

Abstract
Ongoing research has gradually recognized and understood the importance of adipose tissue (AT) angiogenesis as a key modulating factor of adipogenesis in the development of obesity. Previously, we carried out the first in vitro demonstration of the down-regulation of hypoxic angiogenesis during adipogenesis using cell-permeable chemical conjugates composed of antisense hypoxia-inducible factor 1α (HIF1α) oligonucleotide (ASO) and low-molecular weight protamine (LMWP). To further confirm the in vivo feasibility, we administered ASO-LMWP conjugates (AL) to diet-induced obese (DIO) mice by intraperitoneal injection (IP). Results showed that the AL conjugates significantly reduced the body weight, total fat tissue weight, and plasma lipid concentrations in the mice. Moreover, the AL conjugates not only decreased liver weight and hepatic triglyceride concentration but also significantly attenuated subcutaneous adipocyte cell size, which was conversely increased in the AL-untreated high-fat diet (HFD) group. Interestingly, more blood vessels were observed in the HFD group than in the lean group, indicating that blood vessel development could induce growth of the fat mass. This pattern was reversed in the AL-treated groups, which displayed a decrease in blood vessel density compared to the AL-untreated HFD group. This study presents the first in vivo evidence, in an obese mouse model, of the feasibility of achieving a biological treatment modality for obesity by blocking the angiogenic transcriptional factor HIF1α, thereby limiting angiogenesis, via the use of an adipose tissue-permeable ASO-LMWP.
Keywords: Antisense oligonucleotide, obesity, high fat diet, hypoxia inducible factor 1α, low molecular weight protamine, angiogenesis, adipogenesis
1. Introduction
Obesity is reaching epidemic proportions worldwide, and it is currently the most prevalent disease among children and young adults in the United States [1]. The World Health Organization identifies individuals with a body mass index (BMI) of 25 kg/m2 or greater as overweight, while those with a BMI of 30 kg/m2 or higher are further classified as obese. It is estimated that over 1.7 billion adults are overweight; more than 300 million are obese. Studies have shown that overweight individuals are more likely to die early, losing an average of 7 years of their lifespan [1]. Obesity is also closely associated with a variety of metabolic disorders, including diabetes, and cardiovascular diseases such as atherosclerosis, hyperlipidemia, and hypertension [2]. This disease burden results in a devastating loss of lives as well as tremendous healthcare costs.
Obesity treatments have thus far focused mainly on lifestyle modifications such as reduction of food intake by influencing the central nervous system (satiety center), or increasing physical activity. While lifestyle change is the ideal solution, this approach has failed to resolve this growing epidemic to date [2]. One of the factors for this failure is the silent nature of this disease, with its insidious effects becoming apparent only when it gives rise to other, more visible health-related problems [1]. The long-term results of these efforts have hitherto been disappointing.
Pharmaco-therapeutic management for obesity is currently limited to 2 drugs: orlistat and sibutramine [3-4]. These therapies, however, are unable to induce substantial weight loss, and are effective only when used in combination with other drugs or strict weight management programs. Additionally, orlistat and sibutramine can cause significant side effects such as gastrointestinal distress and increase of blood pressure and heart rate [3]. Surgical intervention (e.g., gastric bypass), an option that has recently attracted greater attention in the US, is able to produce weight loss of 25 – 30%, but is associated with several contraindications, including schizophrenia, depression, and eating disorders, and requires long-term monitoring of patients after surgery [1]. Overall, current practices are unable to manage this growing epidemic, and there is great urgency and need for a novel, safe, and effective therapy.
Obesity, the expansion and growth of white adipose tissue (WAT) caused by hyperplasia and hypertrophy of adipocytes [5], is dependent on the neovascularization and dilation of existing capillaries, respectively [5-7]. Hence, hypertropic adipocytes are typically found to possess low-oxygen microenvironments—hypoxia [8]. Similar to tumor growth, the inhibition of adipose tissue angiogenesis inhibits WAT growth and ultimately, the development of obesity. Indeed, endogenous angiogenesis inhibitors such as angiostatin [9] and endostatin [10] have been reported to reduce the body weight of obese mice [11]. In addition, it has been demonstrated that angiogenesis inhibitors such as TNP-470 and vascular endothelial growth factor receptor 2 (VEGFr2)-specific inhibitors prevent the development of obesity in genetic mouse models raised on high-fat diets (HFD) [12]. It thus appears that specific treatment of adipose tissues is critical to achieving control of obesity. Therefore, pharmacological manipulation of adipose tissue angiogenesis potentially offers a novel therapeutic option for effective treatment of obesity and related metabolic disorders [12].
To inhibit angiogenesis, we propose the use of an RNA-interfering agent, antisense oligonucleotides (ASO), for the biological treatment of obesity. ASO are novel therapeutics designed to bind to a target messenger RNA (mRNA), resulting in degradation of the mRNA and thereby decreasing the production of specific proteins associated with disease progression [13]. Since the introduction of nucleic acid-based therapeutics in the late 1970s, a wide variety of ASO have been developed that display target gene silencing efficacy [13]. The major barrier to achieving effective ASO therapy lies in the poor intracellular delivery efficacy and stability of ASO drugs [14]. Recently, RNA interfering agents (RNAi), siRNA and shRNA, have been introduced as a new class of agents for targeted gene therapy [15]. While RNAi technology continues to advance, ASO nevertheless offer several advantages over RNAi, including 1) flexibility in chemistry and target site design; 2) lower synthesis costs; 3) longer circulation half-life; and 4) relative ease of delivery to the target cells [16]. Furthermore, ASO produced by current state-of-the-art technologies are far more resistant to degradation by nucleases than current siRNA compounds [14]. Indeed, several ASO compounds have been shown to possess improved pharmacokinetic properties and significant activity towards the expression of targeted genes as well as disease progression in preclinical rodent studies and preliminary clinical trials [17-18].
