

Abstract
Hepatic fibrin(ogen) has been noted to occur after acetaminophen (APAP)-induced liver injury in mice. Deficiency in plasminogen activator inhibitor-1 (PAI-1), an endogenous inhibitor of fibrinolysis, increases APAP-induced liver injury in mice. However, the roles of fibrinogen and fibrinolysis in APAP-induced liver injury are not known. We tested the hypothesis that hepatic fibrin(ogen) deposition reduces severity of APAP-induced liver injury. APAP-induced (300 mg/kg) liver injury in mice was accompanied by thrombin generation, consumption of plasma fibrinogen, and deposition of hepatic fibrin. Neither fibrinogen depletion with ancrod nor complete fibrinogen deficiency [via knockout of the fibrinogen alpha chain gene (Fbg−⧸−)] affected APAP-induced liver injury. PAI-1 deficiency (PAI-1−⧸−) increased APAP-induced liver injury and hepatic fibrin deposition 6 hours after APAP administration, which was followed by marked hemorrhage at 24 hours. As in PAI-1−⧸− mice, administration of recombinant tissue plasminogen activator (tenecteplase, 5 mg/kg) worsened APAP-induced liver injury and hemorrhage in wild-type mice. In contrast, APAP-induced liver injury was reduced in both plasminogen-deficient mice and in wild-type mice treated with tranexamic acid, an inhibitor of plasminogen activation. Activation of matrix metalloproteinase 9 (MMP-9) paralleled injury, but MMP-9 deficiency did not affect APAP-induced liver injury. The results indicate that fibrin(ogen) does not contribute to development of APAP-induced liver injury and suggest rather that plasminogen activation contributes to APAP-induced liver injury.
Acetaminophen (APAP) overdose is a leading cause of drug-induced liver failure in the United States.1 APAP bioactivation by hepatocytes to the highly reactive N-acetyl-p-benzoquinone imine (NAPQI) metabolite results in intracellular macromolecule modification and mitochondrial dysfunction, which are key initiating events of the subsequent hepatocellular necrosis.1 The progression of APAP-induced liver injury is associated with numerous events, including activation of the blood coagulation cascade. Thrombin generation is evident in patients with acute APAP overdose and in mouse models of acute APAP-induced liver injury.2,3 In mice given a hepatotoxic dose of APAP, tissue factor-dependent thrombin generation is associated with consumption of circulating fibrinogen and deposition of insoluble fibrin clots in the liver.2 However, the role of fibrin(ogen) in APAP-induced liver injury is not known.
Fibrin deposition is determined by a balance of procoagulant (ie, thrombin) and fibrinolytic pathways. Tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA) convert plasminogen to the fibrinolytic enzyme plasmin, which degrades fibrin clots.4 The primary physiological inhibitor of uPA and tPA is plasminogen activator inhibitor 1 (PAI-1).5 Hepatic PAI-1 mRNA expression and plasma PAI-1 protein levels increase dramatically after APAP administration.2,6,7 A recent study indicated that APAP-induced liver injury is increased in PAI-1−⧸− mice, suggesting a protective role of PAI-1 in APAP hepatotoxicity.6 Bajt et al6 proposed the intriguing hypothesis that accelerated fibrin degradation in livers of PAI-1−/− mice precipitates APAP-induced hemorrhage and increased parenchymal cell injury, although the effect of PAI-1 deficiency on hepatic fibrin deposition is not known. Moreover, the exact role of fibrin and fibrinolytic enzymes has not been determined in APAP-induced liver injury.
Although exaggerated fibrinolysis and a reduction in hepatic fibrin composition is one mechanism whereby PAI-1 deficiency could increase APAP-induced liver injury, fibrin-independent mechanisms cannot be excluded. For example, plasmin can directly activate pro-matrix metalloproteinase-2 (MMP-2) and pro-MMP-9.8–12 Expression and activity of MMP-2 and MMP-9 were shown to be increased in livers of mice treated with APAP, and an inhibitor of these gelatinases significantly reduced APAP-induced liver injury.13 Accordingly, one potential fibrin-independent mechanism whereby increased plasmin could contribute to APAP-induced liver injury is activation of the MMPs. However, it is not currently known whether the fibrinolytic system contributes to MMP activation after APAP overdose in mice.
