RM, Hillman B, Makkay A, Marzachi C, Matthijnssens J, Mertens PPC, Milne RG, Mohd Jaafar F, Mori H, Noordeloos AA, Omura T, Patton JT, Rao S, Maan M, Stoltz D, Suzuki N, Upadhyaya NM, Wei C, Zhou H. Part II: The Viruses – The Double Stranded RNA Viruses - Family Reoviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus taxonomy: classification and nomenclature: Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press; San Diego: 2012. pp. 541–637. [Google Scholar]
- Bressanelli S, Tomei L, Rey FA, De Francesco R. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J Virol. 2002;76:3482–3492. doi: 10.1128/JVI.76.7.3482-3492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruenn JA. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res. 2003;31:1821–1829. doi: 10.1093/nar/gkg277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher SJ, Grimes JM, Makeyev EV, Bamford DH, Stuart DI. A mechanism for initiating RNA-dependent RNA polymerization. Nature. 2001;410:235–240. doi: 10.1038/35065653. [DOI] [PubMed] [Google Scholar]
- Chen D, Patton JT. De novo synthesis of minus strand RNA by the rotavirus RNA polymerase in a cell-free system involves a novel mechanism of initiation. RNA. 2000;6:1455–1467. doi: 10.1017/s1355838200001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Lambden PR, Lau J, Caul EO, Clarke IN. Human group C rotavirus: completion of the genome sequence and gene coding assignments of a non-cultivatable rotavirus. Virus Res. 2002;83:179–187. doi: 10.1016/s0168-1702(01)00442-7. [DOI] [PubMed] [Google Scholar]
- Estes MK, Kapikian AZ. Rotaviruses. In: Howley PM, editor. Fields Virology. 5. Lippincott, Williams and Wilkins; Philadelphia: 2007. pp. 1918–1974. [Google Scholar]
- Estrozi LF, Navaza J. Ab initio high-resolution single-particle 3D reconstructions: the symmetry adapted functions way. J Struct Biol. 2010;172:253–260. doi: 10.1016/j.jsb.2010.06.023. [DOI] [PubMed] [Google Scholar]
- Ferrer-Orta C, Arias A, Escarmis C, Verdaguer N. A comparison of viral RNA-dependent RNA polymerases. Curr Opin Struct Biol. 2006;16:27–34. doi: 10.1016/j.sbi.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Jiang S, Ji S, Tang Q, Cui X, Yang H, Kan B, Gao S. Molecular characterization of a novel adult diarrhoea rotavirus strain J19 isolated in China and its significance for the evolution and origin of group B rotaviruses. J Gen Virol. 2008;89:2622–2629. doi: 10.1099/vir.0.2008/001933-0. [DOI] [PubMed] [Google Scholar]
- Johne R, Otto P, Roth B, Lohren U, Belnap D, Reetz J, Trojnar E. Sequence analysis of the VP6-encoding genome segment of avian group F and G rotaviruses. Virology. 2011;412:384–391. doi: 10.1016/j.virol.2011.01.031. [DOI] [PubMed] [Google Scholar]
- Kao CC, Singh P, Ecker DJ. De novo initiation of viral RNA-dependent RNA synthesis. Virology. 2001;287:251–260. doi: 10.1006/viro.2001.1039. [DOI] [PubMed] [Google Scholar]
- Lawton JA, Zeng CQ, Mukherjee SK, Cohen J, Estes MK, Prasad BV. Three-dimensional structural analysis of recombinant rotavirus-like particles with intact and amino-terminal-deleted VP2: implications for the architecture of the VP2 capsid layer. J Virol. 1997;71:7353–7360. doi: 10.1128/jvi.71.10.7353-7360.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Baker ML, Jiang W, Estes MK, Prasad BV. Rotavirus architecture at subnanometer resolution. J Virol. 2009;83:1754–1766. doi: 10.1128/JVI.01855-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, McDonald SM, Tortorici MA, Tao YJ, Vasquez Del Carpio R, Nibert ML, Patton JT, Harrison SC. Mechanism for coordinated RNA packaging and genome replication by rotavirus polymerase VP1. Structure. 2008;16:1678–1688. doi: 10.1016/j.str.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain B, Settembre E, Temple BR, Bellamy AR, Harrison SC. X-ray crystal structure of the rotavirus inner capsid particle at 3.8 A resolution. J Mol Biol. 2010;397:587–599. doi: 10.1016/j.jmb.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SM, Aguayo D, Gonzalez-Nilo FD, Patton JT. Shared and group-specific features of the rotavirus RNA polymerase reveal potential determinants of gene reassortment restriction. J Virol. 