

Abstract
Computational methods and experimental data are used to provide structural models for NaChBac, the homo-tetrameric voltage-gated sodium channel from the bacterium Bacillus halodurans, with a closed and partially open pore domain. Molecular dynamics (MD) simulations on membrane-bound homo-tetrameric NaChBac structures, each comprising six helical transmembrane segments (labeled S1 through S6), reveal that the shape of the lumen, which is defined by the bundle of four alpha-helical S6 segments, is modulated by hinge bending motions around the S6 glycine residues. Mutation of these glycine residues into proline and alanine affects, respectively, the structure and conformational flexibility of the S6 bundle. In the closed channel conformation, a cluster of stacked phenylalanine residues from the four S6 helices hinders diffusion of water molecules and Na+ ions. Activation of the voltage sensor domains causes destabilization of the aforementioned cluster of phenylalanines, leading to a more open structure. The conformational change involving the phenylalanine cluster promotes a kink in S6, suggesting that channel gating likely results from the combined action of hinge-bending motions of the S6 bundle and concerted reorientation of the aromatic phenylalanine side-chains.
1. Introduction
Voltage-gated sodium channels (VGSCs) are responsible for the rising phase of the action potential in excitable cells, most notably in neurons and cardiac myocytes, where they play a critical role in neuronal firing and cardiac rhythm. Importantly, mutations in VGSC subunits have been implicated in an array of diseases including: paralysis, myotonia, epilepsy, arrhythmia and chronic neuropathic pain [1]. Notably, VGSCs are targets for drug molecules, including local anesthetics, painkillers, anticonvulsants and antiarrythmics [2], which bind to sites in the VGSC pore domain [3–5]. Moreover, many of these drugs show use-dependence, which reflects channel conformational dependent access to the relevant binding sites 6–9]. Accordingly, elucidating the functional conformations of the channel pore domain will allow a deeper understanding of the roles of VGSCs in physiology and disease.
Activation and inactivation are key voltage-dependent processes by which channels control ion conduction during the rising phase of the action potential. VGSCs preferentially adopt a non-conductive resting (closed) conformation at hyperpolarized transmembrane (TM) potentials. In response to membrane depolarization, VGSCs switch to their active (open) conformation, exhibiting high Na+ ion conductance. Voltage-dependent activation is mediated by TM voltage sensor (VS) domains, each comprised of four helical segments (S1 to S4). The VS is mechanically coupled to the pore domain (S5 and S6) via a linker region (S4–S5 linker). During activation, the S4 helix undergoes a conformational transition resulting in a net displacement towards the extracellular side of the membrane. During this process, a set of conserved arginines in S4 is involved in a sequential breaking and formation of salt-bridges with the acidic residues of neighboring TM segments [10–17]. This conformational transition of the VS changes the position of the linker, which, in turn, induces a rearrangement of the S5 and S6 helical regions. Specifically, this process affects the structure of the helix bundle lining the pore and formed by four S6 helices from different subunits. Ultimately, the displacement of the linker results in the opening of the activation gate; a hydrophobic constriction region located at the crossing point of the four S6 segments [18–21]. Studies on NaChBac have postulated a mechanism, likely preserved in several VGSCs 22,23], in which this opening of the activation gate requires the S6 helix to feature a kink in correspondence to a conserved glycine [24,25]. As a probable ancestor of eukaryotic Na+ and Ca2+ channels 26], NaChBac shares features of activation and gating with the mammalian counterparts. Interestingly, NaChBac also shows sensitivity to mammalian VGSC inhibitors such as the local anesthetic etidocaine and the general anesthetic isoflurane 25,27].
Experimentally, activation and gating of VGSCs has received much attention [28]. However, thus far structural investigations have been limited by the size of the mammalian Na+ channel pore-forming alpha subunit, which consists of twenty-four TM helices forming a pseudo-tetrameric structure. The discovery of homo-tetrameric bacterial VGSCs, each with just six TM helices [24,29,30] has provided an avenue for investigating VGSC gating behavior and structural characterization based on homology to known crystal structures of other tetrameric channels [31–33].
Herein we report a series of molecular dynamics (MD) studies on atomistic structural models of membrane-bound NaChBac based on the NavAb crystal structure. Our main focus of investigation is the characterization of structural properties of the TM S6-segment in different conformations of the channel. Accordingly, we present results on three channel conformations differing by the degree of openness at the activation gate and by the position of the VS-S4 helix.
Anticipating our results, our modeling reveals hinge bending motions around two S6-glycine residues of the channel are the key to modulating the shape of the main channel lumen. Specifically, a kink in S6 at G219 is related to pore opening; and another kink in S6, at G229, seems to favor the closure of the gate. In particular, a cluster of stacked phenylalanine residues in the crossing bundle of S6 helices, which hinder diffusion of water molecules and Na+ ions in the closed pore domain experimental VGSC structure, are critical for the gating process, working together as a major hydrophobic gate on the ionic permeation pathway. Overall, the present MD study identifies the S6-glycines as the crucial molecular hinge points, accounting for the bending motions of the S6 segment, which are associated with VS activation leading to channel opening. We thus hypothesize that these S6-glycines are key players in the mechanism of pore opening and closing during the voltage-gating process.
