

Conspectus
Effective methodology to functionalize C–H bonds requires overcoming the key challenge of differentiating among the multitude of C–H bonds that are present in complex organic molecules. This Account focuses on our work over the past decade toward the development of site-selective Pd-catalyzed C–H functionalization reactions using the following approaches: substrate-based control over selectivity through the use of directing groups (approach 1), substrate control through the use of electronically activated substrates (approach 2), or catalyst-based control (approach 3). In our extensive exploration of the first approach, a number of selectivity trends have emerged for both sp2 and sp3 C–H functionalization reactions that hold true for a variety of transformations involving diverse directing groups. Functionalizations tend to occur at the less-hindered sp2 C–H bond ortho to a directing group, at primary sp3 C–H bonds that are β to a directing group, and, when multiple directing groups are present, at C–H sites proximal to the most basic directing group. Using approach 2, which exploits electronic biases within a substrate, our group has achieved C-2-selective arylation of indoles and pyrroles using diaryliodonium oxidants. The selectivity of these transformations is altered when the C-2 site of the heterocycle is blocked, leading to C–C bond formation at the C-3 position. While approach 3 (catalyst-based control) is still in its early stages of exploration, we have obtained exciting results demonstrating that site selectivity can be tuned by modifying the structure of the supporting ligands on the Pd catalyst. For example, by modulating the structure of N~N bidentate ligands, we have achieved exquisite levels of selectivity for arylation at the α site of naphthalene. Similarly, we have demonstrated that both the rate and site selectivity of arene acetoxylation depend on the ratio of pyridine (ligand) to Pd. Lastly, by switching the ligand on Pd from an acetate to a carbonate, we have reversed the site selectivity of a 1,3-dimethoxybenzene/benzo[h]quinoline coupling. In combination with a growing number of reports in the literature, these studies highlight a frontier of catalyst-based control of site-selectivity in the development of new C–H bond functionalization methodology.
Introduction
C–H bonds are truly ubiquitous, an attribute that makes them attractive starting materials for the elaboration of complex molecules. However, this same characteristic is also a great impediment to developing practical methods for C–H bond functionalization. To achieve useful yields of a single product, functionalization must occur with high site selectivity for one C–H bond over the others within a complex molecule. Finding a solution to this challenge has been a driving force for much of the research in our laboratory over the past decade. This Account describes our work on the development of Pd-catalyzed C–H bond functionalization reactions, with a particular focus on strategies for achieving site selectivity in these transformations.
Background
Our overall goal has been to develop selective methods for oxidatively transforming C–H bonds embedded in complex molecules into a wide variety of other functional groups (e.g., C–O, C–S, C–halogen, C–N, and C–C bonds; Scheme 1). Our first efforts (initiated in 2003) focused on identifying a competent catalyst for these diverse transformations. We noted a number of literature reports demonstrating PdII-mediated oxidative functionalization of simple arenes. These typically employed Pd(OAc)2 in conjunction with an oxidant to effect C–heteroatom1 or C–C bond formation.2,3 However, these methods were typically limited by high catalyst loadings (TON ~ <1–10), modest substrate scope (typically electron-neutral or electron-rich aromatics), and formation of mixtures of product isomers.
Scheme 1.
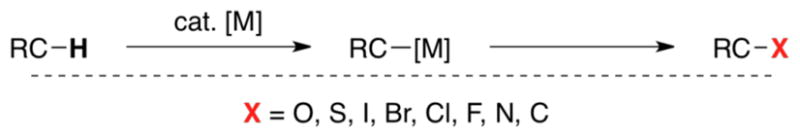
Transformation of C–H bonds into diverse functional groups.
In 1996 Crabtree demonstrated that Pd(OAc)2-catalyzed arene oxidation could be accomplished with substantially lower catalyst loading (TON = 19–127) using PhI(OAc)2 as the terminal oxidant.4 However, like the earlier work discussed above, the transformation proceeded with poor site selectivity and both alkanes and electron-deficient arenes showed low reactivity. Motivated by this precedent, we initiated studies to address these key limitations.
Strategies for Control of Selectivity
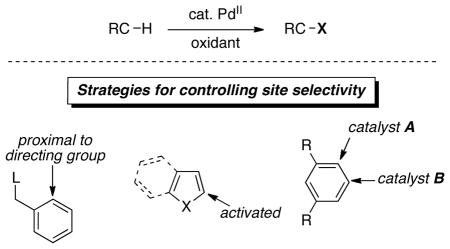
Using the combination of PdII catalysts and oxidants, we have developed a wide variety of C–H functionalization reactions. We have taken three basic strategies to achieve site selectivity (Scheme 2) that involve both ‘substrate-based control’ (approaches 1 and 2) and ‘catalyst-based control’ over selectivity (approach 3). Approach 1 employs substrates that contain coordinating functional groups to direct C–H activation and subsequent functionalization to a proximal site. Approach 2 involves the use of heterocyclic substrates that contain highly activated C–H sites. Finally, approach 3 involves the design of ligands for the Pd catalyst that exert control over site selectivity in C–H functionalization. Each of these approaches is discussed in detail below.
Scheme 2.
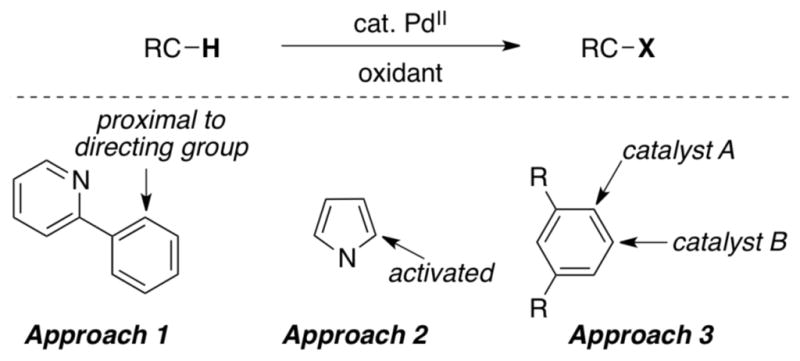
Strategies for controlling site selectivity.
Approach 1. Substrate-Based Control of Selectivity Through the Use of Directing Groups
Catalytic Reaction Development
Our initial efforts focused on developing a ligand-directed version of Crabtree’s Pd(OAc)2-catalyzed arene acetoxylation with PhI(OAc)2.4 We aimed to capitalize on the well-known cyclopalladation reaction (stoichiometric ligand-directed C–H bond activation at PdII) to achieve site-selective C–H cleavage (Scheme 3, step i).5 Subsequent reaction between the cyclopalladated intermediate 1 and PhI(OAc)2 could then release the desired acetoxylated product 2 (steps ii–iii).2,5,6
Scheme 3.
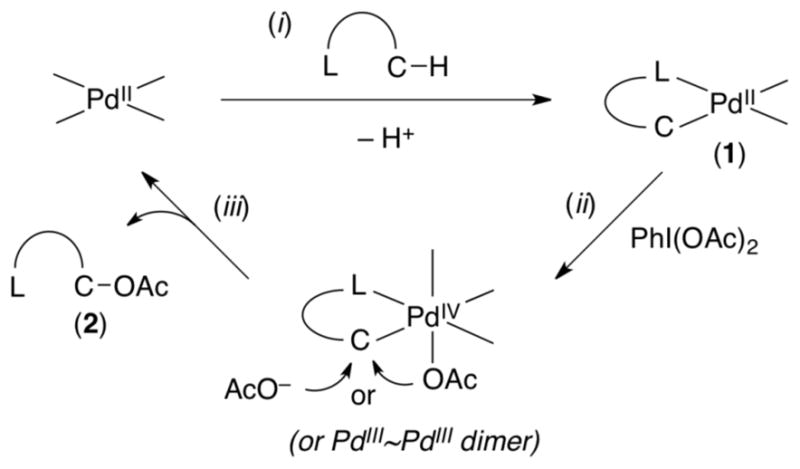
Catalytic cycle for ligand-directed C–H acetoxylation.
We first pursued the Pd(OAc)2-catalyzed C–H acetoxylation of benzo[h]quinoline (3). Gratifyingly, C–H oxygenation occurred in high yield to afford 4 as a single isomer (eq 1).7a Notably, this transformation (like nearly all of those described in this Account) can be conveniently carried out on the bench top without exclusion of ambient air or moisture.
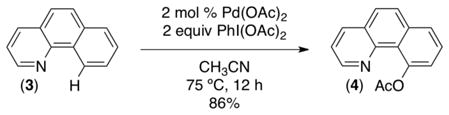 |
(1) |
The Pd(OAc)2-catalyzed ligand-directed C–H acetoxylation could be accomplished at both sp2- and sp3-C–H bonds. Diverse directing groups including pyridine, pyrimidine, pyrazine, pyrazole, azobenzene, imine, pyrrolidinone, oxime ether and acetate, isoxazoline, and amide derivatives were all effective in providing high selectivity for a proximal C–H site (Scheme 4).7 Substrates containing two or three identical sites for functionalization could be di- or tri-oxygenated by using multiple equivalents of oxidant. With many substrates, C–H acetoxylation also proceeds in high yield with polymer-immobilized iodine(III) reagents,7e Oxone,7d or K2S2O87d as oxidants. Furthermore, conducting these reactions in alcohol solvents (e.g., MeOH, iPrOH, CF3CH2OH) results in C–H etherification.7a,d Other groups have subsequently demonstrated related Pd-catalyzed C–H oxygenation reactions directed by carboxylic acids, oxazolines, anilides, and amidoquinolines.8
Scheme 4.

