

Abstract
Copy number variant (CNV) analysis was performed on renal cell carcinoma (RCC) specimens (chromophobe, clear cell, oncocytoma, papillary type 1, and papillary type 2) using high-resolution arrays (1.85 million probes). The RCC samples exhibited diverse genomic changes within and across tumor types, ranging from 106 to 2238 CNV segments in a clear-cell specimen and in a papillary type 2 specimen, respectively. Despite this heterogeneity, distinct CNV segments were common within each tumor classification: chromophobe (seven segments), clear cell (three segments), oncocytoma (nine segments), and papillary type 2 (two segments). Shared segments ranged from a 6.1-kb deletion (oncocytomas) to a 208.3-kb deletion (chromophobes). Among common tumor type–specific variations, chromophobes, clear-cell tumors, and oncocytomas were composed exclusively of noncoding DNA. No CNV regions were common to papillary type 1 specimens, although there were 12 amplifications and 12 deletions in five of six samples. Three microRNAs and 12 mRNA genes had a ≥98% coding region contained within CNV regions, including multiple gene families (chromophobe: amylases 1A, 1B, and 1C; oncocytoma: general transcription factors 2H2, 2B, 2C, and 2D). Gene deletions involved in histone modification and chromatin remodeling affected individual subtypes (clear cell: SFMBT and SETD2; papillary type 2: BAZ1A) and the collective RCC group (KDM4C). The genomic amplifications/deletions identified herein represent potential diagnostic and/or prognostic biomarkers.
The annual incidence of renal cell carcinoma (RCC) has increased steadily in the United States for the past three decades, with approximately 58,000 new cases diagnosed in 2010, representing 3% of all malignancies.1,2 Treatment of RCC is complicated by the fact that it is not a single disease but composes multiple tumor types with different morphological characteristics, clinical courses, and outcomes (ie, clear-cell carcinoma, 82% of RCC cases; type 1 or 2 papillary tumors, 11% of RCC cases; chromophobe tumors, 5% of RCC cases; and collecting duct carcinoma, approximately 1% of RCC cases).2,3 Benign renal neoplasms are subdivided into papillary adenoma, renal oncocytoma, and metanephric adenoma.2,3 Treatment of RCC often involves surgical resection of a large renal tissue component or removal of the entire affected kidney because of the relatively large size of renal tumors on discovery and the availability of a life-sustaining bilateral organ. Renal tumor classification provides predominantly postsurgical information pertinent to follow-up treatment (eg, tumor subtypes, such as papillary type 2, are prone to metastasis, whereas others, such as oncocytoma, are benign). Molecular assays could help classify RCC specimens that are difficult to identify using standard immunohistochemical (IHC) approaches and provide a basis for evaluation of biopsy specimens or fine-needle aspirates from kidney tumors before treatment.
The differential diagnosis of renal cancers relies largely on morphological and IHC analysis of tissue specimens. Recent advances in our understanding of the genomic basis of RCC have come from molecular analysis of hereditary forms, which account for 2% of renal cancer cases.4 The most common hereditary renal cancers are associated with the following: i) Von Hippel–Lindau (VHL) syndrome, usually caused by deletions in the VHL gene on chromosome 3p25-p26, which results in clear-cell tumors; ii) mutations in the MET proto-oncogene on chromosome 7q31, which results in papillary RCCs; iii) familial renal oncocytomas associated with Birt-Hogg-Dube (BHD) syndrome and the BHD gene on chromosome 17p11; and iv) hereditary leiomyomatosis and renal carcinomas with germline mutations in the fumarate hydratase gene on chromosome 1q42.1.5 In addition to these hereditary renal cancers, subjects with germline mutations in succinate dehydrogenase B are predisposed to RCC, whereas patients with tuberous sclerosis complex typically develop renal cancers at a much younger age than the general population.6,7 Molecular diagnostic techniques (cytogenetic karyotyping, fluorescence in situ hybridization, and array comparative genomic hybridization), applied to the genes associated with hereditary forms of RCC, have proved clinically useful for classification of renal tumors. Although sporadic clear-cell carcinomas typically have VHL mutations consistent with hereditary clear-cell forms, other sporadic RCCs often lack somatic mutations typical of hereditary forms, complicating the use of hereditary markers for classification of these spontaneous renal cancers.
The RCCs, both within and across tumor subclassifications, exhibit marked molecular heterogeneity, consistent with the theory that tumorigenesis requires an accumulation of mutations to undergo neoplastic transformation.8 This inherent genomic instability among sporadic tumors complicates the identification of diagnostic, tumor type–specific biomarkers and the search for underlying cancer-causing mutations. In a recent exome sequencing study, more than half of 101 renal clear-cell carcinoma cases contained somatic mutations within the VHL gene, which is an established hallmark of this type of tumor.9 Determination of robust RCC biomarkers for sporadic RCC will likely require interrogation of additional genomic sequences, including noncoding regions to identify tumor type–specific changes that can affect gene expression through modification of chromatin structure and/or alteration of transcriptional regulatory elements, including promoters, enhancers, activators, and repressors.
Oligonucleotide microarrays designed to interrogate single-nucleotide polymorphisms (SNPs) have proved clinically useful as an accessory tool for classification of challenging renal epithelial tumors, including specimens that have undergone formalin fixation and preservation in paraffin.10 Profiling the genome for novel and/or common genetic lesions was limited in these previous studies by both fragmentation of the DNA during fixation and the low density of genomic targets on the arrays (10,000 to 250,000). The current study was designed to evaluate copy number variations (CNVs) in canonical RCC tumors using high-mol.wt. DNA (>10 kb) from fresh specimens and high-resolution arrays (1,852,600 distinct targets) to characterize CNV for direct comparison with our in-house reference library of normal tissues generated using the same platform. We characterized five subtypes of renal cancers (chromophobe, clear cell, oncocytoma, and papillary types 1 and 2) for the distribution of CNV both within and across tumors spanning the entire genome for the presence of diagnostic biomarkers and rearrangements indicative of underlying mechanisms of tumorigenesis. Common tumor type–specific patterns of CNV were associated with four of the five tumor classifications, despite a background of immense CNV variation within each sample.
Materials and Methods
Tissue Samples
Tissue samples were obtained from the University of Pittsburgh Health Sciences Tissue Bank using an honest broker system and according to Institutional Review Board–approved protocol 970480. Samples were acquired as surgical specimens and flash frozen in a 1.8-mL cryotube (NalgeNunc, Inc., Rochester, NY), followed by immediate storage at −80°C. Each tumor sample (n = 27) was classified into one of five renal cancer subtypes (chromophobe, n = 5; clear cell, n = 5; oncocytoma, n = 5; papillary type 1, n = 6; papillary type 2, n = 6) by consensus evaluation of correlative H&E-stained slides performed independently by three anatomical pathologists (S.I.B., A.V.P., R.D.) (Figure 1). Chromophobe tumor cells exhibited a unique microvacuolated appearance, often with a microvesicular cytoplasmic pattern and prominent cell membranes (Figure 1A). Clear-cell specimens typically exhibited a clear cytoplasm, with a low nucleus to cytoplasm ratio and an alveolar pattern of growth (Figure 1B). Oncocytoma tumor cells had a large granular cytoplasm and formed clusters of small aggregates and tubules (Figure 1C). Papillary type 1 tumors consisted of small cuboidal cells, often forming a single line of uniform nuclei covering vascular papillae (Figure 1D), whereas papillary type 2 specimens contained pleomorphic nuclei, with prominent nucleoli in large cells with an eosinophilic cytoplasm (Figure 1E). The three pathologists also confirmed the absence of pathological features in adjacent normal renal samples (n = 9) (Figure 1F), and this normal reference group was expanded by inclusion of 14 normal thyroid samples and eight normal lung specimens. DNA from each of these specimens was analyzed using genotyping microarrays (SNP 6.0; Affymetrix, Sunnyvale, CA). Additional papillary type 2 specimens and matching blood samples were obtained from two patients for direct comparison of the results obtained by microarray analysis to whole genome paired-end sequencing performed on the SOLiD 4 system (sequencing by oligonucleotide ligation and detection; Life Technologies, Foster City, CA). The blood samples were collected by the University of Pittsburgh Health Sciences Tissue Bank in EDTA K2 tubes (Becton, Dickinson and Company, Franklin Lakes, NJ), followed by centrifugation (1500 × g for 10 minutes) for separation of the white blood cell layer. The plasma and buffy coat were serially collected and immediately stored at −80°C.