2. Methods
2.1 Modeling the closed conformation
A structural model for NaChBac in a closed conformation was built starting from the X-ray crystal structure of NavAb (PDB ID: 3RVY), which was crystallized in a closed-pore conformation with the four VS domains partially activated. The VGSCs NaChBac and NavAb are closely related, sharing significant levels of sequence identity (~37%) (Fig. S1). Ten residues at the N-terminus of NaChBac are not present in NavAb and were not included in the model. Five additional residues at the C-terminal region of NaChBac, predicted to be in an alpha-helical configuration by PSIPRED [34] and not resolved in the x-ray structure of NavAb, were added to the structure. We used MODELLERv9 [35] to generate the NaChBac structural homology model.
After energy minimization, the model for NaChBac in a closed conformation was embedded in a fully hydrated lipid bilayer for subsequent equilibration by MD simulation. The homo tetrameric channel was inserted at the center of a 1-palmitoyl–2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid patch. To generate the initial configuration, a set of conserved aromatic side-chains was used as a guide to delimit the TM region of the bilayer. The macromolecular system contained a total of ~ 162,000 atoms, including NaChBac, 434 lipid molecules, 28173 water molecules and 232 ions in solution. Two Na+ ions were initially placed in the channel SF, at sites HFS and IN (Fig. 1), in agreement with a previous computational study of NavAb showing double occupancy of the filter by Na+ ions [36]. All charged amino acids were simulated in their fully ionized state (pH = 7.0).
Figure 1.

Structure of the selectivity filter with two solvated Na+ ions: (A) upper view from the outer vestibule; (B) side-view. Main chain groups from residues Thr189, Leu190 and Glu191 and the side-chain of Glu191 plus water molecules are shown in stick representation, while Na+ ions are rendered as yellow spheres.
The membrane-bound channel system was relaxed following two consecutive steps: (i) membrane equilibration for ~10.0 ns with the channel backbone coordinates restrained around their starting structure with a harmonic force constant of 1 kcal/mol/Å2. This procedure ensured a uniform and tight distribution of lipid molecules around the protein without perturbing the initial conformation; and (ii) 0.3 μs relaxation of the full channel system by means of unconstrained MD simulation. Starting from the relaxed structure of the wild type (WT), additional unconstrained MD simulations were performed on two mutants, G219P and G219A. For each mutant a trajectory of 0.15 μs was collected for subsequent analysis.
2.2 Modeling the early-activated-open conformation
Starting from the closed structure of NaChBac, we generated the pre-open conformation of the channel by means of a steered-MD protocol, which mimics the iris-like dilation mechanism recently proposed for pore opening of the channel [37]. In our protocol, the S6-helical bundle was splayed apart by applying time-dependent harmonic potentials (force constant of 5 kcal/mol/Å2) along 16 inter-subunit distances, defined between the C-alpha atoms of the four pore-lining S6 residues F224, I228, I231, R234. The time-dependent restraints were applied over a 40 ns MD run until the inter-subunit distances approximated the corresponding ones in the open structure of Kv1.2/2.1. After these restraints were satisfied, solvation of the channel lumen and free diffusion of Na+ ions across the activation gate were observed. We then launched a restraint-free 0.300 μs MD trajectory for the membrane-bound channel, which allowed extensive assessment of the structural properties of the channel in a “pre-open” conformation.
2.3 Modeling the activated-partially-open conformation
Starting from the structure of the closed conformation of NaChBac, a model of the activated conformation was built via a steered-MD approach. Harmonic restraints were applied to the center of mass of the voltage-sensing S4 helix and the equilibrium position was shifted over time so as to induce a 6-Å rigid-body motion of the helix over an MD trajectory spanning 70 ns. The choice of a 6-Å displacement is based on an a priori inspection of the VS domain of Kv1.2–2.1. To preserve the alpha-helical conformation and prevent unfolding of the S4 helix, additional restraints were applied to all the main-chain H-bonds within the helical segment, which spans from residues 110 to 126. The total displacement of S4 is the minimal distance allowing interaction between R4 and D60, a salt-bridge shown to be present in the fully activated state [37]. To explore the effect of a depolarizing potential on this partially activated conformation, an additional 0.270 μs MD trajectory was sampled with an imposed TM potential of 1V, which was applied through the addition of an external uniform electrostatic field.
2.4 Molecular dynamics simulations
All MD simulations used the CHARMM22-CMAP force field with torsional cross-terms for the protein and CHARMM27 for the phospholipids [38,39]. A united-atom representation was adopted for the acyl chains of the POPC lipid molecules [40]. The water molecules were described using the TIP3P model [41,42]. Periodic boundary conditions were employed for all of the MD simulations and the electrostatic potential was evaluated using the particle-mesh Ewald method [43]. The lengths of all bonds containing hydrogen were constrained with the SHAKE algorithm [44]. The system was maintained at a temperature of 300 K and pressure of 1 atm using the Langevin thermostat and barostat methods as implemented in the MD code NAMD2.7 [45], which was used for the simulations with a 2.0 fs time step.