Representative products of ligand-directed C–H oxygenation.
Mechanistic studies are consistent with the catalytic cycle depicted in Scheme 3.4,7a,9 This mechanism suggests that the use of oxidants that could transfer alternative groups to the Pd center (oxidant–X in eq 2) should generate high valent Pd complexes of general structure 5. These, in turn, could undergo C–X bond-forming reductive elimination to generate diverse functionalized products.
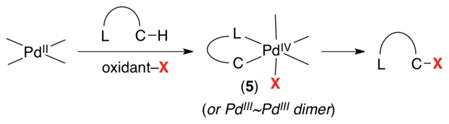 |
(2) |
Consistent with this hypothesis, we found that N-halosuccinimides are effective oxidants for Pd-catalyzed ligand-directed C–H halogenations to generate C–Cl, C–Br, and C–I bonds,7g,10 and that N-fluoropyridinium oxidants can be utilized for related C–H fluorination reactions (Scheme 5).11 Other research groups have reported related C–H halogenation reactions using CuX2,12c,e IOAc,12a,b,d and N-fluoropyridinium reagents.13
Scheme 5.

Representative products of ligand-directed C–H halogenation.
This strategy has also proven successful for achieving ligand-directed C–H arylation. We have shown that either diaryliodonium salts (Ar2I+, Scheme 6) or in situ generated Ph• can be used as oxidants to effect the selective Pd-catalyzed C–H arylation of a variety of substrates.9c,14,15 Numerous related Pd-catalyzed ligand-directed C–H arylation reactions (typically with aryl halide oxidants) have been reported in the literature using diverse directing groups including anilides, pyridines, benzoxazoles, carboxylic acids, amides, oxime ethers, aminoquinolines, and picolinamides.8b,16
Scheme 6.
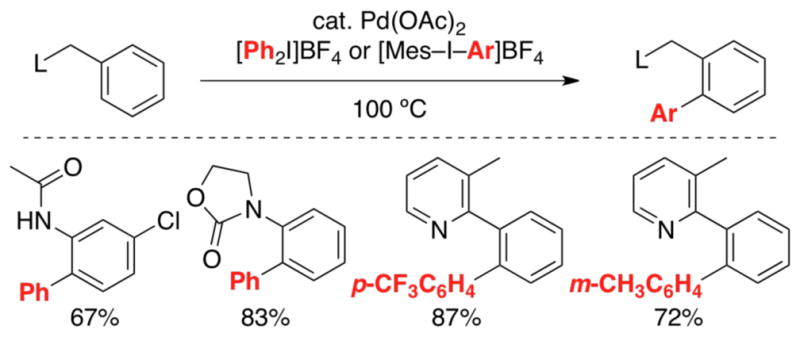
Representative products of ligand-directed C–H arylation.
Selectivity Trends for Ligand-Directed C–H Functionalization
Selectivity in Ligand-Directed sp2-C–H Functionalization
Pd-catalyzed ligand-directed arene C–H functionalization reactions generally afford products functionalized exclusively ortho to the directing group. This selectivity is dictated by the C–H activation step, as only a palladacycle containing a Pd–Cortho bond is geometrically feasible.5 Arene functionalization most commonly occurs via five- or six-membered palladacycles, although reactions proceeding via seven-membered and larger palladacyclic intermediates have also been reported.17
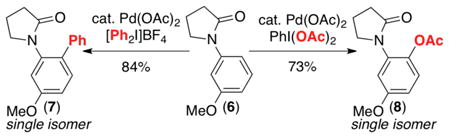
When two sterically inequivalent ortho C–H sites are available, high selectivity is typically observed for functionalization at the less hindered site (eq 3, product A).7c This selectivity stands in contrast to directed ortho-lithiation (DoL). When R can act as a secondary binding site for Li+ (e.g., R = OMe, OMOM), DoL leads to functionalization at the site between the two substituents.18 In contrast, the Pd-catalyzed functionalization of substrate 6 yields products 7 or 8 without detectable formation of isomers (eq 4).7c,14
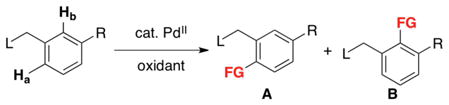 |
(3) |
 |
(4) |
In an attempt to test the limits of this selectivity, we devised a number of substrates that might be biased toward acetoxylation at the more sterically hindered ortho site (Scheme 7).7c Substrate 9 contains a meta-fluorine substituent, which is comparable in size to hydrogen, thereby potentially ameliorating the steric bias. Substrates 10 and 11 contain meta ketone/oxime ether moieties, which could act as secondary ligands for Pd. However, remarkably all of these substrates still provided moderate (6:1) to excellent (>20:1) preference for A. Indeed, only one substrate was identified (12) that afforded modest selectivity for B (2:1).
Scheme 7.