Several delivery methods have been developed to enhance intracellular uptake of ASO, such as the use of viral vectors [19], cationic liposomes [20], polymeric micelles [21], peptides [22], and cationic polymers [23]. Although viral vectors (e.g., adenoviruses) have demonstrated great promise in achieving effective intracellular delivery of gene compounds, the associated adverse effects of cytotoxicity, immunogenicity, and mutagenesis have raised serious safety concerns [24]. Conversely, the use of relatively non-toxic cell-penetrating peptides (CPPs) as drug carriers have displayed several distinctive advantages over other delivery methods without altering the biological properties of the ASO [25]. These benefits are: 1) rapid and efficient intracellular delivery of an ASO with a significant antisense effect; 2) improving ASO stability, resulting in a reduced dose of ASO required for therapeutic efficacy; and 3) reducing ASO-induced side effects [25-26]. In a previous investigation, we reported the development of a cell-permeable ASO-low molecular weight protamine (LMWP) chemical conjugate (AL) by linking hypoxia-inducible factor 1α (HIF1α) (a key regulating transcription factor under hypoxia) ASO with LMWP [27], a nontoxic CPP developed in our laboratory [28]. In vitro cell culture studies demonstrated a significantly enhanced intracellular localization of the AL conjugates, which consequently induced marked down-regulation of adipogenic and angiogenic gene expressions, thereby blocking the angiogenesis during adipogenesis of 3T3-L1 cells under hypoxia as well as reducing fat accumulation in these cells [27].
In the current investigation, we further confirmed the feasibility of this novel AL therapy in vivo by using a clinically relevant diet-induced obese (DIO) mouse model. The findings from these animal studies further confirmed our hypothesis that blocking HIF1α expression and subsequently reducing angiogenesis and adipogenesis in adipocytes as well as fat mass could potentially be a novel and effective means of clinical treatment of obesity.
2. Materials and Methods
2.1.Materials
Salmon protamine and thermolysin were purchased from Sigma (St. Louis, MO). Succinimidyl 3-(2-pyridyldithio)propionate (SPDP) and dithiothreitol (DTT) were purchased from Pierce Biotechnology, Inc. (Rockford, IL). Fetal bovine serum (FBS), phosphate-buffered saline (PBS), and 0.25% (w/v) trypsin-EDTA were purchased from Gibco-BRL (Invitrogen, Carlsbad, CA). Human umbilical vein endothelial cells (HUVEC), F-12K media, heparin, and endothelial cell growth factor (ECGS) were purchased from American Type Culture Collection (ATCC) (Manassas, VA). All other chemicals used were reagent-grade commercial products. Phosphorothioate (PS)-modified antisense HIF1α oligonucleotide (ASO) and mismatch HIF1α oligonucleotide (MMO) were synthesized by IDT Corporation (San Diego, CA). For further conjugation with LMWP, these oligonucleotides were phosphorylated at the 5′-end. An individual sequence of these oligonucleotides was specifically designed to consist of 5′-ACA ACG CGG GCA CCG ATT CGC CAT G-3′ for ASO and 5′-GTG ATC CCC TGC TCT TGC CGT-3′ for MMO.
2.2.Preparation of LMWP
LMWP was derived by the enzymatic digestion of protamine according to a previously described protocol [29]. In brief, protamine was hydrolyzed with thermolysin at room temperature for 1 h, followed by the addition of 50 mM EDTA to quench the reaction. The product was further purified using a heparin affinity column.
2.3.Chemical conjugation of ASO with LMWP
To create a reactive sulfhydryl group at the 5′-phosphated end of the oligonucleotides, both ASO and MMO were reacted with 0.25 M EDC solution. Following the addition of 20 μL of 0.1 M imidazole (pH 6.0) and cystamine, the reaction mixture was incubated overnight at 50°C. Unreacted EDC, imidazole, cystamine, and the reaction byproducts were removed by HPLC using a desalting column eluted with 20 mM sodium phosphate buffer at pH 7.4. Prior to conjugation, LMWP was activated using SPDP according to a previously established protocol [29]. Activated ASO-SH and LMWP-SPDP were then reacted in PBS buffer (pH 7.4) overnight at room temperature to produce a 1:1 (molar ratio) AL conjugate. The chemical conjugates of MMO-LMWP linked with a disulfide bond (ML) were prepared in a similar manner. All conjugates were then mixed with excess LMWP (1:10 molar ratio) to form a protective, ionically stabilized complex around the ASO [30].
2.4.Cell culture and tube formation assay
A spontaneous tube formation assay on a basement membrane matrix, Matrigel, was conducted to examine whether AL exhibited anti-angiogenic potential on HUVEC.
HUVEC cell line was purchased from ATCC and maintained in F-12K medium supplemented with 10% FBS and ECGF in 5% CO2 – 95% O2. Liquid Matrigel solution was used to coat a 24-well plate (500 μg/well) and then allowed to polymerize at 37°C for 30 min. HUVEC was seeded on the Matrigel-coated plates at a density of 2 × 106 cells/well and grown for 18 h. Upon reaching 90% confluence, cells were treated at 37°C for 18 h with the test components; 1) ASO alone, 2) MMO alone, 3) ASO+LMWP mixture (ASO+L), 4) ASO-Lipofectamine complex (ASO-Lipo), 5) AL conjugates, and 6) ML conjugates). The concentration of each component was 1.5 μg/μL ASO for the AL which, that was determined to be an optimum concentration not associated with toxic effects in our previous report [29]. The extent of tube formation was analyzed by tube length, with 4 random fields observed from each well, and the length of tube-like structures was determined with the MetaMorph software, (version 7.6) image analysis program (Molecular Devices, LLC, Sunnyvale, CA). Three independent sets of experiments were conducted for statistical analysis.