In the present study, we characterized the time course of coagulation and hepatic fibrin deposition in a mouse model of APAP overdose and used two strategies to determine the role of fibrin(ogen) in APAP-induced liver injury. Moreover, using a combination of gene-targeted mice and specific pharmacological tools, we explored the role of plasminogen activation on MMP activity and on the progression of APAP hepatotoxicity by using pharmacological and genetic approaches to modulate plasminogen activators.
Materials and Methods
Mice
All studies were performed with male mice, 8 to 16 weeks of age. Wild-type C57Bl/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). PAI-1−⧸− mice (B6.129S2-Serpine1tm1Mlg/J mice), MMP-9−⧸− mice [B6.FVB(Cg)-Mmp9tm1Tvu/J], and age-matched wild-type C57Bl/6J mice were purchased from the Jackson Laboratory. Fibrinogen α-chain-deficient mice (Fbg−⧸−) and heterozygous control mice (Fbg+⧸−) mice back-crossed six generations onto a C57Bl/6J background [kindly provided by Dr. Jay Degen (Cincinnati Children's Hospital Medical Center, Cincinnati, OH)] were maintained at the University of Kansas Medical Center in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Mice were housed at an ambient temperature of 22°C with 14/10-hour light/dark cycles and were provided water and rodent chow ad libitum (Teklad 8604; Harlan, Indianapolis, IN). Plg-deficient mice (Plg−⧸−) or wild-type control mice (Plg+⧸+) backcrossed for eight generations on a C57Bl/6J background were maintained at the Cincinnati Children's Hospital Medical Center. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center or the Cincinnati Children's Hospital Medical Center.
APAP Model and Pharmacological Interventions
Mice fasted for approximately 15 hours were given 300 mg/kg APAP (at 30 μL/g body weight) or vehicle (sterile saline solution) via intraperitoneal injection and food was returned. To deplete fibrinogen, mice were given ancrod (1.5 U i.p.; National Institute for Biological Standards and Control, Potters Bar, UK) or vehicle (300 μL sterile PBS i.p.) 2 hours before APAP treatment. Tenecteplase (TNK; 5 mg/kg at 10 μL/g body weight; a kind gift from Genentech, San Francisco, CA) or saline vehicle was given 2 hours after APAP administration by retro-orbital injection under isoflurane anesthesia. Tranexamic acid (600 mg/kg at 30 μL/g body weight; Spectrum Chemicals & Laboratory Products, New Brunswick, NJ) or saline vehicle was given intraperitoneally 2 hours after APAP treatment. At various times after APAP administration, mice were anesthetized using isoflurane. Blood was collected from the caudal vena cava into sodium citrate (final, 0.38%) or an empty syringe for the collection of plasma and serum, respectively. The liver was removed, washed in saline, and the intact gall bladder removed. The left medial lobe of the liver was affixed to a cork with optimal cutting temperature compound and frozen for 3 minutes in liquid nitrogen-chilled isopentane. Sections of the left lateral lobe were fixed in neutral-buffered formalin for 48 hours before routine processing. The remaining liver was cut into 100-mg pieces and frozen in liquid nitrogen.
Histopathology and Fibrin Staining
Formalin-fixed livers were sectioned at 5 μm, stained with H&E, and evaluated under light microscopy. Three sections of liver from the left lateral lobe were evaluated from each animal. The area of hepatocellular necrosis in several low-magnification (×40) images encompassing the total area of the three sections was determined by J.P.L. in a masked fashion as described previously,14 using Scion Image software version 4.0.2 (Scion Image Corp., Gaithersburg, MD). To determine percent necrotic area, the area of liver occupied by necrosis was compared with the total area of liver in each image. Immunofluorescent staining for insoluble hepatic fibrinogen was performed as described previously,15 using a rabbit anti-human fibrinogen antibody (A0080; Dako, Carpinteria, CA). The primary antibody was detected by the addition of goat anti-rabbit IgG conjugated to Alexa Fluor 488 (A11008; Invitrogen, Carlsbad, CA) and evaluated under fluorescent microscopy.