Major site for C–H acetoxylation of 3′-substituted 2-arylpyridines.
Analogous selectivity trends have been reported for many other Pd-catalyzed ligand-directed arene C–H oxidations10,11,12a,16,19,20,21 with substrates bearing diverse directing groups and aromatic ring substituents (Scheme 8).22
Scheme 8.

Selectivity for isomer A in diverse C–H functionalizations.
Selectivity in Ligand-Directed sp3 C–H Functionalization
Ligand-directed sp3-C–H functionalization typically proceeds with high selectivity for 1° over 2° C–H bonds.5,7b,g In addition, selectivity is observed for C–H bonds that are β versus α or γ to the directing group (i.e, 5-membered palladacycles are strongly favored over their 4- or 6-membered counterparts). These trends are illustrated by the C–H acetoxylation of 13 (Scheme 9), which provides a single detectable product derived from functionalization at the 1° β-C–H site.7b With few exceptions (vide infra), 2° sp3 C–H bonds do not undergo functionalization even in the absence of more preferable sites for oxidation [e.g., substrate 14 (Figure 1)].7b
Scheme 9.

Selectivity in ligand-directed sp3-C–H acetoxylation.
Figure 1.

Unreactive substrates for sp3-C–H oxygenation.
The selectivity for 1° C–H oxidation stands in contrast to free radical and/or electrophilic C–H oxidation reactions.23 These typically afford functionalization at electron rich C–H sites; as such, the opposite order of C–H bond reactivity is observed (3° > 2° > 1°). The difference between the selectivities in Pd-catalyzed versus radical/electrophilic mechanisms likely reflects the strong influence of sterics and of C–H bond pKa24 on the palladation step.
Five-membered palladacyclic intermediates are typically important for achieving high-yielding sp3 C–H functionalization. For example, substrates such as 15 showed no reactivity in the presence of Pd(OAc)2/PhI(OAc)2 (Figure 1).7b The markedly higher reactivity of β-C–H bonds is likely due to the more favorable energy requirements for forming a 5-membered palladacycle.5
To date there are only a few examples of Pd-catalyzed ligand-directed C–H functionalization at 2° sp3-C–H sites (Figure 2). These reactions typically occur in substrates that have steric constraints/geometric biases or are electronically activated. One example involves trans-decalone methyl oxime ether, whose rigid conformation positions the equatorial 2° β-C–H bond near the coordinated PdII, leading to product 16 with high selectivity.7b In a second example, an amidoquinoline ligand binds to Pd in a bidentate fashion, thereby placing a 2° C–H site in close proximity to the Pd center to afford product 17.8b Electronic activation likely contributes to the facility of 2° sp3-C–H acetoxylation to form 18 (α to an activating oxygen atom)7b and arylation to form 19 (benzylic C–H site).14 Finally, 2° C–H bonds on cyclopropanes are often amenable to functionalization (e.g., to afford 2012b and 2125), likely due to the diminished steric requirements for C–H activation, the high rigidity of the substrate, and/or the increased s character of cyclopropyl C–H bonds.
Figure 2.

Products of 2° sp3-C–H functionalization.
Selectivity Between Directing Groups
Many substrates of interest contain multiple basic groups that could potentially bind to the Pd center and direct C–H functionalization. We conducted systematic competition studies to assess the relative propensities of different N- and O-donor groups to direct Pd-catalyzed C–H acetoxylation.7f These investigations revealed a strong correlation between the basicity of the directing group and the relative rate of C–H functionalization proximal to that group. This is exemplified by competition experiments between electronically varied benzylpyridine substrates (eq 5), as well as those between different heterocyclic directing groups (eq 6).
 |
(5) |
| (6) |
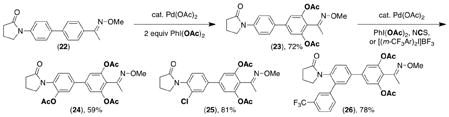
Scheme 10 summarizes the relative reactivity of directing groups in competition studies. Importantly, the trends derived from these experiments have predictive power. For example, they correctly predicted that substrate 22 would undergo selective C–H acetoxylation adjacent to the oxime ether group (eq 7). The resulting product 23 could then be further elaborated to afford products 24–26 via amide-directed oxygenation, chlorination, or arylation.
Scheme 10.

Relative reactivity of directing groups toward C–H acetoxylation in competition studies.
 |
(7) |
Catalyzed versus Uncatalyzed Selectivity
With many aromatic substrates, arene C–H functionalization with an electrophilic oxidant can occur by either a Pd-catalyzed pathway or by uncatalyzed electrophilic aromatic substitution (EAS). In certain cases, these two pathways afford different and complementary site selectivity (Scheme 11). Some examples of this phenomenon include the halogenation of electron rich oxime ether 27 (which selectively affords 28 in the absence of Pd and 29 under Pd catalysis), pyrazole 30, (forming 31 and 32, respectively), and quinoline 33 (generating 34 and 35).10
Scheme 11.