2.5.Development of the DIO mice model and AL treatment (n = 5 per group)
Twenty 12-week-old male C57BL/6J obese mice fed HFD, which contains 60% kcal from fat, were purchased from the Jackson Laboratory (Bar Harbor, ME). Five mice were fed a standard chow diet, while the remaining 15 DIO mice were maintained on HFD. Mice were randomly assigned to 4 groups: 1) control (C); 2) DIO mice injected with PBS (H); 3) H+AL1-treated (0.5 mM of AL conjugates, 3.9 mg/kg mouse); and 4) H+AL2-treated (2.0 mM of AL conjugates, 14.7 mg/kg mouse). The concentrations of AL1 and AL2 were determined by a pilot study using concentrations of AL ranging 0.1 – 2.5 mM; a range determined from other published reports [16, 31-33]. Results from a subgroup of mice injected with non-specific MMO were indistinguishable from the PBS injection group. All injections were administered intraperitoneally (IP) twice weekly for 5 weeks. Body weight was monitored weekly. At the end of the experiment, the mice were euthanized, and the liver and adipose tissues (subcutaneous and retroperitoneal) isolated for analysis. All animal experiment protocols were in accordance with guidelines from the University Committee on Use and Care of Animals (UCUCA) of the University of Michigan animal facility (Approval No. 10130).
2.6.Blood collection and analysis of plasma lipids, alanine transaminase (ALT), and aspartate transaminase (AST)
We collected 200 μL of whole blood from mouse tail vein of mice at the start and end of the experiment using insulin syringes, and were then centrifuged at 3,000 ×g for 15 min at 4°C. All plasma samples obtained after centrifugation were stored at −70°C prior to analysis. Total cholesterol (TC), triglyceride (TG), and glucose concentrations were determined using a commercial diagnostic kit from Sigma. The activities of plasma ALT and AST were measured by the Reitman and Frankel method [34].
2.7Analysis of liver TG concentration
Total liver lipids were extracted from livers by the Folch method [35] and TG concentration was determined colorimetrically using the TG analysis kit mentioned in Section 2.6. The liver TG concentration was expressed as milligrams of TG/g of liver weight.
2.8.Adipose tissue weight and fat cell size and area
Weighed adipose tissues were quickly frozen with dry ice, and then used for adipocyte staining. Frozen subcutaneous adipose tissue was thinly sliced serially at a thickness of 20 μm with a freezing microtome (Cryostat, Leica Microsystems, Nussloch GmbH). Samples were then stained with Mayer's hematoxylin and the fat cell diameters were measured using a light microscope equipped with MetaMorph image analysis software.
2.9.Statistical analyses
Statistical analyses of experimental results were performed using SPSS (Version 11.0) and all measurements are expressed as mean ± S.D. Statistically significant differences among groups were determined using one-way ANOVA and Tukey's analysis as a post-hoc test. The level of significance was set at p < 0.05.
3. Results
3.1. AL-mediated inhibition of angiogenic tube formation
The inhibitory effect of AL conjugates on vascular structures was assessed in vitro using the Matrigel tube formation assay. Both the ASO and MMO-treated groups developed completely enclosed capillary-like tube formations after HUVEC were cultured with native oligonucleotides for 18 h (Fig. 1A). Based on the tube length analysis by MetaMorph, the addition of a physical mixture of ASO and LMWP (ASO+L) exhibited no statistically distinguishable tube forming potential compared to the ASO control group. In sharp contrast, the AL-treated group displayed a remarkable decrease in tube formation (Fig. 1A), and the tube forming potential was estimated to be approximately 39% of that of the ASO control group (Fig. 1B), indicating a significant attenuation of angiogenesis. This reduction was also greater than that of the ASO-Lipo positive control group, in which ASO was delivered via commercial lipofectamine and which exhibited a reduction in tube formation of approximately 67% of the ASO control group (p < 0.05).
Fig. 1.
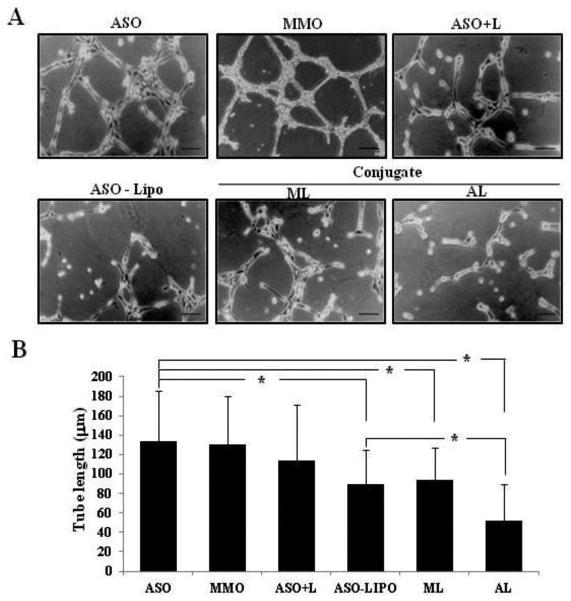
Inhibitory effects of AL conjugates on tube formation of HUVEC. (A) Representative photos (×100, inverted phase contrast microscope) showing that the HUVEC formed a capillary-like tube structure on the Matrigel layer. (B) Tube length quantified by MetaMorph software. (ASO: naked antisense HIF1α oligonucleotide, MMO: mismatch HIF1α oligonucleotide, ASO+L: physical mixture of ASO and LMWP, ASO-Lipo: physical mixture of ASO and lipofectamine, ML: chemical conjugates of MMO-LMWP linked with a disulfide bond, AL: chemical conjugates of ASO-LMWP linked with a disulfide bond) Scale bar = 50 μm. Data are presented as mean ± S.D. from 4 different regions randomly chosen from each group from 3 independent sets of experiments (n = 3). * indicates data were statistically different at p < 0.05.