Clinical Chemistry
The serum activity of alanine aminotransferase (ALT) was determined using a commercially available reagent (Thermo Fisher, Waltham, MA). Plasma thrombin-antithrombin levels were determined using a commercial enzyme-linked immunosorbent assay kit (Siemens Healthcare Diagnostics, Deerfield, IL). For each assay, data were acquired using an Infinite M200 plate reader (Tecan, Durham, NC).
Zymography
Gelatinase (MMP-2 and MMP-9) activity was determined using gelatin zymography. Approximately 100 mg of frozen liver tissue was homogenized in ice-cold 50 mmol/L Tris-HCl buffer (pH 7.4) containing 150 mmol/L NaCl, 5 mmol/L CaCl2, and 1% Triton X-100. The homogenate was subjected to centrifugation at 9000 × g for 30 minutes at 4°C, supernatant was collected, and protein concentration determined using a commercial protein assay kit (DC; Bio-Rad Laboratories, Hercules, CA). Supernatant containing 50 μg total protein was mixed with an equal volume of 2× Laemmli sample buffer lacking β-mercaptoethanol (Bio-Rad Laboratories) and subjected to electrophoresis in Ready Gel zymogram gels with gelatin (Bio-Rad Laboratories). After electrophoresis, SDS was removed from the gels by three 20-minute washes with zymogram renaturation buffer (2.5% Triton X-100; Bio-Rad Laboratories). The gels were then incubated in zymogram development buffer (50 mmol/L Tris-HCl, pH 7.5, 200 mmol/L NaCl, 5 mmol/L CaCl2; Bio-Rad Laboratories) at 37°C without shaking for 48 hours. Gels were stained with Coomassie stain (Invitrogen), and MMPs were identified by their ability to digest gelatin (clear bands) and by their apparent molecular weights. Gel images were captured by a VersaDoc imaging system (Bio-Rad Laboratories) and converted to grayscale. Densitometry was performed using Quantity One software (Bio-Rad Laboratories).
Statistical Analysis
Comparison of two groups was performed using Student's t-test. Comparison of three or more groups was performed using one- or two-way analysis of variance, as appropriate, and the Student-Newman-Keuls post hoc test. Data not conforming to a normal distribution were log10-transformed before statistical evaluation. Statistical outliers identified by Grubb's test (P < 0.05) were excluded from further statistical analysis. The criterion for statistical significance was P < 0.05.
Results
Time Course of Coagulation Cascade Activation and Hepatotoxicity in APAP-Treated Mice
Administration of APAP (300 mg/kg) caused liver injury, as evidenced by increased serum ALT activity, with peak activity evident after 24 hours (Figure 1A). Increased serum ALT activity was accompanied by centrilobular hepatocellular necrosis (data not shown). Plasma thrombin-antithrombin levels, an indicator of thrombin generation, increased dramatically after APAP administration, indicating marked and rapid activation of the coagulation cascade (Figure 1B). In agreement with previous report,2 increased thrombin-antithrombin levels were accompanied by a decrease in plasma fibrinogen levels (Figure 1C) and an increase in hepatic fibrin deposition within areas of hepatocellular necrosis as early as 2 hours (Figure 1, D–H). Taken together, the results indicate that APAP hepatotoxicity in mice is associated with coagulation cascade activation, fibrinogen consumption, and hepatic fibrin deposition.
Figure 1.

Time course of coagulation cascade activation and hepatotoxicity in APAP-treated mice. Fasted wild-type mice were given a toxic dose of APAP (300 mg/kg i.p.) or saline vehicle (time 0); samples were collected at 2, 6, 24, and 48 hours. Levels of serum ALT activity (A), plasma thrombin-antithrombin (TAT) (B), and plasma fibrinogen (C) were determined. D–H: Representative photomicrographs of hepatic fibrin (dark) staining at 0, 2, 6, 24, and 48 hours after APAP treatment. Data are expressed as means ± SEM. *P < 0.05 versus saline-treated control mice. n = 5 mice per group. Original magnification, ×100.