2.5 Free-energy calculations
The free-energy profile or potential of mean force (PMF) of ion conduction was calculated using the adaptive-biasing-force (ABF) method implemented in NAMD [42,46]. Here, we followed a similar strategy previously described to compute the free energy associated with the ion conduction through voltage-gated potassium channels [21]. In brief, the reaction coordinate for conduction through the activation gate was defined as the distance between the ion and the geometric center of the TLESW motif at the selectivity filter. For the early-activated-open conformation, the translocation of the ion follows a path of length 15 Å, ranging from the cytoplasmic entrance of the pore to the entrance of the central cavity, located immediately above the residue F224. The free-energy profile was calculated by means of independent MD runs, each exploring a 5-Å section of the reaction coordinate.
3. Results and Discussion
Three models of NaChBac were built on the basis of the recently determined x-ray crystal structure of the closely related NavAb channel [47] presenting the VS in different stages of activation [12–17], each exhibiting a well-defined electrostatic network between basic amino acids of the helical S4-segment and the acidic binding sites within the domain (Fig. 2). Of note are two salt-bridges (R2-E43 and R3-D60) and the 310-helix conformation of the S4 segment encompassing R3 and R4, which have been shown to be the structural determinants of at least one of the activated conformations of the VSD [37]. Remarkably, the cavities, called fenestrations, which connect the pore lumen to the hydrophobic region of the lipid bilayer are present in all the conformations explored along the MD trajectories and do not experience significant structural rearrangements. In contrast, the main-pore regions differ considerably in the degree of openness of the ionic-conduction pathway with the pore ranging from a closed to an open conformation.
Figure 2.
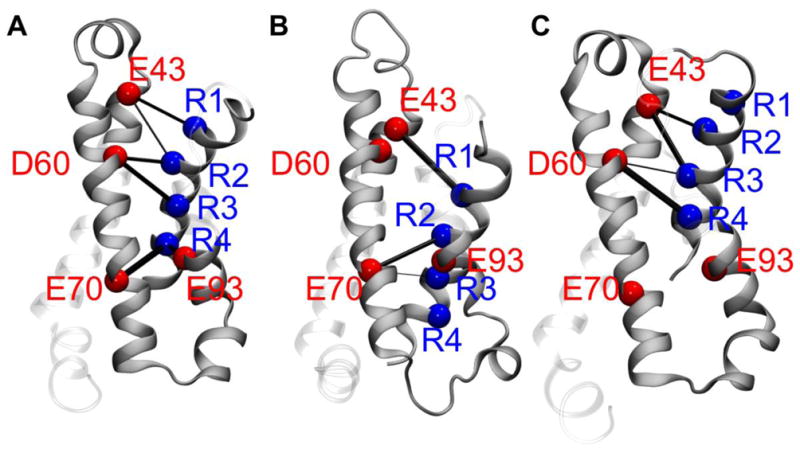
Shown are the patterns of salt-bridges in the voltage-sensing domain (VSD) of NaChBac for selected conformations of the channel: (A) closed; (B) early-activated-open; and (C) activated-partially-open (see text). The C-alpha atoms of the arginines and the acidic residues of the VSD are shown as blue and red spheres, respectively. The thickness of the black cylinders reflects the relative occurrence of each salt-bridge along the MD simulation trajectory.
For all models, the root mean square deviation (RMSD) values for the whole channel as well as for the VS and pore domains range from 1.5 to 4 Å (Fig. 3), which is typical of the structural drift quantified in previous MD simulation studies of ion channels [48,49]. The activated-partially-open conformation has the largest RMSD due to the relaxation of VS on removal of the constraints. To further evaluate our model we compared interatomic distances for 9 specific residue pairs in the final membrane-equilibrated structure of the early-activated-open conformation (Table S1) to distances estimated from luminescence resonance energy transfer (LRET) measurements for these pairs [31]. We found variations of less than 5.4 Å, a result consistent with previously published models of NaChBac [26,33].
Figure 3.

Root mean square deviation (RMSD) of the backbone atoms from the initial structure employed in the MD simulation plotted as a function of time for: (A) closed; (B) early-activated-open; and (C) activated-partially-open conformations, respectively (see text). The RMSD is shown separately for different regions of the channel: entire channel (black), voltage-sensing domains (blue), pore domain (red), and selectivity filter (green).