2009a;83:6135–6148. doi: 10.1128/JVI.00409-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SM, Patton JT. Rotavirus VP2 core shell regions critical for viral polymerase activation. J Virol. 2011;85:3095–3105. doi: 10.1128/JVI.02360-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SM, Tao YJ, Patton JT. The ins and outs of four-tunneled Reoviridae RNA-dependent RNA polymerases. Curr Opin Struct Biol. 2009b;19:775–782. doi: 10.1016/j.sbi.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KK, Arnold JJ, Cameron CE. Structure-function relationships among RNA-dependent RNA polymerases. Curr Top Microbiol Immunol. 2008;320:137–156. doi: 10.1007/978-3-540-75157-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell D, Trowbridge R, Rowlands D, Jager J. Substrate complexes of hepatitis C virus RNA polymerase (HC-J4): structural evidence for nucleotide import and de-novo initiation. J Mol Biol. 2003;326:1025–1035. doi: 10.1016/s0022-2836(02)01439-0. [DOI] [PubMed] [Google Scholar]
- O’Reilly EK, Kao CC. Analysis of RNA-dependent RNA polymerase structure and function as guided by known polymerase structures and computer predictions of secondary structure. Virology. 1998;252:287–303. doi: 10.1006/viro.1998.9463. [DOI] [PubMed] [Google Scholar]
- Ogden KM, Johne R, Patton JT. Rotavirus RNA Polymerases Resolve into Two Phylogenetically Distinct Classes that Differ in Their Mechanism of Template Recognition Virology. doi: 10.1016/j.virol.2012.05.011. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KM, Ramanathan HN, Patton JT. Residues of the rotavirus RNA-dependent RNA polymerase template entry tunnel that mediate RNA recognition and genome replication. J Virol. 2011;85:1958–1969. doi: 10.1128/JVI.01689-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parashar UD, Burton A, Lanata C, Boschi-Pinto C, Shibuya K, Steele D, Birmingham M, Glass RI. Global mortality associated with rotavirus disease among children in 2004. J Infect Dis. 2009;200(Suppl 1):S9–S15. doi: 10.1086/605025. [DOI] [PubMed] [Google Scholar]
- Patton JT. Rotavirus VP1 alone specifically binds to the 3′ end of viral mRNA, but the interaction is not sufficient to initiate minus strand synthesis. J Virol. 1996;70:7940–7947. doi: 10.1128/jvi.70.11.7940-7947.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento JB, Crawford SE, Estes MK, Prasad BV. Rotavirus proteins: structure and assembly. Curr Top Microbiol Immunol. 2006;309:189–219. doi: 10.1007/3-540-30773-7_7. [DOI] [PubMed] [Google Scholar]
- Petterson EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-A Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Small C, Barro M, Brown TL, Patton JT. Genome heterogeneity of SA11 rotavirus due to reassortment with “O” agent. Virology. 2007;359:415–424. doi: 10.1016/j.virol.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz TA. A mechanism for all polymerases. Nature. 1998;391:231–232. doi: 10.1038/34542. [DOI] [PubMed] [Google Scholar]
- Tao Y, Farsetta DL, Nibert ML, Harrison SC. RNA synthesis in a cage--structural studies of reovirus polymerase lambda3. Cell. 2002;111:733–745. doi: 10.1016/s0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- Tortorici MA, Broering TJ, Nibert ML, Patton JT. Template recognition and formation of initiation complexes by the replicase of a segmented double-stranded RNA virus. J Biol Chem. 2003;278:32673–32682. doi: 10.1074/jbc.M305358200. [DOI] [PubMed] [Google Scholar]
- Trojnar E, Otto P, Roth B, Reetz J, Johne R. The genome segments of a group D rotavirus possess group A like conserved termini but encode group specific proteins. J Virol. 2010;84:10254–10265. doi: 10.1128/JVI.00332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk AA, Makeyev EV, Bamford DH. Initiation of viral RNA-dependent RNA polymerization. J Gen Virol. 2004;85:1077–1093. doi: 10.1099/vir.0.19731-0. [DOI] [PubMed] [Google Scholar]
- Yang W, Lee JY, Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]