Fibrin(ogen) Does Not Contribute to Acute APAP-Induced Liver Injury
To determine the role of fibrin(ogen) in APAP-induced liver injury, we used two independent approaches. First, circulating fibrinogen was depleted before APAP treatment by administering ancrod, a component of pit viper venom that enzymatically cleaves circulating fibrinogen.16 Compared with PBS vehicle-pretreated mice, pretreatment of mice with ancrod reduced hepatic fibrin deposition after APAP administration at 24 hours (Figure 2, B and C). However, ancrod pretreatment had no effect on serum ALT activity at this time in APAP-treated mice (Figure 2A). Next, we used mice lacking the fibrinogen α-chain gene (Fbg−⧸− mice), which completely lack circulating fibrinogen.17 As expected, hepatic fibrin deposition was not detected in Fbg−⧸− mice after APAP administration (Figure 2, E and F), confirming the absence of fibrinogen protein and the specificity of the immunofluorescent staining. In agreement with our fibrinogen depletion studies, complete fibrinogen deficiency did not affect APAP-induced liver injury, as indicated by serum ALT activity (Figure 2D). Overall, these studies suggest that fibrin(ogen) is not a critical mediator of APAP-induced liver injury in mice.
Figure 2.

Fibrin(ogen) does not contribute to acute APAP-induced liver injury. A–C: Fasted wild-type mice were treated with ancrod (1.5 U) or vehicle (300 μL sterile PBS) at 2 hours before treatment with APAP (300 mg/kg); samples were collected at 24 hours and serum ALT activity (A) was determined. D–F: Fasted Fbg+⧸− control mice and Fbg−⧸− were treated with APAP (300 mg/kg); samples were collected at 24 hours. Serum ALT activity was determined (D). B, C, E, and F: Representative photomicrographs of fibrin (dark) staining in livers after APAP treatment. Data are expressed as means + SEM. n = 7 to 12 mice per group. Original magnification, ×100.
PAI-1 Deficiency Enhances Liver Injury, Hemorrhage, and Fibrin Deposition in APAP-Treated Mice
Bajt et al6 reported that APAP-induced liver injury was increased in PAI-1-deficient (PAI-1−⧸−) mice. In agreement with their findings, we found that APAP-induced liver injury, as indicated by increased serum ALT activity (Figure 3A) and centrilobular necrosis (data not shown), was significantly increased at 6 hours after APAP administration in PAI-1−⧸− mice. In association with increased necrosis, hepatic fibrin deposition was increased in PAI-1−⧸− mice at this time point (Figure 3, B and C). Serum ALT activity also trended higher in APAP-treated PAI-1−⧸− mice relative to wild-type control mice at 24 hours (Figure 3D). In agreement with a previous study,6 marked hemorrhage was evident in livers of PAI-1−⧸− mice by 24 hours (see Supplemental Figure S1 at http://ajp.amjpathol.org). Fibrin was evident in areas of necrosis in both wild-type and PAI-1−⧸− mice at 24 hours (Figure 3, E and F). These results indicate that increased liver injury in APAP-treated PAI-1−⧸− mice is not a consequence of reduced hepatic fibrin deposition.
Figure 3.

PAI-1-deficiency enhances liver injury and fibrin deposition in APAP-treated mice. Fasted wild-type (PAI-1+/+) or PAI-1−⧸− mice were treated with APAP (300 mg/kg); samples were collected at 6 or 24 hours. Levels of serum ALT activity (A and D) were determined. B, C, E, and F: Representative photomicrographs of fibrin (dark) staining in livers from APAP-treated wild-type (B and E) and PAI-1−⧸− mice (C and F). Data are expressed as means + SEM. *P < 0.05 versus wild-type mice. n = 3 or 4 mice per group. Original magnification, ×100.