Figure 1.

Tumors with classic morphological characteristics from the five most common renal cell tumor subtypes were used to establish copy number profiles for each group: chromophobe (A), clear cell (B), oncocytoma (C), papillary type 1 (D), papillary type 2 (E), and normal adjacent (F). Specimens were chosen based on classification consensus by three independent pathologists (S.I.B., A.V.P., R.D.), combined with clinical outcomes data. Original magnification, ×20.
DNA Isolation, Hybridization, and Array Processing
Genomic DNA was purified from frozen samples, following the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). Briefly, 0.25 mg of frozen tissue was placed in Qiagen buffer and proteinase K (>600 mAU/mL) and incubated for 3 hours at 150 rpm. After tissue lysis and protein removal, genomic DNA was eluted in 100 μL of Buffer AE (Qiagen). Only samples with a spectrophotometric absorption ratio of 260/280 > 1.8 (Nanodrop ND-1000 spectrophotometer; NanoDrop, Wilmington, DE) were included in subsequent assays. Purified genomic DNA was evaluated using agarose gels (2%) or microfluidic chips (12,000 DNA Chip, Bioanalyzer 2100; Agilent Technologies, Santa Clara, CA) to confirm that the DNA from each specimen had a high mol.wt. (≥10 kb), with minimal detectable degradation. Samples were assayed following the Affymetrix Genome-Wide Human SNP Array 6.0 protocol (Affymetrix) comprising 1.8 million genetic markers with a similar distribution of SNPs (906,600) and nonpolymorphic probes (946,000). StyI and NspI restriction enzymes (New England Biolabs, Ipswich, MA) were used to separately digest 250 ng of DNA, resulting in a total digest of 500 ng of whole genomic DNA per sample. Each sample was ligated with the appropriate adaptor (Sty or Nsp) provided in the Affymetrix 6.0 protocol, and a total of seven PCRs were performed per sample (three StyI and four NspI restriction digest/ligation DNA reactions) and pooled into one tube, with a total volume of 700 μL. Purification of the resulting PCR products was performed using the protocol associated with the Affymetrix Cytogenetics Copy Number Assay, which required the use of Agencourt AMPure beads (Beckman Coulter, Danvers, MA). Each pooled sample was fragmented with DNAse1, biotinylated, and end labeled. Samples were subsequently hybridized on Genome-Wide SNP 6.0 arrays for 18 hours at 50°C with rotation (60 rpm) in an Affymetrix Gene Chip Hybridization Oven (model 640). Arrays were washed, stained, and scanned according to the Affymetrix Genome-Wide SNP 6.0 protocol using the Affymetrix GeneChip Fluidics Station (model 450), GeneChip Scanner 3000 7G, and GeneChip Command Console software version 3.0.2 (Affymetrix, Sunnyvale, CA). The entire study data set has been deposited under accession number GSE34676 at the Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo, last accessed December 22, 2011).
Paired-End Sequencing
DNA was purified from matched patient tumor and blood specimens using the Qiagen Genomic Tip protocol (Genomic-tip 100/G). Briefly, cells were lysed and DNA was bound to anion exchange resin under low salt and pH conditions. Impurities were eluted, followed by DNA precipitation and suspension in low Tris-EDTA (10 mmol/L Tris and 0.1 mmol/L EDTA, pH 8.0). Fragment libraries were generated according to the manufacturer's instructions (SOLiD 4 System Library Preparation Guide, part 4445673; Applied Biosystems, Foster City, CA) using the SOLiD Fragment Library Construction Kit with 2% Size Select E-gels (Invitrogen Corp, Carlsbad, CA) and SOLiD Fragment Library Oligos Kit (Applied Biosystems, Carlsbad). Quantification was performed with the SOLiD Library TaqMan Quantitation Kit (Invitrogen Corp).
In short, 5 μg of genomic DNA from each sample was sheared to a peak size distribution approximating 150- to 180-bp fragments (Covaris S2; Covaris Inc., Woburn, MA). The DNA fragments underwent end repair and ligation with double-stranded P1 and P2 adaptors, followed by size selection using the E-Gel iBase Power System and the E-Gel Safe Imager Real-time Transilluminator (Invitrogen Corp). DNA was collected, pooled, and purified, and sizes were confirmed to be in a range of 200 to 250 bp on the BioAnalyzer 2100 with the DNA 1000 chip kit (Agilent Technologies). The library was amplified and nick translated (GeneAmp PCR System 9700; Applied Biosystems) using the Platinum PCR Amplification Mix (SOLiD Fragment Library Construction Kit; Life Technologies). The DNA was purified, and both samples and quantitative PCR standards were run in triplicate in the 7900HT Fast Real-Time PCR System (Applied Biosystems), including nontemplate controls (40 cycles at 95°C for 2 minutes, 95°C for 15 seconds, and 60°C for 1 minute), to determine library concentration. Based on the quantitative PCR results for each library, we used the SOLiD EZ bead system (Life Technologies) to generate beads with attached templates following the manufacturer's protocol (Quick Reference Cards; Life Technologies). A PCR emulsion was generated in the SOLiD EZ bead Emulsifier, followed by PCR amplification in the EZ bead PCR Amplifier using the E80 protocol. Beads were collected in the EZ Bead Enricher, and the concentration of P2-positive beads was determined on the Nanodrop spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE).
A work flow analysis was performed on the SOLiD 4 system to determine bead quality and optimal dilution for whole genome sequencing. Beads were deposited at low density (20 × 106 beads). The work flow analysis slide containing one quadrant with beads was placed on the flow cell chamber, and a single strip of sequencing ligation enzymes (SOLiD ToP Workflow Analysis Reagents) was added to the chilling block. Sequencing proceeded through one ligation to generate a focal map and determine quality metrics (P2 to P1 ratio and noise to signal ratio) and median number of P2-positive beads per panel. Based on the results (noise to signal ratio ≤6%, 90 < P2/P1 < 100), an entire slide was loaded and paired-end sequencing was performed by oligonucleotide ligation and detection of clonally amplified DNA fragments linked to beads. Dye-labeled probes were sequentially ligated, interrogating a two-base position at a time. Images were captured corresponding to unique emission spectra for each of the four different dyes. Forward sequencing covered 50 bp from the P1 adaptor end, and reverse sequencing covered 35 bp from the P2 adaptor end, of the DNA template. Images of the 2357 panels in each of four channels per cycle were saved for analysis.
Data Analysis
The Affymetrix Genotyping Console (version 3.0.2) was used to generate an SNP call for each probe set on each array. SNP calls were determined for an average of 96.78% (SD, 4.27%) of the array probe sets per specimen throughout the study. Affymetrix CEL and CHP files were transferred to Partek Genomics Suite version 6.5 (Partek Inc., St Louis, MO) for copy number and loss-of-heterozygosity (LOH) analysis, respectively. The CEL files were imported using the default setting, which adjusts hybridized intensities based on fragment length and probe sequence. Copy number was generated by comparing the hybridized intensities of each array with the laboratory normal tissue reference set (n = 31), eliminating interlaboratory variability introduced by comparison to other databases, such as the HapMap.11 Copy number measurements were smoothed based on local guanine-cytosine content using a 1-megabase window.12 The Partek segmentation algorithm was used to identify regions of significant CNV from normal, consisting of a minimum of 30 genomic markers and P < 0.001, with signal to noise and expected range set to 0.3. The median length of intermarker spacing on the SNP 6.0 array is 680 bp, with a median length of a 30-marker region approximately 20 kb. Regions of LOH were identified for tumors with a matched normal pair (two chromophobe, one clear-cell, one papillary type 1, and one papillary type 2 sample) using the Hidden Markov Model in Partek Genomics Suite.
The de novo paired-end papillary type 2 specimens and blood whole genome sequences were mapped against the human reference sequence (HG18) using the Mapping program of Bioscope software version 1.3 (Life Technologies). These files were entered into the Pairing program to generate binary sequence alignment map files for tertiary analysis. The binary sequence alignment map files were interrogated for global and local CNV for correlation with the array results. A direct comparison of global and local CNV data for tumor versus blood was obtained using a Perl script. A text file was generated from the comparison of the two general feature format files, which was loaded into Partek for tertiary comparisons.