The major structural differences between the conformations of the pore domain are found in the pore-lining S6 helices. In the closed conformation, below the SF, the channel pore features a large hydrated central cavity, disconnected from the intracellular aqueous environment by a cluster of S6-phenylalanine residues, F224 and F227, located at the activation gate region (Fig. 4A–B). These phenylalanine side chains project into the channel lumen, producing inter-subunit stacking interactions, thereby occluding the pore. In contrast, in the early-activated-open conformation, the F224 side chains are buried in the inter-subunit helical interface rather than being stacked in the lumen, and pairs of F227 side chains interact in an “edge-face” conformation (Fig. 4A–B). The latter arrangement of the phenylalanine cluster allows hydration of the activation gate, a key structural modification required for ionic transportation [21]. The free-energy profile associated with conduction of Na+ through the activation gate of the early-activated-open conformation shows an energy barrier of ~ 15 kcal/mol (Fig. S2), indicating that, although the permeant Na+ ion retains an almost intact shell of solvation, the structure is not fully open for diffusive transport. However, this barrier is significantly smaller than the energy cost (~ 50 kcal/mol) estimated for ion translocation across the closed gate of homologous voltage-gated K+ channels [21] suggesting the early-activated-open conformation as an intermediate partially open structure on the closed-to-open transition pathway of NaChBac.
Figure 4.
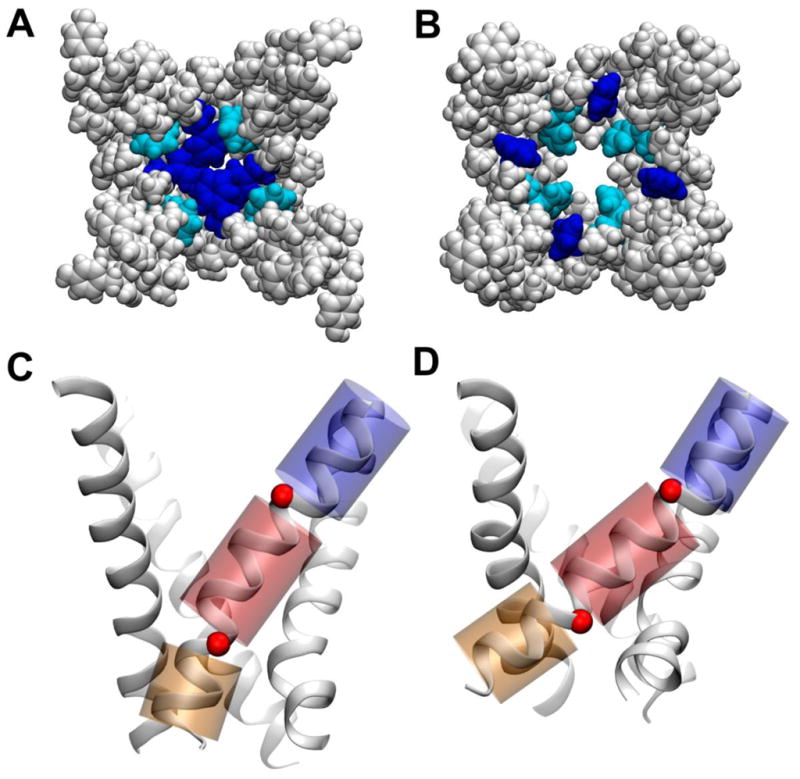
Conformations of the NaChBac pore domain along the activation pathway as determined in the present study, with only the S6-helix bundle shown for clarity. Upper panels: (A, B) space-filling representation of the channel lumen for the closed and activated-partially-open conformations, respectively. Phenyl groups Phe224 and Phe227 are shown in blue and cyan, respectively. Bottom panels: (C, D) cartoon representation of the S6-helix bundle highlighting the hinge-bending motions around residues G219 and G229 (red spheres) involving the three helical regions (blue, red, and orange) for the closed and activated-partially-open conformations, respectively.
The presence of two glycine residues endows the S6-helix with conformational flexibility at positions 219 and 229 and suggests a mechanism underlying pore domain gating transitions in which these glycine residues act as hinges and decouple the motions of the three resulting helical regions (Fig. 4C–D). Notably the angle at position 219 is considerably different between the two conformations (Fig. 5). On passing from closed- to open-pore, the kink angle between the two helical segments upstream and downstream of G219 (residues 210–218 and 220–228) changes from an average value of 10 deg to 20 deg (Fig. 5). The opposite behavior is observed for the kink angle at position 229 (defined as the angle between the helical regions 218–228 and 230–240), for which a more pronounced kink is found in this case for the closed conformation: 20 deg and 10 deg, for closed and open, respectively.
Figure 5.