TNK (Recombinant Human tPA) Administration Enhances APAP-Induced Liver Injury in Mice
Enhanced APAP-induced liver injury in PAI-1−⧸− mice suggests that increased plasminogen activator activity could worsen APAP-induced liver injury. To test this hypothesis, we used TNK, a recombinant human tPA.18 Administration of TNK (5 mg/kg) alone to wild-type mice had no effect on serum ALT levels (Figure 4A) or liver histopathology (data not shown). To minimize potential effects of TNK on metabolism of APAP, we administered TNK 2 hours after APAP injection. TNK administration did not affect serum ALT activity at 6 hours after APAP administration (Figure 4A). However, increased hemorrhage was evident in areas of necrosis at 6 hours in APAP-treated mice given TNK (Figure 4, B and C). Serum ALT activity was significantly increased at 24 hours in mice treated with APAP and TNK, compared with mice treated with APAP alone (Figure 4A). The increase in serum ALT activity at 24 hours in TNK-treated mice was accompanied by a dramatic increase in hemorrhage (Figure 4, D and E), which closely resembled that observed in livers of PAI-1-deficient mice treated with APAP for 24 hours (see Supplemental Figure S1 at http://ajp.amjpathol.org; see also Bajt et al6). These data suggest that exaggerated activation of the plasminogen activators increases APAP-induced liver injury and hemorrhage.
Figure 4.

TNK (recombinant human tPA) administration enhances APAP-induced liver injury in mice. Fasted wild-type mice were treated with APAP (300 mg/kg) or saline vehicle, and 2 hours later were given TNK (5 mg/kg retro-orbital injection) or saline vehicle. A: Serum levels of ALT activity were determined at 6 and 24 hours after APAP treatment. B–E: Representative photomicrographs of H&E-stained liver sections at 6 or 24 hours from mice treated with APAP and given TNK or saline vehicle. Data are expressed as means +SEM. *P < 0.05 versus APAP-treated mice given saline vehicle. n = 3 to 8 mice per group. Original magnification, ×100.
Pharmacological Inhibition of Plasminogen Activation and Genetic Plasminogen Deficiency Reduces APAP-Induced Liver Injury
One mechanism whereby plasminogen activators could contribute to APAP-induced liver injury is through conversion of plasminogen to plasmin.8,9 To identify whether plasmin participates in APAP-induced liver injury, we used tranexamic acid, which binds plasminogen and inhibits its activation by plasminogen activators.19 Administration of tranexamic acid alone did not alter serum ALT levels or liver histopathology (Figure 5, A and B). To minimize potential effects of tranexamic acid on metabolism of APAP, tranexamic acid was administered 2 hours after APAP injection. Compared with saline vehicle-treated mice, tranexamic acid administration reduced serum ALT activity at 6 and 24 hours after APAP administration (Figure 5A), although this difference did not achieve statistical significance at 6 hours (P = 0.058). Of importance, tranexamic acid treatment significantly reduced the area of centrilobular necrosis in APAP-treated mice at 24 hours (Figure 5, B–D). Similar to pharmacological inhibition of plasminogen activation by tranexamic acid, complete plasminogen deficiency significantly reduced APAP-dependent liver injury at 24 hours, as indicated by a reduction in serum ALT activity (Figure 5E) and area of centrilobular necrosis (Figure 5, F–H). These results suggest that plasminogen contributes to APAP-induced liver injury.
Figure 5.