Results
CNV in RCC Tumor Subclassifications
There were pronounced disparities in the number of CNV regions identified as statistically different from normal in each of the tumor samples. For example, only 106 CNV regions were detected in one clear-cell specimen, whereas there were 2238 CNV segments in a papillary type 2 specimen [identification (ID) numbers 513 and 532; Table 1]. This variability was present both within and across tumor subclassifications. The greatest amount of global CNV was present in chromophobe tumors, with an average of 23.5% of the chromophobe genome exhibiting significant copy number differences compared with normal specimens (Figure 2). In contrast, oncocytomas had the smallest amount of their genome affected by copy number changes, with an average of 8.4% of their genome reflecting significant changes. Clear-cell tumors had the largest global copy number losses, with an average of 14.4% of their genome containing deletions, whereas papillary type 1 tumors had the greatest amount of genomic copy number gains (ie, 12.3% of the genome). Papillary type 1 tumors were also the only tumors to have a larger amount of copy number amplification than deletion. Detailed information about the number and length of CNV segments is available in Table 1.
Table 1.
CNVs of the RCC Sample Types
| Group | ID no. | No. of amplified segments | No. of deleted segments | Genome CN gain (%) | Genome CN loss (%) | Minimum length (bases) | Maximum length (megabases) |
|---|---|---|---|---|---|---|---|
| Chromophobe | 508 | 174 | 441 | 3.88 | 15.00 | 4742 | 34.3 |
| 509 | 1011 | 532 | 39.91 | 7.29 | 4803 | 16.6 | |
| 510 | 42 | 128 | 0.24 | 1.83 | 6735 | 21.7 | |
| 511 | 64 | 139 | 0.49 | 0.26 | 4686 | 2.0 | |
| 512 | 149 | 367 | 5.42 | 43.30 | 6257 | 47.9 | |
| Mean | 288 | 321 | 9.99 | 13.54 | 5445 | 24.5 | |
| SD | 408 | 181 | 16.87 | 17.61 | 975 | 17.5 | |
| Clear cell | 513 | 17 | 89 | 0.08 | 11.28 | 6894 | 65.4 |
| 514 | 104 | 155 | 3.01 | 2.55 | 5845 | 25.2 | |
| 516 | 109 | 288 | 9.98 | 17.27 | 4246 | 54.7 | |
| 517 | 43 | 163 | 0.23 | 38.02 | 4686 | 122.6 | |
| 230 | 93 | 103 | 0.63 | 2.78 | 6975 | 59.3 | |
| Mean | 73 | 160 | 2.78 | 14.38 | 5729 | 65.4 | |
| SD | 41 | 79 | 4.19 | 14.59 | 1246 | 35.5 | |
| Oncocytoma | 524 | 73 | 148 | 0.24 | 0.68 | 4782 | 3.3 |
| 525 | 81 | 357 | 1.12 | 12.83 | 2935 | 54.7 | |
| 526 | 74 | 361 | 0.37 | 3.35 | 3110 | 20.3 | |
| 527 | 81 | 440 | 0.39 | 9.61 | 5401 | 12.0 | |
| 221 | 49 | 171 | 0.79 | 12.62 | 2908 | 87.9 | |
| Mean | 72 | 295 | 0.58 | 7.82 | 3827 | 35.6 | |
| SD | 13 | 129 | 0.36 | 5.53 | 1177 | 35.1 | |
| Papillary type 1 | 528 | 242 | 354 | 16.22 | 5.40 | 6453 | 54.7 |
| 529 | 152 | 181 | 16.98 | 0.72 | 8103 | 42.9 | |
| 531 | 485 | 645 | 24.22 | 6.91 | 2935 | 54.7 | |
| 222 | 54 | 114 | 1.16 | 0.47 | 7360 | 25.4 | |
| 223 | 113 | 232 | 3.92 | 7.94 | 2929 | 45.1 | |
| 224 | 157 | 308 | 11.52 | 21.43 | 2957 | 44.0 | |
| Mean | 201 | 306 | 12.33 | 7.14 | 5123 | 44.5 | |
| SD | 152 | 187 | 8.65 | 7.67 | 2447 | 10.7 | |
| Papillary type 2 | 530 | 32 | 113 | 0.07 | 1.21 | 9971 | 21.7 |
| 532 | 739 | 1499 | 13.84 | 17.68 | 5196 | 54.7 | |
| 226 | 80 | 148 | 7.93 | 5.83 | 3122 | 80.1 | |
| 227 | 207 | 464 | 6.66 | 20.05 | 5616 | 23.9 | |
| 228 | 235 | 317 | 15.90 | 6.43 | 6943 | 30.6 | |
| 229 | 125 | 249 | 5.32 | 6.38 | 2494 | 27.4 | |
| Mean | 236 | 465 | 8.29 | 9.60 | 5557 | 39.7 | |
| SD | 258 | 522 | 5.79 | 7.47 | 2715 | 23.1 | |
| Group ANOVA | P value | 0.45 | 0.53 | 0.55 | 0.96 | 0.54 | 0.18 |
Data include amplifications and deletions, percentage of the genome with CNV amplifications and deletions, and smallest and largest CNV segments in each sample. None of the P values (bottom row) obtained by ANOVA for the effect of cancer subtype on the value in each column indicate the presence of a statistically significant difference.
ANOVA, analysis of variance.
Figure 2.

The percentage of the genome affected by CNV for chromophobe (Chromo.; n = 5), clear cell (n = 5), oncocytoma (Onco.; n = 5), papillary type (Pap.) 1 (n = 6), and Pap. 2 (n = 6) subtypes. The total represents the average of copy number segments obtained within a classification. The error bars represent the SEM. The X and Y chromosomes were excluded because of sex imbalance, and the percentages were calculated using 3071.36 megabases as the size of the whole genome. There were no statistically significant differences among the groups for total, deleted, or amplified segments.
CNV Segments Common to RCC
The segmentation results were evaluated for the presence of CNV regions shared across all tumor subtypes but not present in normal specimens. The most common CNV amplifications in renal cancers were shared in 20 (74%) of 27 tumor samples composing two distinct segments, including one on chromosome 7 (58798621 to 158819766) and a second within the mitochondrial genome (MT 10101 to 16150). Reducing the stringency to commonality within 19 samples yielded five CNV segments shared within the entire tumor cohort. The most frequently shared deletions were detected as two segments on chromosome 9 (6685744 to 6703681 and 6714105 to 6733969) that were deleted in 19 (70%) of 27 tumors. A list is given of common segments shared among RCC samples with decreasing levels of stringency (see Supplemental Table S1 at http://ajp.amjpathol.org).
CNV Segments Specific to Individual Tumor Subclassifications
Individual tumor subclassifications were evaluated for the presence of significant CNV segments common to all members within that specific group. Chromophobe samples exhibited an overall total of 1440 amplifications (mean ± SD = 288 ± 408) and 1607 deletions (mean ± SD = 321 ± 181) among the five specimens represented in this group (Table 1; Figure 3A). Chromosomes 1, 10, and 19 had the greatest amount of CNV among chromophobe samples; each sample had CN losses covering 44.8% ± 49.9% (mean ± SD) of chromosome 1, a 43.5% ± 51.9% loss of chromosome 10 and a 31.7% ± 41.6% loss of chromosome 19. However, four regions exhibited common, statistically significant CN deletions in every chromophobe sample tested: a 38.7-kb region on chromosome 1 (1p21.1), a 26.3-kb region on chromosome 6 (6q14.1), and two regions on chromosome 10, including a 208.3-kb region (10p12.33) and a 6.8-kb region (10q22.3) (Figure 4). Comparing the LOH regions found in the two paired chromophobe samples (ID numbers 509 and 512) with the copy number regions yielded three additional regions that exhibited events in 100% of the samples. These three regions include a 15.6-kb region on chromosome 1 (1p36.13), a 30-kb region on chromosome 2 (2p16.3), and a 17-kb region on chromosome 11 (11p15.1) (Figure 4).
Figure 3.

The location and frequency of CNVs in samples: chromophobe (A), clear cell (B), oncocytoma (C), papillary type 1 (D), and papillary type 2 (E). The abscissa is divided into chromosomes delineated horizontally in line with the p arm of the chromosome to the left and the q arm to the right. Copy number gains (amplifications; red) are indicated by positive values. Copy number losses (deletions; blue) are indicated by negative values, which correspond to the percentage of samples containing the CNV. Chromosomes are highlighted yellow if the average values within each subclassification contain significant CNVs comprising at least 30% of the chromosome.