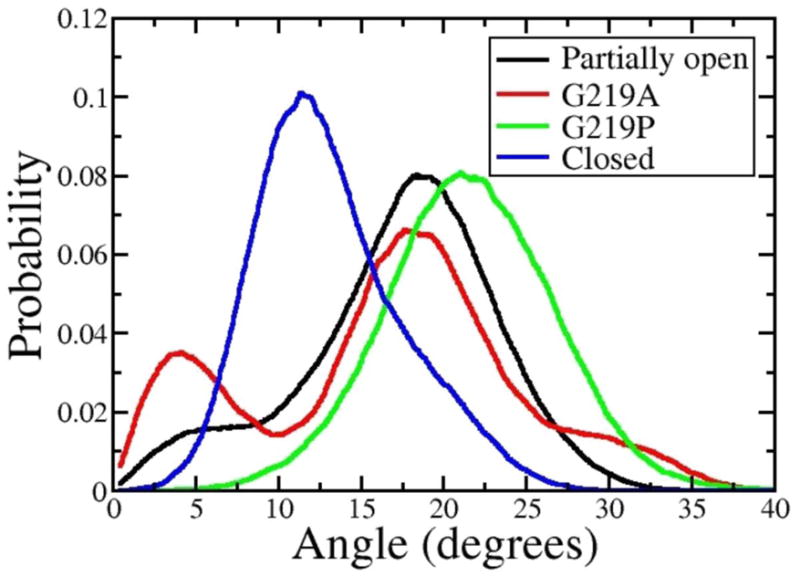
Probability distributions of S6 kink angles around G219 (A) and G229 (B) for the activated-partially-open (black) and closed (blue) conformations, along with results for the mutants G219A (red) and G219P (green). The angles are measured between the helical segments adjacent to the glycine residues: segments 210–218 and 220–228 for G219 and 218–228 and 230–240 for G229.
The rigid-body motions of these helical S6-segments modulate the packing at the inter-subunit interfaces, which in turn, determine the shape of the pore. Indeed an increased tilt of the C-term region of S6 allows the four S6 helices to be splayed apart, which results in an expansion of the constriction region located at the phenylalanine cluster (Fig. 4A–B). Therefore a stable kink at position 219 is seemingly the structural determinant of the open state. Moreover, this structural feature is consistent with mutagenesis data [24] showing that the G219A mutant, with decreased flexibility at position 219, inactivates faster than the WT. The data suggest that a significant destabilization of the open state can be achieved by decreasing the propensity of S6 for kinked configurations. Furthermore, independent experimental studies have shown that mutation of G219 into proline stabilizes a kinked S6-segment, and therefore the open conformation [25].
Additional independent MD simulations were performed on the G219P and G219A mutants to characterize the effect of these mutations on the kink propensity and bending flexibility of the S6-segment. The most dramatic effect is observed for the proline mutant: compared to the WT, where the distribution of kink angles shifts significantly toward larger values (Fig. 5). Furthermore, configurations featuring angles smaller than 10 deg are never explored. As expected, the less perturbing mutation of glycine into alanine results in smaller differences compared to the WT: two peaks are apparent in the distribution around 5 degrees and 20 degrees, respectively corresponding to the straight and kinked configurations (Fig. 5). Despite showing a slightly decreased probability for kinked configurations, the distribution is very similar to that of the WT, suggesting that mutation of glycine into alanine at position 219 does not dramatically affect the overall structure of the S6 bundle and that the increased rate of inactivation may result from the slightly decreased conformational flexibility.
To further characterize the role of the phenylalanine cluster, we investigated the early events of the gating transition. To this end, we have modeled a conformation featuring fully activated VS domains and monitored the response of the pore over a time-scale of 0.270 μs. Simulations were started from the closed-pore configuration and S4 helix was moved to the fully activated state (as described in ref. [37]) by means of steered MD. As expected, the most relevant structural rearrangement occurs at the S4–S5 linker, where the helical region of the linker is displaced by 6 Å along the normal to the membrane surface (Fig. 6). As a result, the steric constraints on the bundle-crossing region of S6 are weaker than in the early-activated state and in-plane movements of the C-term region of S6 are less restricted. It is worth noting that this configuration of the S4–S5 linker is coupled to a conformational change in the S6 bundle. Specifically, an increased tilt of the N-term region of S6 allows the helices to be splayed apart without the need for a kink at position 219. Although the S6 bundle was not able to reach the fully open conformation over the relatively short MD time-scale, a significant rearrangement was observed in the phenylalanine cluster: rather than being stacked in the pore-lumen, the bulky aromatic side-chains are found, on average, to be buried in the helix-helix interfaces in configurations remarkably similar to those found in the early-activated-open state.
Figure 6.

Shown is a superposition of structures of the NaChBac S4–S5 linker for closed (red), early-activated-open (blue) and activated-partially-open (green) conformations (see text), highlighting the displacement of the linker along the direction normal to the membrane.
The major consequence of the above-mentioned rearrangement is the inability of the cluster of phenylalanines to act as a hydrophobic seal preventing waters, and thus solvated ions, from traversing the bundle cross. Over the course of simulation we observe sufficient opening to allow water flux in the presence of a TM potential. This change in hydration can be seen in Fig. 7 in which the intermittent formation of water wires spanning the entire pore is highlighted by the presence of density in the -7 Å to -15 Å region. The ability of water molecules to occupy this section of the pore suggests that hydration of the hydrophobic constriction region may be the early response to activation leading to destabilization of the closed conformation and inducing, ultimately, the conformational transition to the fully open state.
Figure 7.

Shown are the positions of water oxygen atoms projected onto the membrane normal (Z). Blue shading highlights the portion of the MD trajectory sampled before an external electrostatic field was applied. Under the electric field, the frequency of wetting/dewetting transitions at the hydrophobic gate (located approximately between -7 Å and -15 Å) increases dramatically.