Inhibition of plasminogen activation and complete plasminogen deficiency reduces APAP-induced liver injury. A–D: Fasted wild-type mice were treated with APAP (300 mg/kg) or saline vehicle. Two hours later, mice were treated with tranexamic acid (TA; 600 mg/kg i.p.) or saline vehicle. Serum ALT activity (A) was determined at 6 and 24 hours after APAP treatment; area of necrosis (B) was determined 24 hours after APAP treatment. C and D: Representative photomicrographs of H&E-stained liver sections at 24 hours from mice treated with APAP and then treated with saline vehicle or tranexamic acid (TA). E–H: Fasted wild-type (Plg+⧸+) or plasminogen-deficient (Plg−⧸−) mice were treated with APAP (300 mg/kg) or saline vehicle; samples were collected at 24 hours. Serum ALT activity (E) and area of necrosis (F) were determined. G and H: Representative photomicrographs of H&E-stained liver sections from APAP-treated Plg+⧸+ or Plg−⧸− mice at 24 hours after APAP treatment. Data are expressed as means + SEM. *P < 0.05 versus APAP-treated mice given saline vehicle (A and B) or versus APAP-treated Plg+⧸+ mice (E and F). n = 3 to 10 mice per group. Original magnification, ×100.
Role of Plasminogen Activators in MMP-9 Activation in APAP-Treated Mice
One mechanism whereby plasmin could contribute to APAP-induced liver injury is by activating pro-MMP-2 and pro-MMP-9, enzymes implicated in the progression of APAP-induced liver injury.8,9,13 Accordingly, we determined the time course of gelatinase (MMP-2/9) activation in APAP-treated mice and the effect of genetic and pharmacological modulation of the plasminogen activators on MMP-2/9 activity. As indicated by gelatin zymography, MMP-9 activity was increased in livers of APAP-treated mice as early as 6 hours and remained elevated at 48 hours (Figure 6, A and B). However, we found no detectable increase in MMP-2 activity in mice treated with APAP (data not shown). Of note, MMP-9 activity in livers of APAP-treated mice at 6 hours was increased by PAI-1 deficiency (Figure 6, C and D) and by TNK treatment (Figure 6, E and F), suggesting that enhanced activation of plasminogen activators increases MMP-9 activation in APAP-treated mice. Tranexamic acid administration significantly reduced hepatic MMP-9 activity in APAP-treated mice at 6 hours (Figure 6, G and H). These data indicate that MMP-9 is activated after APAP-induced liver injury.
Figure 6.
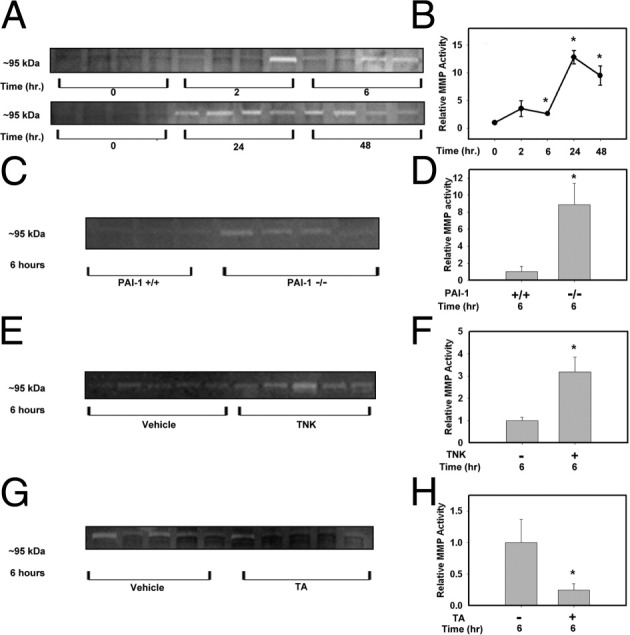
Role of plasminogen activators in MMP-9 activation in APAP-treated mice. Representative zymogram images (A, C, E, and G) and their respective zymogram quantification (B, D, F, and H) from liver homogenates of APAP-treated mice. A: Gelatinase activity was determined from homogenates of APAP-treated wild-type mice various times after APAP treatment. C–H: Gelatinase activity was determined at 6 hours in liver homogenates from PAI-1+⧸+ and PAI-1−⧸− mice(C and D), wild-type mice treated with TNK or vehicle (E and F), and wild-type mice treated with tranexamic acid (TA) or vehicle(G and H). Data are expressed as means ± SEM (B) or as means + SEM (D, F, and H). *P < 0.05 versus the respective control. n = 3 to 5 mice per group.