3.2. Change in body weight and fat mass
DIO mice were fed HFD for 7 weeks to maintain their obese status. The obese mice, with an average body weight of 30.05 ± 1.89 g, were assigned in equal numbers to 4 groups (n = 5 per group): 1) Control (C-mice), whose diet was switched from HFD to a normal diet (Fig. 2A: a and e); 2) H-mice injected with PBS (Fig. 2A: b and f); 3) H+AL1-mice injected with low-dose (0.5 mM) AL conjugates (Fig. 2A: c and g); and 4) H+AL2-mice injected with high-dose (2.0 mM) AL conjugates (Fig. 2A: d and h). Except for the C-mice, all other animal groups maintained HFD. The body weight in the control mice (Fig. 2B: C-group) increased from 29.77 ± 2.12 g to 32.87 ± 3.12 g over the 5-week test period, while the average body weight of the obese mice maintained on HFD (i.e., H-mice) increased dramatically to 41.86 ± 3.58 g at the experimental endpoint. As can be observed in the figures, obese mice treated with AL conjugates for 5 weeks displayed a significant, dose-dependent reduction of intra-abdominal fat (Fig. 2A: c, d, g, and h) and body weight (Fig. 2B). At a higher AL treatment dose, the body weights of the mice were markedly reduced (p < 0.05) to below those of the PBS-treated mice (Fig. 2B: H+AL2-group vs. H-group), eventually approaching the level of the control group fed the normal diet (Fig. 2B: H+AL2-group vs. C-group). To a lesser extent, mice treated with the lower AL dose also exhibited a significantly decreased mean body weight gain of 38.80 ± 3.11 g compared to that of the control (Fig. 2B: H+AL1-group, p < 0.05).
Fig. 2.
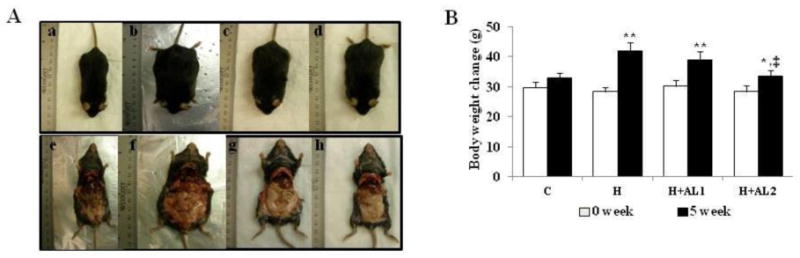
Effect of AL treatments on body weight and adiposity of mice. (A) Photographs of whole body images of experimental mice groups. a and e) Control mice (C-mice) that were switched from HFD to a normal diet during the experimental period; b and f) obese mice (H-mice) fed HFD and intraperitoneally injected with PBS buffer; c and g) obese mice fed with HFD and injected with low-dose (3.9 mg/kg) AL (H+AL1-mice); and d and h) mice fed with HFD and injected with high-dose (14.7 mg/kg) AL (H+AL2-mice). (B) Body weight changes of mice in all experimental groups at the beginning (0 week) and ending (5 week) of the experiment. Unless otherwise stated, the same abbreviations (C, H, H+AL1, and H+AL2) were used here and for Figures 3 – 5. Data are presented as mean ± S.D. (n = 5). * and **indicate that the differences among the control (C-mice) and experimental mice (H, H+AL1, or H+AL2 mice) were statistically significant at p < 0.05 (*) or p < 0.01 (**). ‡ denotes the significant differences among H and AL treated groups (i.e. H vs. H+AL1 or H+AL2 mice group, p < 0.01).
3.3. Morphological changes in adipose tissues
Treating the obese mice (i.e., the H-group) with either a low (H+AL1) or high (H+AL2) dose of AL resulted in a remarkable, dose-dependent reduction in subcutaneous (SubQ) and retroperitoneal (Retro) adipose tissue fat mass as well as total fat tissue mass (SUM [FAT]) (Fig. 3A).
Fig. 3.
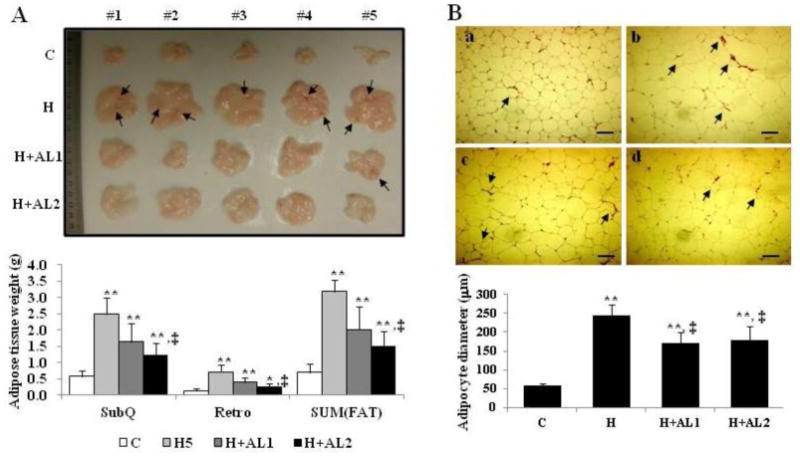
Images of the isolated subcutaneous adipose tissue (A) and adipocytes (B) of mice. (A) Isolated whole subcutaneous adipose tissues (top) and their measured weights (bottom) were analyzed. (B) Cryosectioned adipocytes (top) and their diameters (bottom) were measured. Data are presented as mean ± S.D. (n = 5). Scale bar = 200 μm. * and **indicate that the differences among the control (C-mice) and experimental mice (H, H+AL1, or H+AL2 mice) were statistically significant at p < 0.05 (*) or p < 0.01 (**). ‡ denotes the significant differences among H and AL treated groups (i.e. H vs. H+AL1 or H+AL2 mice group, p < 0.01).
In addition, while a high degree of vascularization was observed in the subcutaneous adipose tissues from the HFD-fed H-mice (Fig. 3B: b, arrows), this increased vascular growth was clearly seen to be retarded in subcutaneous adipose tissues obtained from both the low (Fig. 3B: c) and high-dose groups (Fig. 3B: d) of AL-treated mice. Indeed, the images showed that there was no significant difference in vascularization between the AL-treated mice and the control mice fed a normal diet (Fig. 3B: c and d vs. a). Furthermore, while the adipocyte area in H-mice as measured by MetaMorph was approximately 4.2-fold greater than that of the control group (Fig. 3B: H vs. C), the AL1 and AL2-treated groups displayed a fat cell area reduction amounting to 71% and 73%, respectively, of the AL-untreated group (Fig. 3B: H+AL1 and H+AL2 vs. H).