Complementary site selectivity of halogenation in the presence and absence of Pd.
Approach 2. Substrate-Based Control of Selectivity Through the Use of Electronically Activated Substrates
A second strategy for obtaining site selectivity in Pd-catalyzed C–H functionalization is to utilize substrates that have a significant electronic or steric bias for palladation at a specific site. Electron-rich heterocycles are particularly common targets for this approach. For example, many research groups have demonstrated modest to high site selectivity for Pd-catalyzed C–H functionalization at C-2 of indole and C-2/C-5 of pyrrole derivatives (eq 8).26 This selectivity is believed to derive from an electronic preference for generating Pd-σ-heteroaryl complexes at C-2 (either by initial palladation at this site or by palladation at C-3 followed by palladium migration).26 This area has been the subject of several reviews,26 and this section focuses just on our group’s contributions in this area.
 |
(8) |
Our initial studies focused on the Pd-catalyzed reaction between indole and [Ph2I]BF4 (the oxidant used previously for ligand-directed C–H arylation). (IMes)Pd(OAc)2(H2O) [IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene] proved to be the optimal catalyst, providing 2-phenylindole in excellent (81%) yield and with >20:1 selectivity for functionalization at C-2.27 Comparable yields and site selectivities were obtained with a variety of substituted indole substrates (Scheme 12). Arylation was exclusively observed at C-2 except when this site was blocked (e.g., 1,2-dimethylindole). In the latter case, a modest yield of the C-3 arylation product 36 was obtained.
Scheme 12.

Pd-catalyzed C-2 arylation of indoles and pyrroles with [Ar2I]BF4.
The C-2 arylation of pyrroles also proceeded with high site selectivity under these conditions (e.g., 37). Blocking C-2/C-5 of pyrrole (as in 2,5-dimethylpyrrole) significantly diminished reactivity. Under our original conditions, only 12% yield of 38 was obtained; however, the use of PdCl2(MeCN)2 at an elevated temperature (84 °C) enabled high yielding C-3 phenylation of this substrate.28 Other 2,5-disubstituted pyrroles also underwent arylation under these conditions, and selectivity appears to be dictated by the relative size of the 2- and 5-substituents (Scheme 13). For example, 2-cyclohexyl-1,5-dimethylpyrrole underwent highly selective (29:1) arylation at the less hindered C-4 to form 39. However, when cyclohexyl was replaced by a smaller ethyl group, selectivity for C-4 arylation decreased to 2:1 (40).
Scheme 13.

Pd-catalyzed C-3 arylation of 2,5-disubstituted pyrroles.
Approach 3. Catalyst-Based Control of Selectivity
In addition to substrate-based control strategies, the site selectivity of C–H functionalization can be controlled via modification of the Pd catalyst structure. This approach targets C–H substrates that lack directing and/or activating groups, with the ultimate goal of achieving selective formation of different isomeric products by simply changing the ligands at the Pd center (Scheme 14). This has historically proven difficult because the vast majority of Pd-catalyzed C–H functionalization reactions proceed most efficiently under ‘ligandless conditions’ (involving simple Pd salts like Pd(OAc)2 as catalysts). As such, a key challenge has been to identify ligands that both accelerate these reactions and modulate their site selectivity.
Scheme 14.

Catalyst control of selectivity with ancillary ligands.
This section discusses catalyst control over selectivity in the context of three different C–H oxidation reactions. Research in all three of these areas remains in its infancy, and in most cases the mechanistic origin of the observed selectivity remains to be elucidated. Nonetheless, these efforts represent an exciting frontier for the development of practical and selective Pd-catalyzed C–H functionalization reactions without the requirement for activating and/or directing groups.
Naphthalene Arylation
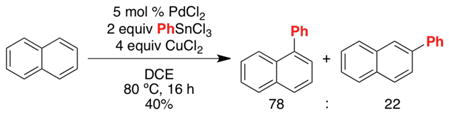
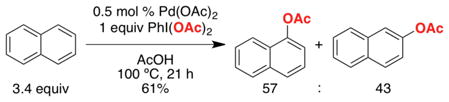
Naphthalene has historically proven a challenging substrate for selective C–H functionalization. For example, the PdCl2-catalyzed C–H arylation of naphthalene with PhSnCl3 proceeds with only modest α selectivity (78:22, eq 9).29 Furthermore, the Pd(OAc)2-catalyzed C–H acetoxylation of naphthalene with PhI(OAc)2 affords a nearly statistical distribution of isomeric products (α:β = 57:43, eq 10).4
 |
(9) |
 |
(10) |
We chose the Pd-catalyzed C–H arylation of naphthalene with diaryliodonium salts as a platform for investigating catalyst control over site selectivity. A survey of Pd sources and ligands revealed that the combination of PdCl2 and bidentate N~N donors (e.g., bipyridine and diimines) afforded catalysts with enhanced reactivity relative to PdCl2 alone. Furthermore, modification of the N~N ligand resulted in significant changes in selectivity (Scheme 15). While no clear trends were observed as a function of the steric/electronic properties of the N~N ligand, catalyst 41 was found to be uniquely effective for this transformation, affording 70% yield with remarkably high site selectivity (α:β = 71:1).30
Scheme 15.

Catalyst control of selectivity in napthalene arylation.
Mechanistic studies of this reaction suggest an unusual pathway in which palladation of naphthalene occurs at a highly electrophilic PdIV center.30 This provides a potential explanation for the high selectivity for phenylation at the most nucleophilic α-site. Further studies will be required to obtain insights into the central role that the 2,6-dichlorodiimine ligand plays in enhancing this α-selectivity. Current efforts are aimed at identifying catalysts capable of reversing the selectivity of this transformation to afford high selectivity for the β-arylated product. In addition, the identification of catalysts that show broader substrate scope is another important future goal.
Arene Acetoxylation
As discussed above, the Pd(OAc)2-catalyzed C–H acetoxylation of arenes with PhI(OAc)2 was initially reported by Crabtree in 1996.4 When applied to substituted aromatic substrates, this original system afforded low yields and poor site selectivity (e.g., eq 11). Our objective was to identify ligands for Pd that could both enhance reactivity as well as control the site selectivity of these transformations.
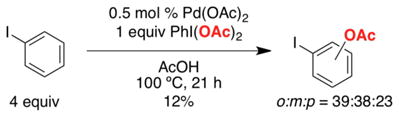 |
(11) |
Crabtree’s early studies showed that the vast majority of common ligands (e.g., pyridine, 1,10-phenanthroline, acetylacetone) dramatically slow the rate of this reaction (eq 12).4 However, we noted that Crabtree had explored all of the ligands in ratios such that they would generate coordinatively saturated complexes with Pd (e.g, 4 mol % Pd(OAc)2/9 mol % pyridine, eq 12). We reasoned that coordinatively unsaturated Pd complexes [e.g., (pyridine)Pd(OAc)2] should be more reactive, 1b,31 and thus explored the influence of L:Pd ratio on catalytic activity with L = pyridine.32 Gratifyingly, moving from L:Pd = 2:1 to 1:1 led to a dramatic rate acceleration for this transformation, demonstrating the feasibility of ligand-accelerated catalysis (Figure 3).
Figure 3.