Figure 4.

Chromophobe samples shared four deleted regions: a 38.7-kb region in 1p21.1 (1: 104066237 to 104104975), a 26.3-kb region in 6q14.1 (6: 77494919 to 77521221), a 208.3-kb region in 10p12.33 (10: 17792529 to 18000842), and a 6.8-kb region in 10q22.3 (10: 77924962 to 77931730). Chromophobe samples have three additional regions that contain CN events in four of five samples and a region of copy-neutral LOH in the remaining sample. The three regions include the following: a 15.6-kb region in 1p36.13 (1: 17119213 to 17134826), a 30-kb region in 2p16.3 (2: 52612264 o 52638249), and a 17-kb region in 11p15.1 (11: 18901547 to 18918504). The numbers at the top of each lane are sample ID numbers consistent with Tables 1 and 2. The black window demarcated on each chromosome (Chr.) indicates the area expanded to the right. The black window located within the expanded region indicates the location of a CNV common to all samples in the tumor type.
Clear-cell tumor samples had the fewest number of CNV regions detected among each of the tumor subtypes, with a total of 366 CN amplifications (mean ± SD = 73 ± 41) and 798 deletions (mean ± SD = 160 ± 79) (Table 1; Figure 3B). Typical variant segments in the clear-cell specimens were among the longest and most contiguous CNV regions encountered throughout the study (Table 1). Clear-cell samples also exhibited vast differences in genotype among specimens, despite their common phenotype. For example, there were as few as two small deletions present on the short arm of chromosome 3 in one specimen, whereas nearly the entire short arm region was deleted in all four of the other specimens (Figure 5). Chromosomes 6, 3, and 10 contained the greatest amount of CNV among clear-cell samples; each sample had CN losses covering 40.4% ± 54.1% of chromosome 6, 35.8% ± 23.9% of chromosome 3, and 34.1% ± 47.4% of chromosome 10 (Figure 3B). There were three common CN deletion regions on chromosome 3 in every clear-cell sample, consisting of a 12.5-kb region in 3p26.1, a 57.1-kb region in 3p23, and a 13.7-kb region in 3p21.1 (Figure 5).
Figure 5.
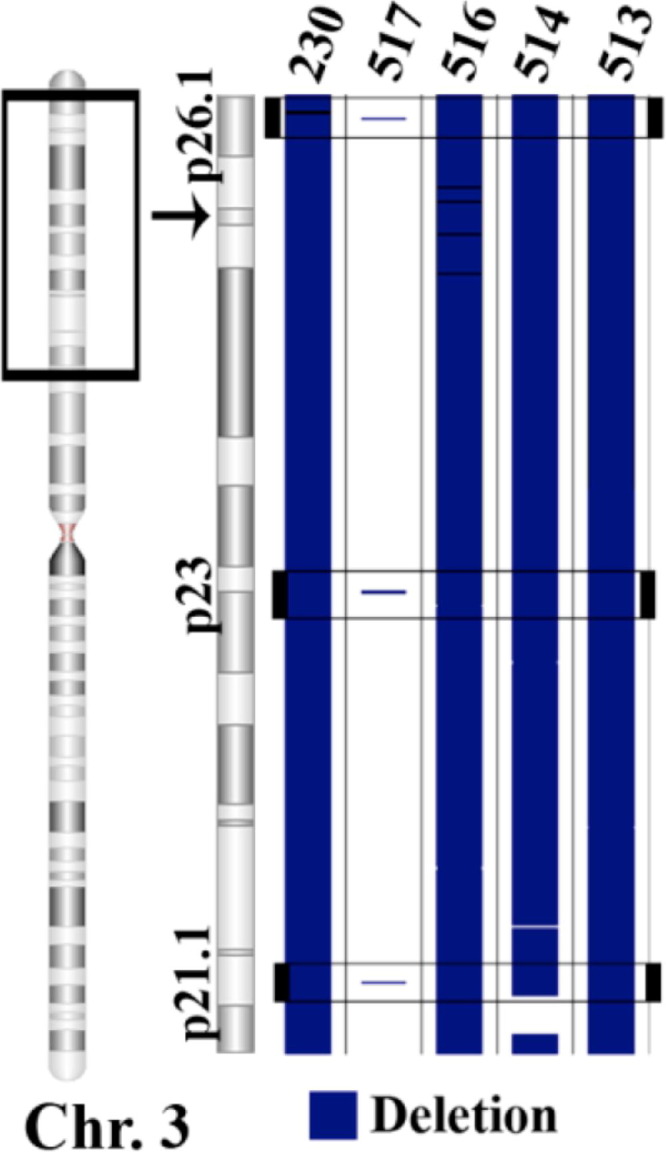
All clear-cell samples shared three deleted regions: a 12.5-kb region in 3p26.1 (3: 6617676 to 6630163), a 57.1-kb region in 3p23 (3: 32029310 to 32086396), and a 13.7-kb region in 3p21.1 (3: 53000833 to 53014512). The black window demarcated on the chromosome (Chr.) indicates the area expanded to the right. The black window located within the expanded region indicates the location of a CNV common to all samples in the tumor type.
Oncocytoma specimens had a total of 358 CN amplifications and an average mean ± SD = 72 ± 13 is comparable to clear-cell specimens (Table 1; Figure 3C). The total number of deletions in oncocytoma specimens was 1477 (mean ± SD = 295 ± 129) (Figure 3C). The greatest amount of CNV among oncocytoma specimens occurred on chromosome 1: each oncocytoma sample had CN losses that totaled 49.7% ± 48.6% (mean ± SD) of the entire length of chromosome 1 (Figure 3C). There were three regions that were amplified and six regions that were deleted in every oncocytoma sample (Figure 6). The three amplified regions included two regions of chromosome 3 comprising a 6.1-kb region and a 25.8-kb region (both in 3q29). The third common amplification was 98.9 kb, found on chromosome 22 at 22q13.33. The six deletions consisted of three regions on chromosome 1, including a 49.6-kb region in 1p31.3, a 70-kb region in 1q25.2, and a 43.5-kb region in 1q44. Additional CN deletions included a 114.3-kb region in 5q13.2 of chromosome 5, a 14.9-kb region in 10q21.3 of chromosome 10, and a 9.9-kb region in 13q12.2 of chromosome 13 (Figure 6).
Figure 6.

Oncocytoma samples shared six deleted regions: a 49.6-kb region in 1p31.3 (1: 62139703 to 62189290), a 70-kb region in 1q25.2 (1: 177623986 to 177693964), a 43.5-kb region in 1q44 (1: 244911496 to 244955034), a 114.3-kb region in 5q13.2 (5: 68892554 to 69006891), a 14.9-kb region in 10q21.3 (10: 66977949 to 66992866), and a 9.9-kb region in 13q12.2 (13: 27526162 to 27536024). There were also three shared amplifications: a 6.1-kb region in 3q29 (3: 194363224 to 194369329), a 25.8-kb region in 3q29 (3: 196911130 to 196936886), and a 98.9-kb region in 22q13.33 (22: 49482441 to 49581322). The black window demarcated on each chromosome (Chr.) indicates the area expanded to the right. The black window located within the expanded region indicates the location of a CNV common to all samples in the tumor type.
The six papillary type 1 samples had a total of 1203 amplified segments (mean ± SD = 201 ± 152) and 1834 deleted segments (mean ± SD = 306 ± 187) (Table 1; Figure 3D). The greatest amount of CNV between papillary type 1 samples was found on chromosomes 7, 9, 12, 16, and 17. For each sample, CN gains composed 54.9% ± 45.4% (mean ± SD) of the length of chromosome 17, 46.1% ± 50.3% of the length of chromosome 7, 35.5% ± 39.6% of the length of chromosome 16, and 34.2% ± 43.4% of the length of chromosome 12. Copy number losses totaled 36.5% ± 45.9% of the length of chromosome 9 (Figure 3D). Unlike the other tumor types, no CNV regions were identified in common to all papillary type 1 samples, although there were 12 regions that were amplified and 12 regions that were deleted in five of six samples.