In light of our observations, the following activation scheme can be envisioned for NaChBac: in response to VS activation, the S4–S5 linker cuff is displaced along the normal to the membrane; this movement increases the tilt of the coupled C-term regions of S6 helices and thus induces an expansion of the bundle. A flexible hinge movement at G219 facilitates this conformational transition and results in an opening of the pore.
4. Conclusions
The bacterial Na+ channel NaChBac is evolutionarily related to eukaryotic Na+ channels and [26,29] has been the subject of extensive experimental and computational investigations [10,12–14,17,24,25,31–33,50,51] aimed at revealing general mechanistic aspects of gating in VGSCs. The structural rearrangement of the pore domain during the gating process is one of the most significant issues to be addressed due to the role of the pore in binding a number of clinically relevant drugs. The present computational study builds upon extensive experimental data and previously derived models of NaChBac gating [12–14,17,52] to reveal significant conformational changes in the S6 pore-lining helix bundle during gating.
By comparing structural models of the closed, early-activated-open, and activated-partially-open conformations of the channel, we identify hinge-bending motions around two glycine residues in S6, which are able to modulate the shape of the channel lumen along the activation pathway. Specifically, starting from the closed state, a tilt of the C-term region of S6 allows pore opening and induces the formation of a kink at position 219; full activation of the VS domains entails a structural rearrangement of the linker, which, in turn, is coupled to tilting of the N-term region of S6 and a relaxation from the kinked to the unkinked state. Significantly, a cluster of stacked phenylalanine residues in the pore, which hinder diffusion of water molecules and Na+ ions in the resting and activated-closed states, undergoes a structural rearrangement upon expansion of the S6 helical bundle in the open state. This scheme is in agreement with previous structural descriptions of a hydrophobic-gate controlled mechanisms of ion conduction in other channels [53–57].
In addition to creating structural models of NaChBac that will be useful for investigating interactions with commonly used VGSC-modulatory drugs, our findings suggest interesting features of the pore domain that merit to be investigated experimentally: the function of gate phenylalanines, the nature of VS-gate coupling, and the role of fenestrations in gating and drug access to internal sites. The hydrophobic seal formed by phenylalanines is not found in the NavAb crystal structure and thus demand further experimental investigation. Mutation of these residues could cause shifts in voltage dependence by changing the ease of opening the gate. Additionally, mutation to charged residues could create constitutively open or closed channels. How the movement of these phenylalanines is energetically coupled to VS conformational changes can also be explored by mutational analysis.
The fenestrations found in the NavAb crystal structure are preserved in the NaChBac homology model in all of the explored conformations. These fenestrations have not been seen in other tetrameric channel crystal structures, but may play a role in allowing pore domain flexibility necessary for gating and could also provide an access path for drugs. Mutations that occlude the fenestration would permit investigation of whether they are necessary for gating and the observation that they are permanently open in all the conformations examined reinforces the hypothesis that small drugs could reach internal sites via fenestrations. Further investigation of drug access pathways through fenestrations and a search for fenestration-like formations in mammalian VGSC sequences via further modeling and electrophysiological experimentation will be necessary to determine whether they represent the “lipophilic” pathway proposed by Hille [6,9].
Given that the action of VGSC modulating drugs is known to shift the relative stability of specific gating conformations [3,4,27,58–64] our structures will enable further computational investigations, which will likely shed light on the nature of drug binding modalities in NaChBac as well as the more general issue of the mechanism of drug-sensitivity of VGSCs.
Supplementary Material
Highlights.
We build structural models of NaChBac inserted in a phospholipid bilayer.
We investigate the properties of three conformations of NaChBac via MD simulations.
Hinge-bending motions around two glycine residues affect the shape of the pore.
Mutations of the glycine residues affect the structure and the conformational flexibility of the S6 bundle.
A cluster of Phe residues undergoes a structural transition upon activation.