Influence of pyridine (pyr) to Pd(OAc)2 ratio on the rate of benzene acetoxylation.
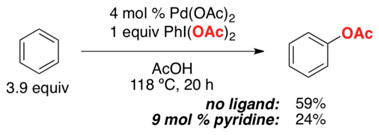 |
(12) |
The pyridine ligand also had a significant influence on the site selectivity with substituted arene substrates. For example, the α:β selectivity of the Pd(OAc)2-catalyzed acetoxylation of 1,2-dichlorobenzene changed from 41:59 without pyridine to 29:71 upon the addition of 0.9 equiv pyridine relative to Pd. Selectivity for the less hindered β site could be further enhanced by employing the bulky oxidant MesI(OAc)2 in place of PhI(OAc)2, resulting in an α:β ratio of 11:89 (eq 13).
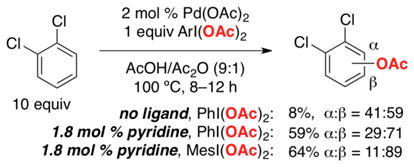 |
(13) |
A similar trend was observed for other aromatic substrates, where the addition of 0.9 equiv of the pyridine ligand in conjunction with MesI(OAc)2 enhanced selectivity for acetoxylation of the less sterically hindered aromatic C–H sites (eq 14).32
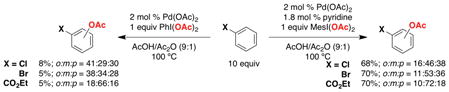 |
(14) |
The selectivities remain modest in most of these systems; furthermore, the mechanism by which pyridine/oxidant dictates selectivity requires further investigation. Nonetheless, the demonstration that pyridine ligands, in the correct proportions, can favorably influence activity and selectivity in arene C–H oxidation provides an exciting path forward for the field.31
Oxidative Cross-Coupling of Aryl–H with Benzo[h]quinoline
Another set of studies has focused on catalyst controlled selectivity in Pd-catalyzed oxidative cross-coupling reactions.33 In particular, we have examined the coupling of benzo[h]quinoline with aromatic substrates to generate a new biaryl linkage (eq 15). The C–H activation of benzo[h]quinoline proceeds with high site selectivity due to the presence of the pyridine directing group, while arene activation is subject to catalyst control.
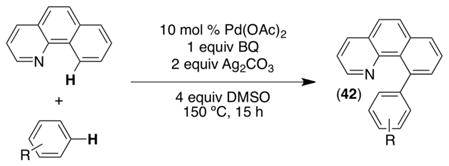 |
(15) |
Mechanistic investigations of this transformation implicated a reaction pathway involving: (i) ligand directed C–H activation of benzo[h]quinoline to generate dimer 43, (ii) reversible arene activation to form monomer 44, (iii) benzoquinone (BQ) complexation to form 45, (iv) C–C bond-forming reductive elimination to release 42 and Pd0, and (v) reoxidation of Pd0 with AgI to regenerate the catalyst (Scheme 16).33b The rate limiting step of this sequence changed from ii to iii as a function of the concentration of BQ. At low [BQ], step iii was rate determining; however, saturation kinetics were observed at high [BQ], suggesting that step ii becomes rate-limiting when BQ is abundant.
Scheme 16.