3.4. Liver toxicity of AL conjugates
Protection against liver toxicity by AL in obese mice was illustrated in Fig. 4. As seen in the figure, mice maintained on the HFD (i.e., H-mice) without treatment displayed hepatic steatosis symptoms and significantly higher liver weights compared to the control (C-mice) (Fig. 4A), with average liver weights of the H and C groups being 1.57 ± 0.60 g and 1.14 + 0.78 g, respectively (p < 0.05) (Fig. 4B: H vs. C group). Treatment with AL was found to retard this increase in liver weight towards the basal level of value (Fig. 4A: H+AL1 and H+AL2 vs. C). As expected, chronic HFD feeding led to an approximate 2.12-fold increase in liver TG content (Fig. 4C: H vs. C). However, treating these mice with a low or high dose of the AL conjugate was able to bring down the liver TG content to approximately 76% and 64%, respectively, of that of the obese mice (Fig. 4C: H+AL1 and H+AL2 vs. H). In addition, AL treatment significantly reduced plasma transaminase concentrations such as the circulating AST and ALT values (Fig. 4D and 4E, respectively), which function as surrogates of liver toxicity. These findings indicated that the symptoms of liver steatosis were alleviated in obese mice treated with the AL conjugates, which conferred improved protection against liver toxicity.
Fig. 4.
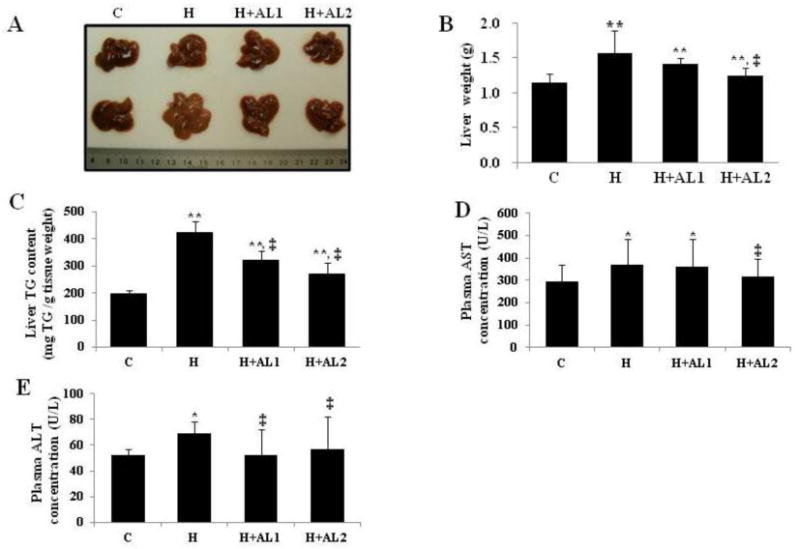
Effects of AL treatment on the liver weight, hepatic steatosis, and plasma ALT and AST concentrations. (A) Images of livers isolated from different groups, (B) Liver weights isolated from all experimental groups, (C) total liver TG concentration, (D) plasma AST concentrations, and (E) plasma ALT concentrations. Data are presented as mean ± S.D. (n = 5). * and **indicate that the differences among the control (C-mice) and experimental mice (H, H+AL1, or H+AL2 mice) were statistically significant at p < 0.05 (*) or p < 0.01 (**). ‡ denotes the significant differences among AL-untreated H group and AL treated groups (i.e. H vs. H+AL1 or H+AL2 mice group, p < 0.01).
3.5. Effects of AL on plasma lipids and glucose concentrations
HFD-induced hyperglycemia was evident by the presence of elevated TC and TG concentrations in the H-mice. While IP injection of the AL conjugates to the control mice did not provoke detectable alterations of plasma TC, TG, and glucose concentrations (Fig. 5: C), it nevertheless produced a significant diminution of the hyperlipidemic effect in the obese mice (Fig. 5: H). While the plasma TC and TG concentrations in the obese mice were elevated by approximately 2.0 and 1.6-fold over those of the control (Fig. 5A and 5B: H vs. C, respectively), these levels were trimmed by nearly 20% following AL treatment (Fig. 5A and 5B: H+AL1 and H+AL2 vs. H, respectively). Although a trend of decreasing plasma glucose concentrations in the obese mice by AL injection appeared to exist (Fig. 5C), such changes were not statistically significant.
Fig. 5.
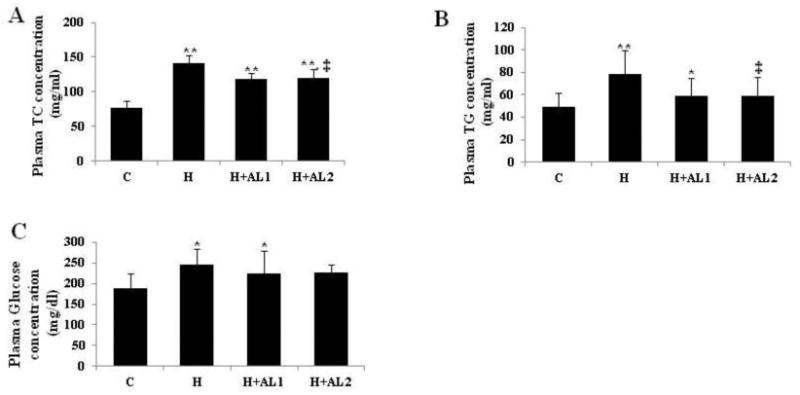
Effect of AL treatment on plasma lipid concentrations. (A) Plasma TC concentrations, (B) plasma TG concentrations, and (C) plasma glucose concentrations. Data are presented as mean ± S.D. (n = 5). * and ** denote the differences among the control (C-mice) and experimental mice (H, H+AL1, or H+AL2 mice) were statistically significant at p < 0.05 (*) or p < 0.01 (**).‡ denotes the significant differences among AL-untreated H group and AL treated groups (i.e. H vs. H+AL1 or H+AL2 mice group, p < 0.01).
4. Discussion
Angiogenesis under local hypoxia in adipose tissue has been known as a major factor that accelerates obesity by supplying nutrients and oxygen to the growing adipose tissue [36]. Since the growth of adipose tissue mass is angiogenesis-dependent, this process may be inhibited by angiogenesis inhibitors [11].