MMP-9 Deficiency Does Not Protect Against APAP-Induced Liver Injury
Ito et al13 reported that APAP-induced liver injury was reduced in mice given a MMP-2/9 inhibitor (2-[(4-biphenylsulfonyl)amino]-3-phenyl-propionic acid). In that light, and because we identified an increase in MMP-9 activity in livers of APAP-treated mice, we further evaluated whether MMP-9 could contribute to APAP-induced liver injury. Hepatic MMP-9 activity was not detectable by gelatin zymography in MMP-9−⧸− mice, confirming specificity of this assay (Figure 7A). Of note, APAP-induced liver injury was similar in MMP-9−⧸− mice and in wild-type control mice, as indicated by serum ALT activity (Figure 7B). These results indicate that MMP-9 does not contribute to the development of acute APAP-induced liver injury.
Figure 7.
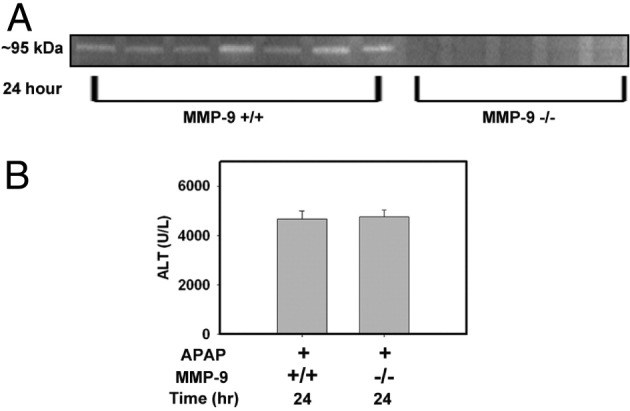
MMP-9 deficiency does not protect against APAP-induced liver injury. Fasted wild-type (MMP-9+⧸+) and MMP-9-deficient (MMP-9−⧸−) mice were treated with APAP (300 mg/kg); samples were collected at 24 hours. A: Representative gelatin zymogram (∼95 kDa) from liver homogenates of APAP-treated MMP-9+⧸+ or MMP-9−⧸− mice at 24 hours after APAP treatment. B: Serum ALT activity was determined 24 hours after APAP treatment. n = 5 to 9 mice per group.
Discussion
The role of PAI-1 in liver injury is model dependent. For example, whereas PAI-1 deficiency reduces liver injury after bile duct ligation,20 PAI-1−/− mice are more susceptible to carbon tetrachloride-induced liver injury.21 The expression of PAI-1 mRNA is increased rapidly in liver after APAP overdose in mice,6,7 and PAI-1 protein levels increase markedly in the plasma.2 In agreement with the report of Bajt et al,6 we found that APAP-induced liver injury was increased in PAI-1−⧸− mice, suggesting that PAI-1 is a protective factor in APAP hepatotoxicity. Of importance, generation of the toxic metabolite NAPQI was unaffected by PAI-1 deficiency,6 indicating that this genotype does not affect APAP metabolism. It has been suggested that the presence of marked hemorrhage in livers of APAP-treated PAI-1−⧸− mice could occur as a consequence of reduced hepatic fibrin deposition and a failure to maintain normal hemostasis.6 Of importance, we found that hepatic fibrin levels actually increased in association with increased hepatocellular injury in PAI-1−⧸− mice at 6 hours, and fibrin remained present in livers of APAP-treated PAI-1−⧸− mice at 24 hours. These findings suggest that increased APAP hepatotoxicity in PAI-1−⧸− mice is not a consequence of reduced fibrin levels in liver.