Proposed mechanism for oxidative coupling between benzo[h]quinoline and aryl–H.
We hypothesized that this change in rate determining step might result in a change in site selectivity, and this possibility was tested using 1,3-dimethoxybenzene as the arene.33c,34 With this substrate, functionalization of C–HA would provide product A, while that of C–HB would afford the isomeric compound B. We were pleased to find that the ratio of products A:B ratio changed from 16:1 to 1.1:1 (eq 16) upon moving from 0.2 equiv of BQ (where step iii is rate determining) to 20 equiv of BQ (where step ii is rate limiting).
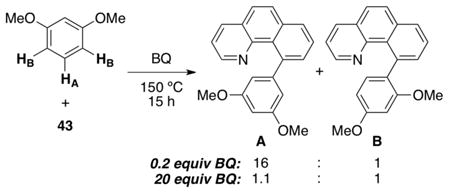 |
(16) |
These results suggest that Pd(OAc)2-mediated C–H activation of DMB (step ii) exhibits low selectivity for C–HA versus C–HB. In contrast, the BQ-promoted reductive elimination (step iii) appears to be highly selective for A. We believe that this is because BQ coordination to PdII aryl intermediate 44 is very sensitive to sterics.
Further exploration showed that a complete reversal in selectivity to favor isomer B could be achieved by simply changing the X-type ligand on Pd.33c When carboxylate was replaced with carbonate (eq 17), a strong preference for isomer B was observed (A:B = 1:11). Thus, it appears that although steric effects control the selectivity of arylation under conditions involving a carboxylate ligand, different factors dominate when X = carbonate. Ongoing studies are directed at elucidating the origin of the B selectivity in the carbonate system, but preliminary evidence suggests that the selectivity is not due to an electrophilic palladation step or a thermodynamically controlled deprotonation step.
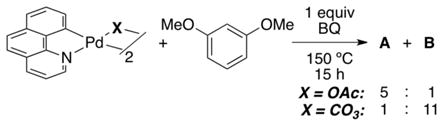 |
(17) |
Summary and Outlook
Substrate-based strategies provide extremely powerful methods for controlling the site selectivity of transition metal-catalyzed C–H bond functionalization and have been extensively explored over the past decade. In contrast, the ability to modulate the site selectivity of a Pd-catalyzed C-H functionalization reaction via variation of the catalyst structure/reaction conditions is an exciting emerging field. Along with the work described above, a number of recent literature reports reflect burgeoning activity in this area. For example, the Pd-catalyzed C–H arylation of N-acetylindoles has been demonstrated to occur selectively at C-3 in the presence of Cu(OAc)2, but at C-2 when AgOAc is used as the oxidant.35a C–H arylation of azine N-oxides has been demonstrated at both sp2 and sp3 sites; either site can be accessed selectively depending on the identity of added base.35b Finally, a highly para-selective C–H arylation is attributed to an [ArPdIV–F] catalytic species formed in the presence of F+ oxidants.35c All of these transformations exemplify the potential power of catalyst control for modulating both reactivity and site selectivity in Pd-catalyzed C–H functionalizations. We anticipate that this will be an area of tremendous growth and activity in the future.
Acknowledgments
This work was supported by the NIH (GM073836) and DOE (DE-FG02-08ER 15997).
Biographies
Sharon R. Neufeldt was born in Tucson, AZ in 1985. She received her B.S. degree in chemistry from Northern Arizona University, performing research in the laboratory of Professor Clinton F. Lane. She has been working toward her Ph.D. at the University of Michigan under the guidance of Professor Melanie Sanford since 2007, where she is studying ligand-directed Pd-catalyzed C–H and C=C functionalization reactions.
Prof. Melanie S. Sanford was born in New Bedford, MA in 1975. She received a B.S. and M.S. in chemistry from Yale University in 1996 before obtaining her Ph.D. at California Institute of Technology with Professor Robert Grubbs in 2001. She then completed post-doctoral studies at Princeton University working with Professor John Groves. In 2003, she joined the faculty of the University of Michigan where she is currently the Moses Gomberg Collegiate Professor of Chemistry as well as Arthur F. Thurnau Professor of Chemistry. Her research program focuses on the development of new catalysts and catalytic transformations for applications in the synthesis of useful organic molecules.
References
- 1.For examples, see: Henry PM. Palladium(II)-Catalyzed Aromatic Substitution. J Org Chem. 1971;36:1886–1890.Eberson L, Gomez-Gonzalez L. Palladium(II) Catalyzed Aromatic Acetoxylation II Nuclear Acetoxylation of Aromatic Compounds: A Reversal of the Usual Isomer Distribution Pattern in Aromatic Substitution. Acta Chem Scand. 1973;27:1249–1254.Stock LM, Tse K-t, Vorvick LJ, Walstrum SA. Palladium(II) Acetate Catalyzed Aromatic Substitution Reaction. J Org Chem. 1981;46:1757–1759.
- 2.(a) Kakiuchi F, Chatani N. Catalytic Methods for C–H Bond Functionalization: Application in Total Synthesis. Adv Synth Catal. 2003;345:1077–1101. [Google Scholar]; (b) Ritleng V, Sirlin C, Pfeffer M. Ru-, Rh-, and Pd-Catalyzed C–C Bond Formation Involving C–H Activation and Addition on Unsaturated Substrates: Reactions and Mechanistic Aspects. Chem Rev. 2002;102:1731–1769. doi: 10.1021/cr0104330. [DOI] [PubMed] [Google Scholar]; (c) Kakiuchi F, Murai S. Catalytic C–H/Olefin Coupling. Acc Chem Res. 2002;35:826–834. doi: 10.1021/ar960318p. [DOI] [PubMed] [Google Scholar]
- 3.Le Bras J, Muzart J. Intermolecular Dehydrogenative Heck Reactions. Chem Rev. 2011;111:1170–1214. doi: 10.1021/cr100209d. [DOI] [PubMed] [Google Scholar]
- 4.Yoneyama T, Crabtree RH. Pd(II) Catalyzed Acetoxylation of Arenes with Iodosyl Acetate. J Mol Catal A. 1996;108:35–40. [Google Scholar]
- 5.Reviews on cyclopalladation: Alacid E, Alonso DA, Botella L, Nájera C, Pacheca MC. Oxime Palladacycles Revisited: Stone-Stable Complexes Nonetheless Very Active Catalysts. Chem Rec. 2006;6:117–132. doi: 10.1002/tcr.20077.Dunina VV, Zalevskaya OA, Potapov VM. General Principles and Characteristics of Cyclopalladation Reactions. Russ Chem Rev. 1988;57:250–269.Ryabov AD. Mechanisms of Intramolecular Activation of C–H Bonds in Transition-Metal Complexes. Chem Rev. 1990;90:403–424.
- 6.Examples of stoichiometric oxygenation of palladacycles: Baldwin JE, Jones RH, Najera C, Yus M. Functionalisation of Unactivated Methyl Groups Through Cyclopalladation Reactions. Tetrahedron. 1985;41:699–711.Carr K, Saxton HM, Sutherland JK. The 4α-Demethylation of Lanostenone. J Chem Soc Perkin Trans. 1988;1:1599–1601.Valk J-M, Boersma J, van Koten G. Oxidation of Heteroleptic Diarylpalladium Complexes with tert-ButylHydroperoxide. Substituent Effects in Aromatic Oxidation Reactions. Organometallics. 1996;15:4366–4372.