In our previous report, we successfully demonstrated the in vitro feasibility of this approach by down-regulating adipocyte differentiation via control of the expression of both leptin and VEGF genes by blocking HIF1α gene using an AL conjugates [27]. A chemically modified ASO with LMWP could improve the stability of the ASO itself, thereby significantly reducing the ASO dose needed for acquiring the highest efficacy and lowest cytotoxicity in vitro system.
Here, we further confirmed the in vivo capability of this ASO delivery system in regulating both adipogenesis and angiogenesis in a clinically relevant DIO mouse model for obesity treatment. An ionically stabilized complex was synthesized by the reaction of chemically conjugated AL with an excess amount of LMWP at a 1:10 molar ratio. Based on our previous report [27], it was effective for the formation of a protective complex network around the ASO. The molecular size of the ionically stabilized conjugates used was 189 ± 39 nm. The zeta potential of AS-L as measured by a Zeta analyzer was 25.23 ± 3.49 mV. The size and surface charge of a conjugated ASO can be considered sufficient for conjugates to penetrate into cells, thereby inducing the activities of ASO.
Our results indicated that treating obese mice with either low (3.9 mg/kg) or high doses (14.7 mg/kg) of the AL conjugates by IP injection not only resulted in a significant diminution of experimental mice body weights (Fig. 2), fat mass weights (Fig. 3), and plasma lipid concentrations (Fig. 5) but also demonstrated improvement against hepatic steatosis without inducing liver toxicity or other side effects (Fig. 4).
To the best of our knowledge, this is the first demonstration of the in vivo therapeutic use of chemically modified ASO for the treatment of obesity. It should be noted that the doses of the LMWP-linked ASO conjugates employed in this study were merely 3.9 and 14.7 mg/kg, or concentrations of 0.5 and 2.0 mM, respectively. These doses were significantly lower than the previously reported concentrations of non-chemically conjugated but physically mixed ASO/TGFβ1 (25 or 50 mg/kg) for regulating diabetic retinopathy [32], ASO/Foxo1 (50 mg/kg) for managing insulin resistance in DIO mice [31], and ASO/JNK1 (25 mg/kg) for treating obesity [40]. Interestingly, when used to treat obese mice, even considerably lower concentrations of cell-permeable AL conjugates yielded similar improvement of liver steatosis and plasma lipid profiles as those in other report using ASO/JNK1 mixtures for obesity treatment [40]. This phenomenon can be explained in terms of reduced hepatic lipogenesis [40] induced from decreased plasma lipids and glucose concentrations presumably caused by the AL treatment (Fig. 5).
Those adipogenic phenomena can be hypothesized by decreased total blood vessel distribution presented in Fig. 3B and inhibited tube formation effect by AL treatment (Fig. 1).
Consistent with our findings presented in Fig. 1, Vorgos and co-workers [41] also demonstrated that the total blood vessel area in subcutaneous or gonadal adipose tissue was positively correlated with fat pad mass suggesting the strategy of utilizing pro- and anti-angiogenic agents for the treatment of obesity. The increased hypoxia during adipose tissue development would trigger blood vessel formation through the up-regulation of several mRNAs such as that for type 1 plasminogen activator inhibitor (PAI-1), thrombospondin-1 (TSP)-1, and VEGF [41]. In line with this strategy, blocking agents the HIF1α, key master regulator of angiogenesis, can be utilized as a powerful target to inhibit both angiogenesis and adipogenesis, via the use of by a cell-permeable ASO.
Antisense therapy has demonstrated the potential to reduce the drug development time/cost and failure rate in clinical trials when compared to traditional drug therapies [16]. It interferes with the expression of disease-inducing proteins by binding with a specifically designed oligonucleotide, effecting selective intervention in the progression of the disease [16]. Since Vitravene (Fomivirsen), the first-generation FDA-approved antisense drug, was launched in the market in 1998, several ASO products were developed for clinical trials, but all failed to reach the anticipated outcome. The primary reason for this failure is believed to be poor cellular uptake of the drug, and toxicities [40].
To overcome this limitation, a number of carrier-based delivery systems, most noticeably using liposomal vesicles, have been developed to facilitate intracellular ASO delivery via the process of endocytosis [40]. Despite the improvement, this cell-entry method requires invagination and vasculation of the membrane lipid bilayer to form free cytoplasmic vesicles. Using a CPP as a carrier, we could reduce the dosage of injected ASO needed to treat obesity. Another report of ASO injected without a carrier for obesity had to use dosages that were 3.4–12.8-fold compared to that of the present study (50 mg/kg vs. 3.9 mg/kg or 14.7 mg/kg) to produce similar improvement of blood glucose and lipid concentrations in DIO mice [31]. From this result, we could confirm that using LMWP as a delivery carrier of ASO can have a beneficial effect on obesity therapy by reducing the dosages without cytotoxic effects (Fig. 4D and 4E).
The recent discovery of potent CPPs, such as TAT obtained from the HIV protein [44] and LMWP derived directly from native protamine by our laboratory [29], has shed light on the final resolution of the intracellular delivery dilemma. Both in vitro and in vivo studies showed that a CPP is able to ferry almost any attached species, including large proteins [29] or nano-carriers (e.g., liposomes) [45], across the cell membrane barrier of all organ types, including the brain. Although the mechanism of CPP-mediated cell internalization remains unclear, it is nevertheless being recognized to not follow, or at least only partially follow the standard endocytosis process [44]. Indeed, cellular translocation mediated by CPP is so potent that none of the existing endocytosis methods can match it. These observations have fostered the concept of the current work, i.e., linking a cell-impermeable drug with a CPP to achieve highly efficient intracellular ASO delivery.
Recently, several reports regarding the systemic delivery of siRNA for obesity and diabetes using different delivery methods were published [38-39]. Intraperitoneal and hydrodynamic injections of non-modified native siRNA itself have been reported as ineffective and/or inappropriate for in vivo gene targeting in DIO and ob/ob mice [38]. Meanwhile, adenovirus-mediated delivery of shRNA provided a relatively benign and effective method for target gene silencing, although results have varied widely [39].