Acknowledgments
We are grateful for support from the National Institutes of Health, and the Commonwealth of Pennsylvania. We also thank Manuel Covarrubias for very useful scientific discussion. A.F.B. acknowledges the National Institutes of Health (Grants NIH-NIAAA T32 AA007463 and NIH-NINDS F31 NS077689) for financial support. The National Science Foundation supported this work through XSEDE resources provided by the National Institute for Computational Sciences (Grant TG-MCA93S020).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.George AL. Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clare JJ, Tate SN, Nobbs M, Romanos MA. Voltage-gated sodium channels as therapeutic targets. Drug Discov Today. 2000;5:506–520. doi: 10.1016/s1359-6446(00)01570-1. [DOI] [PubMed] [Google Scholar]
- 3.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 4.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci USA. 1996;93:9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yarov-Yarovoy V, McPhee JC, Idsvoog D, Pate C, Scheuer T, Catterall WA. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J Biol Chem. 2002;277:35393–35401. doi: 10.1074/jbc.M206126200. [DOI] [PubMed] [Google Scholar]
- 6.Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strichartz GR. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J Gen Physiol. 1973;62:37–57. doi: 10.1085/jgp.62.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Courtney KR. Mechanism of frequency-dependent inhibition of sodium currents in frog myelinated nerve by the lidocaine derivative GEA. J Pharmacol Exp Ther. 1975;195:225–236. [PubMed] [Google Scholar]
- 9.Hille B. Ion channels of excitable membranes. Sinauer; Sunderland, Mass: 2001. [Google Scholar]
- 10.Kuzmenkin A, Bezanilla F, Correa AM. Gating of the bacterial sodium channel, NaChBac: voltage-dependent charge movement and gating currents. J Gen Physiol. 2004;124:349–356. doi: 10.1085/jgp.200409139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanchet J, Pilote S, Chahine M. Acidic residues on the voltage-sensor domain determine the activation of the NaChBac sodium channel. Biophys J. 2007;92:3513–3523. doi: 10.1529/biophysj.106.090464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeCaen PG, Yarov-Yarovoy V, Zhao Y, Scheuer T, Catterall WA. Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci USA. 2008;105:15142–15147. doi: 10.1073/pnas.0806486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, Catterall WA. Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci USA. 2009;106:22498–22503. doi: 10.1073/pnas.0912307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paldi T, Gurevitz M. Coupling between residues on S4 and S1 defines the voltage-sensor resting conformation in NaChBac. Biophys J. 2010;99:456–463. doi: 10.1016/j.bpj.2010.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimomura T, Irie K, Nagura H, Imai T, Fujiyoshi Y. Arrangement and mobility of the voltage sensor domain in prokaryotic voltage-gated sodium channels. J Biol Chem. 2011;286:7409–7417. doi: 10.1074/jbc.M110.186510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Decaen PG, Yarov-Yarovoy V, Scheuer T, Catterall WA. Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc Natl Acad Sci USA. 2011;108:18825–18830. doi: 10.1073/pnas.1116449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 19.Townsend C, Horn R. Interaction between the pore and a fast gate of the cardiac sodium channel. J Gen Physiol. 1999;113:321–332. doi: 10.1085/jgp.113.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuo CC, Liao SY. Facilitation of recovery from inactivation by external Na+ and location of the activation gate in neuronal Na+ channels. J Neurosci. 2000;20:5639–5646. doi: 10.1523/JNEUROSCI.20-15-05639.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Treptow W, Tarek M. Environment of the gating charges in the Kv1.2 Shaker potassium channel. Biophys J. 2006;90:L64–L66. doi: 10.1529/biophysj.106.080754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 23.Webster SM, Del Camino D, Dekker JP, Yellen G. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 2004;428:864–868. doi: 10.1038/nature02468. [DOI] [PubMed] [Google Scholar]
- 24.Irie K, Kitagawa K, Nagura H, Imai T, Shimomura T, Fujiyoshi Y. Comparative study of the gating motif and C-type inactivation in prokaryotic voltage-gated sodium channels. J Biol Chem. 2010;285:3685–3694. doi: 10.1074/jbc.M109.057455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Scheuer T, Catterall WA. Reversed voltage-dependent gating of a bacterial sodium channel with proline substitutions in the S6 transmembrane segment. Proc Natl Acad Sci USA. 2004;101:17873–17878. doi: 10.1073/pnas.0408270101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charalambous K, Wallace BA. NaChBac: the long lost sodium channel ancestor. Biochemistry. 2011;50:6742–6752. doi: 10.1021/bi200942y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.OuYang W, Hemmings HC. Isoform-selective effects of isoflurane on voltage-gated Na+ channels. Anesthesiology. 2007;107:91–98. doi: 10.1097/01.anes.0000268390.28362.4a. [DOI] [PubMed] [Google Scholar]
- 28.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 29.Ren D, Navarro B, Xu H, Yue L, Shi Q, Clapham DE. A prokaryotic voltage-gated sodium channel. Science. 2001;294:2372–2375. doi: 10.1126/science.1065635. [DOI] [PubMed] [Google Scholar]
- 30.Koishi R, Xu H, Ren D, Navarro B, Spiller BW, Shi Q, Clapham DE. A superfamily of voltage-gated sodium channels in bacteria. J Biol Chem. 2004;279:9532–9538. doi: 10.1074/jbc.M313100200. [DOI] [PubMed] [Google Scholar]