- 7.(a) Dick AR, Hull KL, Sanford MS. A Highly Selective Catalytic Method for the Oxidative Functionalization of C–H Bonds. J Am Chem Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; (b) Desai LV, Hull KL, Sanford MS. Palladium-Catalyzed Oxygenation of Unactivated sp3 C–H Bonds. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; (c) Kalyani D, Sanford MS. Regioselectivity in Palladium-Catalyzed C–H Activation/Oxygenation Reactions. Org Lett. 2005;7:4149–4152. doi: 10.1021/ol051486x. [DOI] [PubMed] [Google Scholar]; (d) Desai LV, Malik HA, Sanford MS. Oxone as an Inexpensive Safe and Environmentally Benign Oxidant for C–H Bond Oxygenation. Org Lett. 2006;8:1141–1144. doi: 10.1021/ol0530272. [DOI] [PubMed] [Google Scholar]; (e) Kalberer EW, Whitfield SR, Sanford MS. Application of Recyclable, Polymer-Immobilized Iodine(III) Oxidants in Catalytic C–H Bond Functionalization. J Mol Catal A. 2006;251:108–113. [Google Scholar]; (f) Desai LV, Stowers KJ, Sanford MS. Insights into Directing Group Ability in Palladium-Catalyzed C–H Bond Functionalization. J Am Chem Soc. 2008;130:13285–13293. doi: 10.1021/ja8045519. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Neufeldt SR, Sanford MS. O-Acetyl Oximes as Transformable Directing Groups for Pd-Catalyzed C–H Functionalization. Org Lett. 2010;12:532–535. doi: 10.1021/ol902720d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For examples, see: Giri R, Liang J, Lei JG, Li JJ, Wang DH, Chen X, Naggar IC, Guo C, Foxman BM, Yu JQ. Pd-Catalyzed Stereoselective Oxidation of Methyl Groups by Inexpensive Oxidants under Mild Conditions: A Dual Role for Carboxylic Anhydrides in Catalytic C–H Bond Oxidation. Angew Chem Int Ed. 2005;44:7420–7424. doi: 10.1002/anie.200502767.Reddy BVS, Reddy LR, Corey EJ. Novel Acetoxylation and C-C Coupling Reactions at Unactivated Positions in α-Amino Acid Derivatives. Org Lett. 2006;8:3391–3394. doi: 10.1021/ol061389j.Wang GW, Yuan TT, Wu XL. Direct Ortho-Acetoxylation of Anilides via Palladium-Catalyzed sp2 C–H Bond Oxidative Activation. J Org Chem. 2008;73:4717–4720. doi: 10.1021/jo8003088.Zhang J, Khaskin E, Anderson NP, Zavalij PY, Vedernikov AN. Catalytic Aerobic Oxidation of Substituted 8-Methylquinolines in PdII-2,6-pyridinedicarboxylic Acid Systems. Chem Commun. 2008:3625–3627. doi: 10.1039/b803156h.Zhang YH, Yu J-Q. Pd(II)-Catalyzed Hydroxylation of Arenes with 1 Atm of O2 or Air. J Am Chem Soc. 2009;131:14654–14655. doi: 10.1021/ja907198n.
- 9.For discussions of the intermediacy of monomeric PdIV or closely related dimeric PdIII~PdIII/PdIV~PdIV intermediates in these reactions, see Dick AR, Kampf JW, Sanford MS. Platinum Model Studies for Palladium-Catalyzed Oxidative Functionalization of C–H Bonds. Organometallics. 2005;24:482–485.Racowski JR, Dick AR, Sanford MS. Detailed Study of C–O and C–C Bon-Forming Reductive Elimination from Stable C2N2O2-Ligated Palladium(IV) Complexes. J Am Chem Soc. 2009;131:10974–10983. doi: 10.1021/ja9014474.Deprez NR, Sanford MS. Synthetic and Mechanistic Studies of Pd-Catalyzed C–H Arylation with Diaryliodonium Salts: Evidence for a Bimetallic High Oxidation State Pd Intermediate. J Am Chem Soc. 2009;131:11234–11241. doi: 10.1021/ja904116k.Ye Y, Ball ND, Kampf JW, Sanford MS. Oxidation of a Cyclometalated Pd(II) Dimer with “CF3+”: Formation and Reactivity of a Catalytically Competent Monomeric Pd(IV) Aquo Complex. J Am Chem Soc. 2010;132:14682–14687. doi: 10.1021/ja107780w.Ariafard A, Hyland CJT, Canty AJ, Sharma M, Yates BF. Theoretical Investigation into the Mechanism of Reductive Elimination from Bimetallic Palladium Complexes. Inorg Chem. 2011;50:6449–6457. doi: 10.1021/ic102323s.Ariafard A, Hyland CJT, Canty AJ, Sharma M, Brookes NJ, Yates BF. Ligand Effects in Bimetallic High Oxidation State Palladium Systems. Inorg Chem. 2010;49:11249–11253. doi: 10.1021/ic1020912.Powers DC, Ritter T. Bimetallic Synergy in Oxidative Palladium Catalysis. Acc Chem Res. doi: 10.1021/ar2001974. Article ASAP.
- 10.Kalyani D, Dick AR, Anani WQ, Sanford MS. Scope and Selectivity in Palladium-Catalyzed Directed C–H Bond Halogenation Reactions. Tetrahedron. 2006;62:11483–11498. [Google Scholar]
- 11.Hull KL, Anani WQ, Sanford MS. Palladium-Catalyzed Fluorination of Carbon-Hydrogen Bonds. J Am Chem Soc. 2006;128:7134–7135. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]
- 12.For examples, see: Giri R, Chen X, Yu JQ. Palladium-Catalyzed Asymmetric Iodination of Unactivated C–H Bonds under Mild Conditions. Angew Chem Int Ed. 2005;44:2112–2115. doi: 10.1002/anie.200462884.Giri R, Chen X, Hao XS, Li JJ, Liang J, Fan ZP, Yu JQ. Catalytic and Stereoselective Iodination of Prochiral C–H Bonds. Tetrahedron: Asymmetry. 2005;16:3502–3505.Wan X, Ma Z, Li B, Zhang K, Cao S, Zhang S, Shi Z. Highly Selective C–H Functionalization/Halogenation of Acetanilide. J Am Chem Soc. 2006;128:7416–7417. doi: 10.1021/ja060232j.Mei TS, Giri R, Maugel N, Yu JQ. PdII-Catalyzed Monoselective ortho Halogenation of C–H Bonds Assisted by Counter Cations: A Complementary Method to Directed ortho Lithiation. Angew Chem Int Ed. 2008;47:5215–5219. doi: 10.1002/anie.200705613.Bedford RB, Engelhart JU, Haddow MF, Mitchell CJ. Solvent-Free Aromatic C–H Functionalisation/Halogenation Reactions. Dalton Trans. 2010;39:10464–10472. doi: 10.1039/c0dt00385a.
- 13.(a) Wang X, Mei TS, Yu JQ. Versatile Pd(OTf)2–2H2O-Catalyzed ortho Fluorination Using NMP as a Promoter. J Am Chem Soc. 2009;131:7520–7521. doi: 10.1021/ja901352k. [DOI] [PubMed] [Google Scholar]; (b) Chan KSL, Wasa M, Wang X, Yu JQ. Palladium(II)-Catalyzed Selective Monofluorination of Benzoic Acids Using a Practical Auxiliary: A Weak-Coordination Approach. Angew Chem Int Ed. 2011;50:9081–9084. doi: 10.1002/anie.201102985. [DOI] [PubMed] [Google Scholar]
- 14.Kalyani D, Deprez NR, Desai LV, Sanford MS. Oxidative C–H Activation/C–C Bond Forming Reactions: Synthetic Scope and Mechanistic Insights. J Am Chem Soc. 2005;127:7330–7331. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]