Papillary type 2 samples had 1418 amplifications (mean ± SD = 236 ± 258) and 2790 deletions, yielding, by far, the highest average number (mean ± SD = 465 ± 522) of CNV events (Table 1 and Figure 3E). The largest amount of CNV between papillary type 2 samples was found on chromosomes 7, 19, and 17; each sample had CN gains that covered 39.0% ± 42.1% (mean ± SD) of the length of chromosome 7 and 29.4% ± 41.9% of the length of chromosome 17. Each sample also had CN losses that covered 29.6% ± 29.6% of the length of chromosome 19 (Figure 3E). In contrast to the papillary type 1 subtype, there were two regions with CNV deletions common to all six of papillary type 2 samples. These included a 19.9-kb region on chromosome 9 (9p24.1) and a 52-kb deletion on chromosome 14 (14q13.2) (Figure 7).
Figure 7.

Papillary type 2 samples shared two deleted regions among all samples: a 19.9-kb region in 9p24.1 (9: 6714105 to 6733969) and a 52-kb region in 14q13.2 (14: 34360232 to 34412171). The black window demarcated on each chromosome (Chr.) indicates the area expanded to the right. The black window located within the expanded region indicates the location of a CNV common to all samples in the tumor type.
CNV Analysis by Paired-End Sequencing
Two additional papillary type 2 specimens were obtained for post hoc SNP array analysis and paired-end genomic sequencing to validate the presence of deletions identified on chromosomes 9 (19.9-kb region) and 14 (52-kb region), in common among all previous papillary type 2 specimens. SNP 6.0 array analysis confirmed the presence of CNV deletions encompassing and extending beyond these regions compared with our reference database of normal samples. Genomic sequencing provided maps of chromosomes 9 and 14 for both blood and tumor specimens, achieving an average resolution for each base call ranging from 3.3 to 5.7 times in these regions. Areas of CNV in the sequencing data were derived by direct comparison of copy number values in tumor specimens with blood samples compiled across each entire chromosome. The two chromosomal regions containing deletions common to all papillary type 2 specimens in the array analysis were identified in these additional specimens via SNP arrays and were verified by genomic sequencing of chromosomes 9 and 14 (Figure 8). Not surprisingly, there were differences in the resolution of the CNV results when SNP array segmentation and paired-end sequencing were directly compared because of the high resolution of sequencing technology (single-base calls versus 680–base marker array spacing between SNP probes) and direct comparison of tumor to blood CNV ratios after sequencing versus CNV calculation from SNP data compared with a pool of normal tissue samples as a reference database.
Figure 8.

The rows labeled SEQUENCE and SNP Array indicate copy number deletions (blue) and amplifications (red) detected in the identical papillary type 2 tumor sample via paired-end genomic sequencing and SNP 6.0 array analysis (see Materials and Methods for details). The regions depicted correlate with the locations indicated below each plot on chromosome (Chr.) 9 or 14. The copy number plot provides copy number calculations for each marker obtained from the comparison of the SNP data to the reference database of normal samples, with blank regions indicative of the absence of probes.
Genes Located within Common RCC CNV Regions
The total number of genes for mRNAs and microRNAs (miRNAs) encoded within CNV regions ranged from as few as 190 unique mRNA coding regions and one miRNA in a papillary type 1 sample (ID number 222) to as many as 12,542 mRNAs and 348 miRNAs in a single chromophobe sample (ID number 512) (Table 2). We evaluated the genes contained within common CNV regions for the overall classification of RCC, as well as associated with a specific type of tumor (Figure 8). The most commonly amplified genes among all RCC samples [ie, amplified in 17 (63%) of the 27 tumor samples] were as follows: i) rhomboid family member 1 (RHBDF1), ii) DNA-directed RNA polymerase III subunit K (POLR3K), iii) small nuclear ribonucleoprotein 25kDa (SNRNP25), and iv) Wiskott Aldrich Syndrome protein family homolog 1 pseudogene (LOC100288778). The gene most commonly contained within a CNV deletion was lysine-specific demethylase 4C (KDM4C), which was deleted in 19 (70%) of 27 samples. KDM4C is alternatively known as GASC1 or the gene amplified in squamous cell carcinoma 1 because of its overexpression in several esophageal squamous cell carcinomas.13 KDM4C was the only gene with an altered copy number state in at least 50% of the samples in each subtype (Figure 9; see also Supplemental Figure S1 and Supplemental Table S2 at http://ajp.amjpathol.org).
Table 2.
mRNAs and miRNAs Mapped to CNV Segments Detected in Each Tumor Sample Using the NCBI Reference Sequence Transcript Database
| Group | Sample ID no. | mRNA amplified | mRNA deleted | miRNA amplified | miRNA deleted |
|---|---|---|---|---|---|
| Chromophobe | 508 | 616 | 3467 | 28 | 125 |
| 509 | 8920 | 3169 | 258 | 73 | |
| 510 | 91 | 124 | 1 | 4 | |
| 511 | 175 | 124 | 1 | 2 | |
| 512 | 2415 | 10,127 | 51 | 297 | |
| Clear cell | 513 | 21 | 2073 | 0 | 40 |
| 514 | 722 | 714 | 17 | 24 | |
| 516 | 1967 | 3311 | 48 | 131 | |
| 517 | 45 | 7886 | 0 | 193 | |
| 230 | 453 | 646 | 22 | 14 | |
| Oncocytoma | 524 | 116 | 183 | 1 | 4 |
| 525 | 416 | 3398 | 10 | 77 | |
| 526 | 152 | 1021 | 4 | 16 | |
| 527 | 135 | 2428 | 3 | 97 | |
| 221 | 88 | 3168 | 1 | 113 | |
| Papillary type 1 | 528 | 4174 | 1471 | 99 | 31 |
| 529 | 3909 | 309 | 93 | 9 | |
| 531 | 4931 | 2368 | 115 | 109 | |
| 222 | 94 | 96 | 1 | 0 | |
| 223 | 1352 | 1774 | 27 | 101 | |
| 224 | 3763 | 4773 | 142 | 124 | |
| Papillary type 2 | 530 | 35 | 165 | 0 | 4 |
| 532 | 3257 | 6226 | 93 | 171 | |
| 226 | 1577 | 1269 | 46 | 73 | |
| 227 | 2215 | 4873 | 106 | 148 | |
| 228 | 4322 | 2206 | 115 | 47 | |
| 229 | 2026 | 1492 | 58 | 55 |
Numbers of mRNAs and miRNAs in CNV segments are separated into the number amplified and the number deleted.
Figure 9.

The lists of mRNAs and miRNAs for each subtype were compared using a Venn diagram. The total number of mRNAs and miRNAs located within CNV segments in at least 60% of the samples in a subtype are provided in parentheses, next to the name of the corresponding tumor subtype. The number contained within the outer circle for a specific subtype indicates genes found in at least 60% of the samples for that group. Overlapping areas represent the number of mRNAs or miRNAs found in at least 60% of the samples of more than one subtype. There was only one gene, KDM4C, affected by CNV in at least 60% of the samples of all subtypes. (For annotation of individual gene transcripts expressed within each area of overlap, please see Supplemental Figure S1 and Supplemental Table S2 at http://ajp.amjpathol.org).
Genes in CNV Regions Associated with Subclassification of Renal Tumors
CNV segments shared within an individual tumor classification were curated for the presence of protein-encoding and miRNA genes. All chromophobe specimens had eight genes that were entirely lost because of CN deletions, including amylases α 1A, 1B, and 1C (AMY1A, AMY1B, and AMY1C, respectively) on chromosome 1 (Table 3) and the mannose receptor C-type gene (MRC1), the mannose receptor C-type like gene (MRC1L1), and family with sequence similarity 23, member A (FAM23A), located on chromosome 10 within the 10p12.33 CN deletion. A small part of the signal-transducing adapter molecule 1 (STAM) gene (7.5%) was also contained in this chromosome 10 CN deletion, along with the entire coding region for miRNAs 511–1 and 511–2. Down-regulation of miR-511-1 has been previously reported in glioblastoma tumors.14 The evaluation of regions of copy-neutral LOH yielded two additional genes, MAS-related GPR, member X1 (MRGPRX1), and ciliary rootlet coiled-coil, rootletin (CROCC), that were contained within regions affected in 100% of chromophobe samples.
Table 3.