In contrast, Samuel et al. also reported the effective delivery of non-modified Foxo1-ASO itself to DIO and insulin-resistant mice by IP injection, resulting in reduced hepatic TG concentration and improved insulin resistance [31]. We currently adopted a CPP as an ASO carrier; enabling the effective delivery of ASO to the target organ.
We can further hypothesize the delivery mechanism of AL in the adipose tissue of mice with IP injection as follows. First possibility of delivery mechanism is direct penetration of AL into the adipocyte. Because the AL conjugate is a nano-sized (189 ± 39 nm) structure, which is favorable size range (100 – 200 nm) for cellular drug delivery [46], it can be easily delivered to the adipocytes of adipose tissue. Second, as the adipose tissue does not have its own specific boundary that can separate it from other tissues, once the drug molecule is injected into the local area neighboring the adipose tissue, the injected drug can easily penetrate and be delivered by the carrier CPP. Third, once the AL is delivered to abdominal adipose tissue, where blood vessels have become highly developed as the adipose tissue builds up, the injected AL can travel in the bloodstream and be delivered to other adipose tissues by repetitive injection of AL. Finally, IP injection of chemically modified, ionically stabilized AL conjugates can be effectively delivered to abdominal adipose tissue thereby resulted in the HIF1α knockdown effect on highly vascularized adipose tissue in obese mice and the reduction of fat accumulation, adipogenesis. The detailed exact mechanism of AL delivery by IP injection still remains to be further elucidated.
Taken together, we could confirm that LMWP-mediated ASO delivery is a promising strategy for adipogenesis inhibition by silencing the related genes and preventing hypoxic angiogenesis, and has potential in obesity control in in vivo system.
5. Conclusions
Our findings are the first in vivo experimental evidence, in a clinically relevant mouse model, for the potential retardation of the progression of obesity via biological means. Although the overall prospects and clinical outcomes for treating obesity with this approach remain to be further explored, the current investigation nevertheless, at least, sheds light on the possibility of eradicating fat cells “locally” and inhibiting adipogenesis by direct injection or via other administrative routes, using AL conjugates to specifically target fat cells. In addition, the technology developed in this study could open up opportunities to treat a variety of other diseases via the application of a specifically designed cell-permeable ASO or siRNA. Hence, the impact of this investigation could be far-reaching and widespread.
Acknowledgments
This work was supported in part by NIH R01 Grant CA114612. This research was alsopartially sponsored by Grant R31-2008-000-10103-01 from the World Class University (WCU) project of the MEST and NRF of South Korea. Victor C. Yang is currently a Participating Faculty in the Department of Molecular Medicine and Biopharmaceutical Sciences, College of Medicine & College of Pharmacy, Seoul National University, South Korea.
Abbreviations
- HIF1α
Hypoxia Inducible Factor 1α
- ASO
Antisense-HIF1α-oligonucleotide
- AL
ASO-LMWP conjugates
- ALT
Alanine Transaminase
- AST
Aspartate Transaminase
- CPP
Cell Permeable Peptide
- DIO
Diet-Induced Obesity
- HFD
High Fat Diet
- MMO
mismatch-HIF1α-oligonucleotide
- ML
MMO-LMWP conjugates
- LMWP
Low Molecular Weight Protamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haslam DW, James WP. Obesity. Lancet. 2005;366(9492):1197–209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 2.Carr MC, Brunzell JD. Abdominal obesity and dyslipidemia in the metabolic syndrome: importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J Clin Endocrinol Metab. 2004;89(6):2601–7. doi: 10.1210/jc.2004-0432. [DOI] [PubMed] [Google Scholar]
- 3.Padwal R, et al. Long-term persistence with orlistat and sibutramine in a population-based cohort. Int J Obes (Lond) 2007;31(10):1567–70. doi: 10.1038/sj.ijo.0803631. [DOI] [PubMed] [Google Scholar]
- 4.Sari R, et al. Comparison of efficacy of sibutramine or orlistat versus their combination in obese women. Endocr Res. 2004;30(2):159–67. doi: 10.1081/erc-200027356. [DOI] [PubMed] [Google Scholar]
- 5.Brook CG. Relation between age of onset of obesity and size and number of adipose cells. British medical journal. 1972;2(5804):25–7. doi: 10.1136/bmj.2.5804.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat Rev Drug Discov. 9(2):107–15. doi: 10.1038/nrd3055. [DOI] [PubMed] [Google Scholar]
- 7.Lijnen HR. Angiogenesis and obesity. Cardiovascular research. 2008;78(2):286–293. doi: 10.1093/cvr/cvm007. [DOI] [PubMed] [Google Scholar]
- 8.Helmlinger G, et al. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3(2):177–82. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 9.O'Reilly MS, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79(2):315–28. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 10.O'Reilly MS, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 11.Rupnick M, et al. Adipose tissue mass can be regulated through the vasculature. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(16):10730–10735. doi: 10.1073/pnas.162349799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao R, et al. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(11):6390–6395. doi: 10.1073/pnas.101564798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hnik P, et al. Antisense oligonucleotide therapy in diabetic retinopathy. J Diabetes Sci Technol. 2009;3(4):924–30. doi: 10.1177/193229680900300440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu L, Anchordoquy T. Drug delivery trends in clinical trials and translational medicine: challenges and opportunities in the delivery of nucleic acid-based therapeutics. J Pharm Sci. 2011;100(1):38–52. doi: 10.1002/jps.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rayburn ER, Zhang R. Antisense, RNAi, and gene silencing strategies for therapy: mission possible or impossible? Drug Discov Today. 2008;13(11-12):513–21. doi: 10.1016/j.drudis.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu B, et al. Targeted delivery systems for oligonucleotide therapeutics. AAPS J. 2009;11(1):195–203. doi: 10.1208/s12248-009-9096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leonetti C, Zupi G. Targeting different signaling pathways with antisense oligonucleotides combination for cancer therapy. Curr Pharm Des. 2007;13(5):463–70. doi: 10.2174/138161207780162917. [DOI] [PubMed] [Google Scholar]