- 15.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. Room-Temperature C–H Arylation: Merger of Pd-Catalyzed C–H Functionalization and Visible-Light Photocatalysis. J Am Chem Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For recent reviews, see: Daugulis O, Do HQ, Shabashov D. Palladium and Copper-Catalyzed Arylation of Carbon–Hydrogen Bonds. Acc Chem Res. 2009;42:1074–1086. doi: 10.1021/ar9000058.Chen X, Engle KM, Wang DH, Yu JQ. Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality. Angew Chem Int Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273.McGlacken GP, Bateman LM. Recent Advances in Aryl–Aryl Bond Formation by Direct Arylation. Chem Soc Rev. 2009;38:2447–2464. doi: 10.1039/b805701j.
- 17.For examples, see: Li JJ, Giri R, Yu JQ. Remote C–H Bond Functionalization Reveals the Distance-Dependent Isotope Effect. Tetrahedron. 2008;64:6979–6987.Hennings DD, Iwasa S, Rawal VH. Anion-Accelerated Palladium-Catalyzed Intramolecular Coupling of Phenols with Aryl Halides. J Org Chem. 1997;62:2–3. doi: 10.1021/jo961876k.
- 18.Snieckus V. Directed Ortho Metalation. Tertiary Amide and O-Carbamate Directors in Synthetic Strategies for Polysubstituted Aromatics. Chem Rev. 1990;90:879–933. [Google Scholar]
- 19.(a) Yu WY, Sit WN, Lai KM, Zhou Z, Chan ASC. Palladium-Catalyzed Oxidative Ethoxycarbonylation of Aromatic C–H Bond with Diethyl Azodicarboxylate. J Am Chem Soc. 2008;130:3305–3307. doi: 10.1021/ja710555g. [DOI] [PubMed] [Google Scholar]; (b) Jia X, Zhang S, Wang W, Luo F, Cheng J. Palladium-Catalyzed Acylation of sp2 C-H Bond: Direct Access to Ketones from Aldehydes. Org Lett. 2009;11:3120–3123. doi: 10.1021/ol900934g. [DOI] [PubMed] [Google Scholar]
- 20.Thu H-Y, Yu W-Y, Che C-M. Intermolecular Amidation of Unactivated sp2 and sp3 C–H Bonds via Palladium-Catalyzed Cascade C–H Activation/Nitrene Insertion. J Am Chem Soc. 2006;128:9048–9049. doi: 10.1021/ja062856v. [DOI] [PubMed] [Google Scholar]
- 21.Zhao X, Dimitrijevíc E, Dong VM. Palladium-Catalyzed C–H Bond Functionalization with Arylsulfonyl Chlorides. J Am Chem Soc. 2009;131:3466–3467. doi: 10.1021/ja900200g. [DOI] [PubMed] [Google Scholar]
- 22.Lyons TW, Sanford MS. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newhouse TR, Baran PS. If C–H Bonds Could Talk – Selective C–H Bond Oxidation. Angew Chem Int Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Davies DL, Donald SMA, Macgregor SA. Computational Study of the Mechanism of Cyclometalation by Palladium Acetate. J Am Chem Soc. 2005;127:13754–13755. doi: 10.1021/ja052047w. [DOI] [PubMed] [Google Scholar]; (b) Gorelsky SI, Lapointe D, Fagnou K. Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation Across a Broad Range of Aromatic Substrates. J Am Chem Soc. 2008;130:10848–10849. doi: 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]
- 25.Stowers KJ, Fortner KC, Sanford MS. Aerobic Pd-Catalyzed sp3 C–H Olefination: A Route to Both N-Heterocyclic Scaffolds and Alkenes. J Am Chem Soc. 2011;133:6541–6544. doi: 10.1021/ja2015586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Seregin IV, Gevorgyan V. Direct Transition Metal-Catalyzed Functionalization of Heteroaromatic Compounds. Chem Soc Rev. 2007;36:1173–1193. doi: 10.1039/b606984n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beck EM, Gaunt MJ. Pd-Catalyzed C–H Bond Functionalization on the Indole and Pyrrole Nucleus. Top Curr Chem. 2009;292:85–121. doi: 10.1007/128_2009_15. [DOI] [PubMed] [Google Scholar]
- 27.Deprez NR, Kalyani D, Krause A, Sanford MS. Room Temperature Palladium-Catalyzed 2-Arylation of Indoles. J Am Chem Soc. 2006;128:4972–4973. doi: 10.1021/ja060809x. [DOI] [PubMed] [Google Scholar]
- 28.Wagner AM, Sanford MS. Palladium-Catalyzed C–H Arylation of 2,5-Substituted Pyrroles. Org Lett. 2011;13:288–291. doi: 10.1021/ol102734g. [DOI] [PubMed] [Google Scholar]
- 29.Kawai H, Kobayashi Y, Oi S, Inoue Y. Direct C–H Bond Arylation of Arenes with Aryltin Reagents Catalyzed by Palladium Complexes. Chem Commun. 2008:1464–1466. doi: 10.1039/b717251f. [DOI] [PubMed] [Google Scholar]
- 30.Hickman AJ, Sanford MS. Catalyst Control of Site Selectivity in the PdII/IV-Catalyzed Direct Arylation of Naphthalene. ACS Catal. 2011;1:170–174. [Google Scholar]
- 31.(a) Zhang YH, Shi BF, Yu JQ. Pd(II)-Catalyzed Olefination of Electron-Deficient Arenes Using 2,6-Dialkylpyridine Ligands. J Am Chem Soc. 2009;131:5072–5074. doi: 10.1021/ja900327e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Izawa Y, Stahl SS. Aerobic Oxidative Coupling of o-Xylene: Discovery of 2-Fluoropyridine as a Ligand to Support Selective Pd-Catalyzed C–H Functionalization. Adv Synth Catal. 2010;352:3223–3229. doi: 10.1002/adsc.201000771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emmert MH, Cook AK, Xie YJ. Remarkably High Reactivity of Pd(OAc)2/Pyridine Catalysts: Nondirected C–H Oxygenation of Arenes. Angew Chem Int Ed. 2011;50:9409–9412. doi: 10.1002/anie.201103327. [DOI] [PubMed] [Google Scholar]
- 33.(a) Hull KL, Sanford MS. Catalytic and Highly Regioselective Cross-Coupling of Aromatic C–H Substrates. J Am Chem Soc. 2007;129:11904–11905. doi: 10.1021/ja074395z. [DOI] [PubMed] [Google Scholar]; (b) Hull KL, Sanford MS. Mechanism of Benzoquinone-Promoted Palladium-Catalyzed Oxidative Cross-Coupling Reactions. J Am Chem Soc. 2009;131:9651–9653. doi: 10.1021/ja901952h. [DOI] [PubMed] [Google Scholar]; (c) Lyons TW, Hull KL, Sanford MS. Controlling Site Selectivity in Pd-Catalyzed Oxidative Cross-Coupling Reactions. J Am Chem Soc. 2011;133:4455–4464. doi: 10.1021/ja1097918. [DOI] [PubMed] [Google Scholar]
- 34.Because site selectivity on DMB is independent of the cyclometallation or oxidation steps of the catalytic cycle, we chose to simplify the reaction system by studying the stoichiometric arylation of cyclopalladated benzoquinoline complex 43.
- 35.(a) Potavathri S, Dumas AS, Dwight TA, Naumiec GR, Hammann JM, DeBoef B. Oxidant-Controlled Regioselectivity in the Oxidative Arylation ofN-Acetylindoles. Tetrahedron Lett. 2008;49:4050–4053. doi: 10.1016/j.tetlet.2008.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schipper DJ, Campeau LC, Fagnou K. Catalyst and Base Controlled Site-Selective sp2 and sp3 Direct Arylation of Azine. N-Oxides Tetrahedron. 2009;65:3155–3164. [Google Scholar]; (c) Wang X, Leow D, Yu JQ. Pd(II)-Catalyzed para-Selective C–H Arylation of Monosubstituted Arenes. J Am Chem Soc. 2011;133:13864–13867. doi: 10.1021/ja206572w. [DOI] [PMC free article] [PubMed] [Google Scholar]