Genes Located Within CNV Segments in 100% of the Samples
| Group | Cytoband | Region length | CN status | Genes within the region⁎ |
|---|---|---|---|---|
| Chromophobe | 1p21.1 | 38,738 | Deletion | AMY1A (100%), AMY1B (100%), AMY1C (100%) |
| 1p36.13 | 15,613 | Deletion, LOH | CROCC (27%) | |
| 2p16.3 | 25,985 | Deletion, LOH | ||
| 6q14.1 | 26,302 | Deletion | ||
| 10p12.33 | 208,313 | Deletion | STAM (7.5%), FAM23A (100%), MRC1 (100%), MRC1L1 (100%), MIR511-1 (100%), MIR511-2 (100%) | |
| 10q22.3 | 6768 | Deletion | ||
| 11p15.1 | 16,957 | Amplification, LOH | MRGPRX1 (100%) | |
| Clear cell | 3p26.1 | 12,487 | Deletion | |
| 3p23 | 57,086 | Deletion | ZNF860 (40%) | |
| 3p21.1 | 13,679 | Deletion | SFMBT1 (29%) | |
| Oncocytoma | 1p31.3 | 49,587 | Deletion | INADL (12%) |
| 1q25.2 | 69,978 | Deletion | ||
| 1q44 | 43,538 | Deletion | SCCPDH (2%) | |
| 3q29 | 6105 | Amplification | ||
| 3q29 | 25,756 | Amplification | MIR570 (100%), MUC20 (22%) | |
| 5q13.2 | 114,337 | Deletion | GTF2H2 (98%), GTF2H2B (98%), GTF2H2C (98%), GTF2H2D (98%), GUSBP3 (50%) | |
| 10q21.3 | 14,917 | Deletion | ||
| 13q12.2 | 9862 | Deletion | FLT3 (10%) | |
| 22q13.33 | 98,881 | Amplification | RABL2B (100%), SHANK3 (62%), ACR (100%), RPL23AP82 (77%) | |
| Papillary type 2 | 9p24.1 | 19,864 | Deletion | KDM4C (6%) |
| 14q13.2 | 51,939 | Deletion | BAZ1A (85%) |
The percentages in parentheses represent the amount of the gene located in the CNV region. Noncoding regions are blank.
Clear-cell samples had three CNV deletions on chromosome 3 containing 40% of the zinc finger protein 860 (ZNF860) in the 3p23 deletion and 29% of the gene Scm-like with four mbt domains 1 (SFMBT1) in the 3p21.1 CNV region (Table 3). The SFMBT1 gene shares marked similarity with the Drosophila Scm (sex comb on midleg) gene, which encodes a protein containing four malignant brain tumor repeat (mbt) domains. CNV losses of the SFMBT1 gene were associated with ventriculomegaly in elderly persons.15
Oncocytoma samples contained CNV amplifications comprising the entire miRNA 570 gene (miR-570) and 22% of the mucin 20 gene (MUC20) within the 3q29 region of chromosome 3 (Table 3). MUC20 transcripts are abundantly expressed in the kidney and up-regulated in renal tissues of patients with IgA nephropathy.16 An amplified region on chromosome 22 (22q13.33) contained the complete genes for RAB member of RAS oncogene family-like 2B (RABL2B) and acrosin (ACR). Also, partially contained within this region was part (60%) of the SH3 and multiple ankyrin repeat domains 3 (SHANK3) gene, along with three quarters of a ribosomal protein L23a pseudogene 82 (RPL23AP82). None of the genes in the chromosome 22 CNV has been implicated in pathological processes. CNV deletions on chromosome 5 (5q13.2) included four members of the general transcription factors IIH, polypeptide 2 (GTF2H2) gene/pseudogene family, including GTF2H2, GTF2H2B, GTF2H2C, and GTF2H2D. These genes are involved in nucleotide excision repair, and the expression of at least one of these genes (GTF2H2) has been associated with chemoresistance in non–small-cell lung cancer cell lines. Half of the glucuronidase β pseudogene 3 (GUSBP3) was contained within the 5q13.2 region. Finally, a CNV deletion on chromosome 13 (13q12.2) contained 10% of the class III receptor tyrosine kinase FLT3. Ligand binding to the receptor domain of this kinase activates multiple cytoplasmic effector molecules in pathways involved in apoptosis, proliferation, and differentiation of hematopoietic cells in acute myeloid leukemia.17
Papillary type 2 samples contained a zinc finger protein (ZNF214) gene, also known as the bromodomain adjacent to zinc finger domain 1A (BAZ1A), which was largely contained (85%) within the 14q13.2 CNV deletion (Table 3). A small component (6%) of the lysine (K)-specific demethylase 4C gene (KDM4C) was contained within the 9p24.1 CNV deletion.
Discussion
The use of high-density SNP arrays to interrogate high-mol.wt. DNA from prototypical renal tumor specimens provided resolution of CNVs at levels previously unattainable using microarrays. A surprising finding was the identification of distinct CNV segments common to each tumor specimen within chromophobe, clear-cell, oncocytoma, or papillary type 2 subtypes. Papillary type 1 specimens were the only exception among the five RCC subtypes, with no common CNV regions, despite the presence of many statistically significant CNV segments within each sample. However, there were eight regions of CNV gain and nine regions of CNV loss shared among five of six of these specimens. Shared RCC type-specific copy number changes included many previously undetected noncoding segments (ie, DNA not targeted for translation into protein products, suggesting a potentially important role for intronic and intergenic DNA in tumorigenesis). Although each tumor subclassification contained a different set of common chromosomal regions with copy number alterations specific to that phenotype, there was overlap among the five RCC categories, depending on the size and location selected (eg, a 100-megabase segment on chromosome 7 was present in 20 of 27 specimens).
The tumor specimens in this study derived from patients presenting with sporadic RCC and were classified and selected by a panel of pathologists as illustrative samples of their diagnostic classification. The copy number analysis revealed genomic regions near or overlapping regions associated with hereditary RCCs that were often affected in these sporadic tumors. Of the 27 sporadic renal neoplasms analyzed in this study, seven samples (three chromophobes, one clear cell, one papillary type 1, and two papillary type 2) exhibited a copy number decrement overlapping the BHD gene, which exhibits hereditary allelic loss in chromophobe, papillary, and clear-cell renal cancers.18 Copy number losses in the VHL gene typical of inherited clear-cell cancer occurred in nine of the 27 tumors (two chromophobe, four clear cell, and three papillary type 2), whereas amplification of the MET proto-oncogene associated with hereditary papillary RCC occurred in eight of the 27 tumors (two chromophobe, three papillary type 1, and three papillary type 2). Five specimens had copy number deletions (two chromophobe, one clear cell, and two oncocytoma), and one papillary type 1 specimen exhibited amplification of the fumarate hydratase gene, which exhibits germline mutations in renal carcinomas. These data indicated that DNA domains typically associated with inherited RCC were also unstable genomic regions susceptible to changes in sporadic renal cancers. Because these CNV alterations often occurred across vastly different phenotypes of sporadic cancers, it was likely that they represented passenger, rather than driver, mutations. However, these results emphasized the need for mechanistic studies of inherited DNA alterations regarding their impact on the phenotype of hereditary cancers to understand their contribution as genetic determinants of tumorigenesis.