- 18.Rayburn ER, Wang H, Zhang R. Antisense-based cancer therapeutics: are we there yet? Expert Opin Emerg Drugs. 2006;11(2):337–52. doi: 10.1517/14728214.11.2.337. [DOI] [PubMed] [Google Scholar]
- 19.Gao JQ, et al. Effective tumor targeted gene transfer using PEGylated adenovirus vector via systemic administration. J Control Release. 2007;122(1):102–10. doi: 10.1016/j.jconrel.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 20.Ko YT, Bhattacharya R, Bickel U. Liposome encapsulated polyethylenimine/ODN polyplexes for brain targeting. J Control Release. 2009;133(3):230–7. doi: 10.1016/j.jconrel.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 21.Kim Y, et al. Polymersome delivery of siRNA and antisense oligonucleotides. J Control Release. 2009;134(2):132–40. doi: 10.1016/j.jconrel.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wadhwa MS, et al. Peptide-mediated gene delivery: influence of peptide structure on gene expression. Bioconjug Chem. 1997;8(1):81–8. doi: 10.1021/bc960079q. [DOI] [PubMed] [Google Scholar]
- 23.Dong L, et al. Targeting delivery oligonucleotide into macrophages by cationic polysaccharide from Bletilla striata successfully inhibited the expression of TNF-alpha. J Control Release. 2009;134(3):214–20. doi: 10.1016/j.jconrel.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 24.Miller DG, Adam MA, Miller AD. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol. 1990;10(8):4239–42. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Junghans M, Kreuter J, Zimmer A. Antisense delivery using protamine-oligonucleotide particles. Nucleic Acids Res. 2000;28(10):E45. doi: 10.1093/nar/28.10.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinauer N, et al. Intracellular tracking of protamine/antisense oligonucleotide nanoparticles and their inhibitory effect on HIV-1 transactivation. J Control Release. 2004;96(3):497–507. doi: 10.1016/j.jconrel.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 27.Park YS, et al. Specific down regulation of 3T3-L1 adipocyte differentiation by cell-permeable antisense HIF1alpha-oligonucleotide. J Control Release. 2010;144(1):82–90. doi: 10.1016/j.jconrel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park Y, et al. Nontoxic membrane translocation peptide from protamine, low molecular weight protamine (LMWP), for enhanced intracellular protein delivery: in vitro and in vivo study. The FASEB journal. 2005;19(11):1555–1557. doi: 10.1096/fj.04-2322fje. [DOI] [PubMed] [Google Scholar]
- 29.Huang Y, et al. Synthetic skin-permeable proteins enabling needleless immunization. Angew Chem Int Ed Engl. 2010;49(15):2724–7. doi: 10.1002/anie.200906153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lv W, et al. RNAi-mediated gene silencing of vascular endothelial growth factor inhibits growth of colorectal cancer. Cancer biotherapy & radiopharmaceuticals. 2007;22(6):841–852. doi: 10.1089/cbr.2007.0409. [DOI] [PubMed] [Google Scholar]
- 31.Samuel V, et al. Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes. 2006;55(7):2042–2050. doi: 10.2337/db05-0705. [DOI] [PubMed] [Google Scholar]
- 32.Younis HS, et al. Antisense inhibition of S6 kinase 1 produces improved glucose tolerance and is well tolerated for 4 weeks of treatment in rats. Pharmacology. 2011;87(1-2):11–23. doi: 10.1159/000322526. [DOI] [PubMed] [Google Scholar]
- 33.Yu X, et al. Reduction of JNK1 expression with antisense oligonucleotide improves adiposity in obese mice. American journal of physiology: endocrinology and metabolism. 2008;295(2):E436–E445. doi: 10.1152/ajpendo.00629.2007. [DOI] [PubMed] [Google Scholar]
- 34.Reitman S, Frankel S. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am J Clin Pathol. 1957;28(1):56–63. doi: 10.1093/ajcp/28.1.56. [DOI] [PubMed] [Google Scholar]
- 35.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497–509. [PubMed] [Google Scholar]
- 36.Hosogai N, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56(4):901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, et al. Adipose tissue-specific inhibition of hypoxia-inducible factor 1{alpha} induces obesity and glucose intolerance by impeding energy expenditure in mice. Journal of biological chemistry. 2010;285(43):32869–32877. doi: 10.1074/jbc.M110.135509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sorensen DR, Leirdal M, Sioud M. Gene silencing by systemic delivery of synthetic siRNAs in adult mice. J Mol Biol. 2003;327(4):761–6. doi: 10.1016/s0022-2836(03)00181-5. [DOI] [PubMed] [Google Scholar]
- 39.Wilcox DM, et al. Delivery of RNAi reagents in murine models of obesity and diabetes. J RNAi Gene Silencing. 2006;3(1):225–36. [PMC free article] [PubMed] [Google Scholar]
- 40.Alam MR, et al. Intracellular delivery of an anionic antisense oligonucleotide via receptor-mediated endocytosis. Nucleic Acids Res. 2008;36(8):2764–76. doi: 10.1093/nar/gkn115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voros G, et al. Modulation of angiogenesis during adipose tissue development in murine models of obesity. Endocrinology. 2005;146(10):4545–4554. doi: 10.1210/en.2005-0532. [DOI] [PubMed] [Google Scholar]
- 42.Pandya N, Dhalla N, Santani D. Angiogenesis--a new target for future therapy. Vascular pharmacology. 2006;44(5):265–274. doi: 10.1016/j.vph.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Nishimura S, et al. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes. 2007;56(6):1517–1526. doi: 10.2337/db06-1749. [DOI] [PubMed] [Google Scholar]
- 44.Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release. 2005;102(1):247–53. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 45.Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57(4):637–51. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 46.Midoux P, Monsigny M. Efficient gene transfer by histidylated polylysine/pDNA complexes. Bioconjug Chem. 1999;10(3):406–11. doi: 10.1021/bc9801070. [DOI] [PubMed] [Google Scholar]