- 31.Richardson J, Blunck R, Ge P, Selvin PR, Bezanilla F, Papazian DM, Correa AM. Distance measurements reveal a common topology of prokaryotic voltage-gated ion channels in the lipid bilayer. Proc Natl Acad Sci USA. 2006;103:15865–15870. doi: 10.1073/pnas.0607532103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shafrir Y, Durell SR, Guy HR. Models of the structure and gating mechanisms of the pore domain of the NaChBac ion channel. Biophys J. 2008;95:3650–3662. doi: 10.1529/biophysj.108.135327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shafrir Y, Durell SR, Guy HR. Models of the structure and gating mechanisms of the pore domain of the NaChBac ion channel. Biophys J. 2008;95:3650–3662. doi: 10.1529/biophysj.108.135327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices1. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 35.Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 36.Carnevale V, Treptow W, Klein ML. Sodium ion binding sites and hydration in the lumen of a bacterial ion channel from molecular dynamics simulations. J Phys Chem Lett. 2011 [Google Scholar]
- 37.Yarov-Yarovoy V, Decaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D, Catterall WA. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1118434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacKerell AD, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 39.Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 40.Hénin J, Shinoda W, Klein ML. United-atom acyl chains for CHARMM phospholipids. J Phys Chem B. 2008;112:7008–7015. doi: 10.1021/jp800687p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. [Google Scholar]
- 42.Darve E, Rodríguez-Gómez D, Pohorille A. Adaptive biasing force method for scalar and vector free energy calculations. J Chem Phys. 2008;128:144120. doi: 10.1063/1.2829861. [DOI] [PubMed] [Google Scholar]
- 43.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. The Journal of Chemical Physics. 1995;103:8577. [Google Scholar]
- 44.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comp Phys. 1977;23:327–341. [Google Scholar]
- 45.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comp Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henin J, Fiorin G, Chipot C, Klein ML. Exploring multidimensional free energy landscapes using time-dependent biases on collective variables. J Chem Theory Comp. 2009;6:35–47. doi: 10.1021/ct9004432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011 doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berneche S, Roux B. Molecular dynamics of the KcsA K+ channel in a bilayer membrane. Biophysical Journal. 2000;78:2900–2917. doi: 10.1016/S0006-3495(00)76831-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shrivastava IH, Sansom MSP. Simulations of ion permeation through a potassium channel: molecular dynamics of KcsA in a phospholipid bilayer. Biophys J. 2000;78:557–570. doi: 10.1016/S0006-3495(00)76616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pavlov E, Bladen C, Winkfein R, Diao C, Dhaliwal P, French RJ. The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys J. 2005;89:232–242. doi: 10.1529/biophysj.104.056994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yue L, Navarro B, Ren D, Ramos A, Clapham DE. The cation selectivity filter of the bacterial sodium channel, NaChBac. J Gen Physiol. 2002;120:845. doi: 10.1085/jgp.20028699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chakrapani S, Sompornpisut P, Intharathep P, Roux B, Perozo E. The activated state of a sodium channel voltage sensor in a membrane environment. Proc Natl Acad Sci USA. 2010;107:5435–5440. doi: 10.1073/pnas.0914109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kitaguchi T, Sukhareva M, Swartz KJ. Stabilizing the closed S6 gate in the Shaker Kv channel through modification of a hydrophobic seal. J Gen Physiol. 2004;124:319–332. doi: 10.1085/jgp.200409098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 55.Bright JN, Shrivastava IH, Cordes FS, Sansom MS. Conformational dynamics of helix S6 from Shaker potassium channel: simulation studies. Biopolymers. 2002;64:303–313. doi: 10.1002/bip.10197. [DOI] [PubMed] [Google Scholar]
- 56.Spronk SA, Elmore DE, Dougherty DA. Voltage-dependent hydration and conduction properties of the hydrophobic pore of the mechanosensitive channel of small conductance. Biophys J. 2006;90:3555–3569. doi: 10.1529/biophysj.105.080432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corry B. An energy-efficient gating mechanism in the acetylcholine receptor channel suggested by molecular and Brownian dynamics. Biophys J. 2006;90:799–810. doi: 10.1529/biophysj.105.067868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shiraishi M, Harris RA. Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels. J Pharmacol Exp Ther. 2004;309:987–994. doi: 10.1124/jpet.103.064063. [DOI] [PubMed] [Google Scholar]
- 59.Ouyang W, Jih TY, Zhang TT, Correa AM, Hemmings HC. Isoflurane inhibits NaChBac, a prokaryotic voltage-gated sodium channel. J Pharmacol Exp Ther. 2007;322:1076–1083. doi: 10.1124/jpet.107.122929. [DOI] [PubMed] [Google Scholar]
- 60.Hemmings HC. Sodium channels and the synaptic mechanisms of inhaled anaesthetics. Br J Anaesth. 2009;103:61–69. doi: 10.1093/bja/aep144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dib-Hajj SD, Black JA, Waxman SG. Voltage-gated sodium channels: herapeutic targets for pain. Pain Medicine. 2009;10:1260–1269. doi: 10.1111/j.1526-4637.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- 62.Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010;9:413–424. doi: 10.1016/S1474-4422(10)70059-4. [DOI] [PubMed] [Google Scholar]
- 63.Cohen CJ. Targeting Voltage-gated Sodium Channels for Treating Neuropathic and Inflammatory Pain. Curr Pharm Biotechnol. 2011 doi: 10.2174/138920111798357249. [DOI] [PubMed] [Google Scholar]
- 64.Mahdavi S, Gharibzadeh S, Ranjbar B, Javan M. Voltage-gated sodium channel gating modifiers: valuable targets for multiple sclerosis treatment. J Neuropsych and Clin Neurosci. 2011;23:E17. doi: 10.1176/jnp.23.1.jnpe17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.