Deletions in the short arm of chromosome 3 and amplifications in the long arm of chromosome 5 were present in clear-cell tumors, consistent with prior cytogenetic studies19 of sporadic clear-cell carcinoma. Extensive loss of chromosome 3p, including the VHL gene, was observed in 80% of the clear-cell tumors, in agreement with reports9,20 that 82% of clear-cell tumors harbor a VHL mutation. The clear-cell sample that did not have a CN loss of the VHL gene or any large segments of 3p had the greatest amount of overall CNV across the entire genome (38%), compared with the other clear-cell samples. One of the three regions of copy number loss common to all clear-cell samples was a 13.7-kb region of chromosome 3 at 3p21.1. Recently, a histone methyltransferase gene, SETD2/HypB, was identified in this region as a prospective tumor suppressor gene.21,22 In the present study, 80% of the clear-cell tumors had a loss of the SETD2 gene, whereas all specimens had a loss of the SFMBT1 gene at 3p21. Both of these gene products interact with histone H3 (ie, SFMBT1 binds monomethylated and dimethylated forms of H3K9, inducing transcriptional repression, and SETD2 is responsible for trimethylation of H3K36, associated with active/permissive chromatin).23,24 We can only speculate on the interactive effect of losing both SFMBT and SETD2, but these data suggest that histone modifications play an important role in clear-cell RCC. Furthermore, this paradigm may apply broadly to renal neoplasms because KDM4C, a lysine demethylase that modifies both H3K9 and H3K36, was affected in 60% of the RCC specimens analyzed in this study.25
Chromophobe RCCs typically have copy number decrements in chromosomes 1, 2, 6, 10, 13, 17, and/or 21.26 This broad genomic instability was reflected among the chromophobe specimens in this study, with the added observation that chromosome 19 contained copy number losses affecting many (60%) of the chromophobe specimens. Gene mutations specific to chromophobe tumors are yet to be identified, and the regions of CNV common to chromophobe tumors in this study represented prospective target areas for further investigation. In particular, the 208.3-kb deletion at 10p12.33 was interesting because it contained both the STAM gene, which has a role in MYC induction, and the miRNA 511-1/2 gene, which has been associated with adrenocortical tumor formation.27
Papillary renal cell tumors are characterized by gains within two or more chromosomes from among chromosomes 7, 8, 12, 16, 17, and 20 and the long arm of chromosome 3, without 3p loss.19 We identified amplifications in both papillary type 1 and 2 tumors on chromosomes 7 and 17, but only papillary type 1 tumors showed gains on chromosomes 12 and 16. Papillary type 2 specimens exhibited two common regions of copy number loss that contained KDM4C and BAZ1A, which play a role in nuclear receptor–mediated transcription repression. Papillary type 1 specimens had losses on chromosome 9 that were not observed in papillary type 2 specimens, whereas the latter had losses on chromosome 19 that were not observed in type 1 specimens. Papillary type 1 samples contained no CNV regions common to each sample, but there were overlapping CNV regions found in five of six specimens. Twelve genes were contained in these regions, including significantly reduced copy numbers for CDC14B (cell decision cycle 14 homologue B), an essential regulator of the G2 DNA damage checkpoint, and HABP4 (hyaluronic binding protein 4), involved in remodeling of chromatin and transcriptional regulation (GeneCards V3, http://www.genecards.org, last accessed January 6, 2012). Common (five of six) papillary type 1 copy number gains composed BCAS3 (breast carcinoma–amplified sequence 3), expressed in breast and brain neoplastic tissue and implicated in chronic kidney disease; WWOX (WW domain–containing oxidoreductase), a tumor suppressor that functions synergistically with p53 in breast, prostate, and ovarian cancers; and TBX2 (T-box transcription factor 2), which has enhanced cell proliferation in multiple cancers, including melanoma, breast, pancreatic, and lung tumors (GeneCards V3, http://www.genecards.org, last accessed January 6, 2012).28–31
Renal oncocytoma specimens contained CNVs within chromosomes 1 and 14, with chromosome 1 exhibiting the most variation, as previously reported.32 Nine CNV regions were common to all oncocytoma specimens, indicating a high degree of shared genomic instability in these benign neoplasms, similar to that observed within the chromophobe classification. Important tumor-related genes in these regions included MUC20 as part of the MET/GRB2-RAS pathway, GTFH2 transcription factors involved in nucleotide excision repair, and the FLT3 gene, which incorporates the most frequently identified mutations associated with acute myeloid and acute lymphoblastic leukemia (GeneCards V3, http://www.genecards.org, last accessed January 6, 2012).33 Many of the CNV regions identified in oncocytomas were also present in chromophobe samples, making it difficult to classify these two subtypes using these nine shared regions exclusively. The overlap in genotypic variations between these classifications was consistent with the historical observation that oncocytomas were sometimes difficult to distinguish from chromophobe specimens on a histological basis. However, multiple unique DNA domains specific to each subclassification were revealed using the high-density SNP arrays, and these segments contributed additional molecular indicators to help distinguish between these tumor types and potentially shed light on their different clinical behaviors.32
The present study revealed substantially more CNV segments than previous SNP studies of RCC (eg, 250K Affymetrix arrays detected an average of 5.8 amplifications and 6.8 deletions in clear-cell tumors, whereas we found an average of 73 distinct amplifications and 160 independent deletions).34 These differences were attributable to implementation of the SNP 6.0 platform and several quality control measures used in our study, including the following: i) use of high-integrity genomic DNA from flash-frozen specimens acquired with minimal warm ischemic time, ii) acquisition of precisely curated RCC specimens classified into distinct tumor phenotypes, and iii) statistical comparison with a group of normal tissues (n = 31) acquired and processed in our laboratory on the same platform following identical quality control standards. These measures resulted in detection of statistically significant tumor-specific CNV in segments as small as 2.9 kb in small tumor sample groups. The enhanced resolution allowed us to extend earlier findings regarding genes associated with renal cancers to potentially important new segments, including noncoding regions, such as the 12.5-kb deletion at 3p26.1 in clear-cell samples. The data also suggest that noncoding DNA regions may contribute to tumorigenesis through alterations in chromatin structure or transcriptional regulation.
We were able to confirm our findings, in part, by genomic sequencing of additional papillary type 2 specimens, extending the results obtained by SNP array analysis. However, the higher acuity provided by fine mapping of regions identified using global SNP assays will likely reveal local differences among samples in the magnitude and specific locations of the copy number changes and the presence of other DNA modifications. Consequently, it is likely that SNP arrays represent a potentially important screening method for identifying genomic hot spots that may serve as biomarkers useful for the detection of RCC and the identification of renal tumor subclassifications. However, direct genomic sequencing efforts will be required to elaborate the range and specific types of changes that occur within these segments and the precise mechanisms by which these regions and other protein-coding regions subserve the process of tumorigenesis.
The finding that four of five subclassifications of renal neoplasms shared common CNVs was unexpected given the prevailing theory that cancers arise from disparate genomic changes that induce tumorigenesis through convergence on common pathways. Although the overall results of this study confirm the widespread occurrence of tumor-related DNA rearrangements, the subset of shared genomic amplifications or deletions that we identified in each renal tumor subclassification could provide critical diagnostic or prognostic biomarkers of RCCs. Our results support the need for large-scale studies of high-quality RCC specimens using high-density SNP arrays and/or targeted sequencing of the tumor type–specific regions delineated in this study to determine their diagnostic efficacy and provide further insight into their role in renal cell tumorigenesis.
Acknowledgments
We thank the staff of the Health Sciences Tissue Bank, including Tina Tomko, Patricia Clark, Brandy Greenawalt, and Michelle Bisceglia. The extra efforts they made to rapidly obtain, preserve, and process the specimens through the Tissue Bank allowed us to perform our studies on DNA of unusually high quality and achieve the unique resolution reported in the article.
Footnotes
Supported by a grant from the National Cancer Institute Cancer Center (P30 CA47904) and Shadyside Hospital Foundation.
Disclosures: M.D.K. was a full-time employee of LifeTechnologies at the time of these studies, providing support for the performance and analysis of the whole genome sequencing studies, which were performed on the SOLiD 4 platform in the laboratory of the primary investigator (W.A.L.). M.D.K.'s employment had no influence on the design, outcome, or interpretation of these studies.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.01.044.
Supplementary data
The total number of mRNAs and microRNAs located within CNV segments in at least 60% of the samples in a subtype are provided in parentheses next to the name of the corresponding tumor subtype. Overlapping areas are marked with letters that refer to gene sets located in Supplemental Table S2.
References
- 1.Chow W.H., Devesa S.S., Warren J.L., Fraumeni J.F. Rising incidence of renal cell cancer in the United States. JAMA. 1999;281:1628–1631. doi: 10.1001/jama.281.17.1628. [DOI] [PubMed] [Google Scholar]
- 2.Cohen H.T., McGovern F.J. Renal-cell carcinoma. N Engl J Med. 2005;353:2477–2490. doi: 10.1056/NEJMra043172. [DOI] [PubMed] [Google Scholar]
- 3.Finley D.S., Pantuck A.J., Belldegrun A.S. Tumor biology and prognostic factors in renal cell carcinoma. Oncologist. 2011;16(Suppl 2):4–13. doi: 10.1634/theoncologist.2011-S2-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linehan W.M., Srinivasan R., Schmidt L.S. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277–285. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiuru M., Kujala M., Aittomaki K. Review: inherited forms of renal cell carcinoma. Scand J Surg. 2004;3:103–111. doi: 10.1177/145749690409300204. [DOI] [PubMed] [Google Scholar]
- 6.Ricketts C., Woodward E.R., Killick P., Morris M., Astuti D., Latif F., Maher E.R. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260–1262. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- 7.Crino P.B., Nathanson K.L., Henske E.P. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 8.Nordling C.O. A new theory on cancer-inducing mechanism. Br J Cancer. 1953;7:68–72. doi: 10.1038/bjc.1953.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalgliesh G.L., Furge K., Greenman C., Chen L., Bignell G., Butler A. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hagenkord J.M., Parwani A.V., Lyons-Weiler M.A., Alvarez K., Amato R., Gatalica Z., Gonzalez-Berjon J.M., Peterson L., Dhir R., Monzon F.A. Virtual karyotyping with SNP microarrays reduces uncertainty in the diagnosis of renal epithelial tumors. Diagn Pathol. 2008;3:44. doi: 10.1186/1746-1596-3-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lisovich A., Chandran U.R., Lyons-Weiler M.A., LaFramboise W.A., Brown A.R., Jakacki R.I., Pollack I.F., Sobol R.W. A novel SNP analysis method to detect copy number alterations with an unbiased reference signal directly from tumor samples. BMC Med Genomics. 2011;4:14. doi: 10.1186/1755-8794-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diskin S.J., Mingyao L., Hou C., Yang S., Glessner J., Hakonarson H., Bucan M., Maris J.M., Wang K. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008;36:e126. doi: 10.1093/nar/gkn556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z.Q., Imoto I., Fukuda Y., Pimkhaokham A., Shimada Y., Imamura M., Sugano S., Nakamura Y., Inazawa J. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res. 2000;60:4735–4739. [PubMed] [Google Scholar]
- 14.Godlewski J., Nowicki M.O., Bronisz A., Williams S., Otsuki A., Nuovo G., Raychaudhury A., Newton H.B., Chiocca E.A., Lawler S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68:9125–9130. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 15.Kato T., Sato H., Emi M., Seino T., Arawaka S., Iseki C., Takahashi Y., Wada M., Kawanami T. Segmental copy number loss of SFMBT1 gene in elderly individuals with ventriculomegaly: a community-based study. Intern Med. 2011;50:297–303. doi: 10.2169/internalmedicine.50.4505. [DOI] [PubMed] [Google Scholar]
- 16.Higuchi T., Orita T., Nakanishi S., Katsuya K., Watanabe H., Yamasaki Y., Waga I., Nanayama T., Yamamoto Y., Munger W., Sun H.W., Falk R.J., Jennette J.C., Alcorta D.A., Li H., Yamamoto T., Saito Y., Nakamura M. Molecular cloning, genomic structure, and expression analysis of MUC20, a novel mucin protein, up-regulated in injured kidney. J Biol Chem. 2004;279:1968–1979. doi: 10.1074/jbc.M304558200. [DOI] [PubMed] [Google Scholar]
- 17.Kindler T., Lipka D.B., Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116:5089–5102. doi: 10.1182/blood-2010-04-261867. [DOI] [PubMed] [Google Scholar]
- 18.Khoo S.K., Kahnoski K., Sugimura J., Petillo D., Chen J., Shockley K., Ludlow J., Knapp R., Giraud S., Richard S., Nordenskjöld M., Teh B.T. Inactivation of BHD in sporadic renal tumors. Cancer Res. 2003;63:4583–4587. [PubMed] [Google Scholar]
- 19.Kovacs G., Akhtar M., Beckwith B.J., Bugert P., Cooper C.S., Delahunt B., Eble J.N., Fleming S., Ljungberg B., Medeiros L.J., Moch H., Reuter V.E., Ritz E., Roos G., Schmidt D., Srigley J.R., Störkel S., van den Berg E., Zbar B. Review: the Heidelberg classification of renal cell tumours. J Pathol. 1997;183:131–133. doi: 10.1002/(SICI)1096-9896(199710)183:2<131::AID-PATH931>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 20.Nickerson M.L., Jaeger E., Shi Y., Durocher J.A., Mahurkar S., Zaridze D., Matveev V., Janout V., Kollarova H., Bencko V., Navratilova M., Szeszenia-Dabrowska N., Mates D., Mukeria A., Holcatova I., Schmidt L.S., Toro J.R., Karami S., Hung R., Gerard G.F., Linehan W.M., Merino M., Zbar B., Boffetta P., Brennan P., Rothman N., Chow W.H., Waldman F.M., Moore L.E. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–4734. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van den Berg A., Dijkhuizen T., Draaijers T.G., Hulsbeek M.M., Maher E.R., van den Berg E., Störkel S., Buys C.H. Analysis of multiple renal cell adenomas and carcinomas suggests allelic loss at 3p21 to be a prerequisite for malignant development. Genes Chromosomes Cancer. 1997;19:228–232. doi: 10.1002/(sici)1098-2264(199708)19:4<228::aid-gcc4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 22.Duns G., van den Berg E., van Duivenbode I., Osinga J., Hollema H., Hofstra R.M., Kok K. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010;70:4287–4291. doi: 10.1158/0008-5472.CAN-10-0120. [DOI] [PubMed] [Google Scholar]
- 23.Wu S., Trievel R.C., Rice J.C. Human SFMBT is a transcriptional repressor protein that selectively binds the N-terminal tail of histone H3. FEBS Lett. 2007;581:3289–3296. doi: 10.1016/j.febslet.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edmunds J.W., Mahadevan L.C., Clayton A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith E., Shilatifard A. The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol Cell. 2010;40:689–701. doi: 10.1016/j.molcel.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Speicher M.R., Schoell B., du Manoir S., Schröck E., Ried T., Cremer T., Störkel S., Kovacs A., Kovacs G. Specific loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am J Pathol. 1994;145:356–364. [PMC free article] [PubMed] [Google Scholar]
- 27.Tömböl Z., Szabó P.M., Molnár V., Wiener Z., Tölgyesi G., Horányi J., Riesz P., Reismann P., Patócs A., Likó I., Gaillard R.C., Falus A., Rácz K., Igaz P. Integrative molecular bioinformatics study of human adrenocortical tumors: microRNA, tissue-specific target prediction, and pathway analysis. Endocr Relat Cancer. 2009;16:895–906. doi: 10.1677/ERC-09-0096. [DOI] [PubMed] [Google Scholar]
- 28.Bärlund M., Monni O., Weaver J.D., Kauraniemi P., Sauter G., Heiskanen M., Kallioniemi O.P., Kallioniemi A. Cloning of BCAS3 (17q23) and BCAS4 (20q13) genes that undergo amplification, overexpression, and fusion in breast cancer. Genes Chromosomes Cancer. 2002;35:311–317. doi: 10.1002/gcc.10121. [DOI] [PubMed] [Google Scholar]
- 29.Köttgen A., Pattaro C., Böger C.A., Fuchsberger C., Olden M., Glazer N.L. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42:376–384. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewandowska U., Zelazowski M., Seta K., Byczewska M., Pluciennik E., Bednarek A.K. WWOX, the tumour suppressor gene affected in multiple cancers. J Physiol Pharmacol. 2009;60(Suppl 1):47–56. [PubMed] [Google Scholar]
- 31.Abrahams A., Parker M.I., Prince S. The T-box transcription factor Tbx2: its role in development and possible implication in cancer. IUBMB Life. 2010;62:92–102. doi: 10.1002/iub.275. [DOI] [PubMed] [Google Scholar]
- 32.Yusenko M.V., Kuiper R.P., Boethe T., Ljungberg B., van Kessel A.G., Kovacs G. High-resolution DNA copy number and gene expression analyses distinguish chromophobe renal cell carcinomas and renal oncocytomas. BMC Cancer. 2009;9:152. doi: 10.1186/1471-2407-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stirewalt D.L., Radich J.P. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3:650–665. doi: 10.1038/nrc1169. [DOI] [PubMed] [Google Scholar]
- 34.Beroukhim R., Brunet J.P., Di Napoli A., Mertz K.D., Seeley A., Pires M.M., Linhart D., Worrell R.A., Moch H., Rubin M.A., Sellers W.R., Meyerson M., Linehan W.M., Kaelin W.G., Jr, Signoretti S. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009;69:4674–4681. doi: 10.1158/0008-5472.CAN-09-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The total number of mRNAs and microRNAs located within CNV segments in at least 60% of the samples in a subtype are provided in parentheses next to the name of the corresponding tumor subtype. Overlapping areas are marked with letters that refer to gene sets located in Supplemental Table S2.