Hepatic fibrin deposition occurs in numerous models of xenobiotic-induced liver injury,2,15,22–25 but the mechanistic contribution of fibrinogen has not been determined in most models. In the present study, we used two independent approaches to evaluate the role of fibrin(ogen) in APAP hepatotoxicity: either depletion of fibrinogen with ancrod or use of Fbg−⧸− mice, which completely lack circulating fibrinogen protein.17 Neither fibrinogen depletion nor complete fibrinogen deficiency affected APAP-induced liver injury at 24 hours, suggesting that fibrinogen does not contribute to acute APAP hepatotoxicity. This is in contrast to other models, in which fibrinogen has been shown to be either damaging or hepatoprotective.14,26 Of importance, the finding that fibrinogen is not required for acute APAP hepatotoxicity supports the hypothesis that a change in hepatic fibrin clearance is not the primary mechanism of increased APAP hepatotoxicity in PAI-1-deficient mice.
To further delineate the effect of enhanced plasminogen activation on APAP-induced liver injury, we used TNK, a recombinant human tPA. We selected to administer TNK at 2 hours after APAP treatment, to avoid the possibility of interfering with APAP metabolism, NAPQI generation, and glutathione depletion, the vast majority of which occurs within 60 minutes of APAP administration.27,28 Mirroring changes in livers of APAP-treated PAI-1−⧸− mice, hepatocellular injury and hemorrhage were increased by TNK administration, consistent with the hypothesis that APAP-induced liver injury is increased by exaggeration of plasminogen activator activity. However, it cannot be concluded from these studies that endogenous tPA is the critical plasminogen activator participating in APAP-induced liver injury. Both uPA and tPA can convert plasminogen to plasmin, but each plasminogen activator generally serves different biological functions. tPA is an important regulator of fibrin degradation and vascular hemostasis, whereas uPA generally participates in tissue repair and extracellular matrix remodeling.5 Of note, tPA directly binds fibrin polymers, and this enhances its enzymatic activity by several hundredfold.5 Insofar as fibrin deficiency did not affect APAP-induced liver injury, this could suggest that uPA rather than tPA is the critical endogenous plasminogen activator during APAP-induced liver injury. Additional studies would be required to determine the relative contribution of uPA and tPA in APAP-induced liver injury.
Studies in PAI-1-deficient mice indicate that PAI-1 limits APAP-induced liver injury, and our studies with TNK administration support the hypothesis that exaggerated plasminogen activation worsens APAP-induced liver injury. To determine whether administration of an inhibitor of plasminogen activation could confer additional protection against APAP-induced liver injury under conditions in which PAI-1 was present, we used tranexamic acid, an antifibrinolytic drug that prevents the conversion of plasminogen to plasmin.19 As in our studies with TNK, tranexamic acid was given 2 hours after APAP, to avoid any potential effect on APAP metabolism. Our results suggest that the activation of plasmin, even in wild-type mice with sufficient levels of PAI-1, contributes to the progression of liver injury in APAP-treated mice. Of importance, we found that complete genetic plasminogen deficiency also reduced APAP-induced liver injury. However, these results do not identify which plasminogen activator (tPA and/or uPA) is responsible for activating plasminogen during APAP-induced liver injury. Additionally, these studies do not test the possibility that tPA and/or uPA also contribute to the progression of APAP-induced liver injury in a plasminogen-independent manner. These studies are the subject of ongoing investigation in our laboratory.
Our findings indicate that fibrinogen is not involved in the development of APAP-induced liver injury. However, the results suggest that plasmin, the primary endogenous fibrinolytic enzyme, contributes to APAP-induced liver injury. One fibrin-independent mechanism whereby plasmin might contribute to APAP toxicity is through activation of pro-MMP-2/9. Plasmin has been shown to contribute to conversion of both pro-MMP2 and pro-MMP9 to their active forms.8–12 Ito et al13 reported that hepatic MMP-2 and MMP-9 expression and activity are increased in livers of mice after APAP overdose.13 In accord with their findings, we found that MMP-9 activity increased in liver as early as 6 hours after APAP administration. However, we were not able to detect an increase in MMP-2 activity after APAP administration (data not shown) and we therefore focused on the possibility that plasmin-dependent MMP-9 activation could contribute to APAP hepatotoxicity. In contrast to a previous study demonstrating that a pharmacological inhibitor of MMP-2/9 reduced APAP hepatotoxicity,10 MMP-9 deficiency did not affect APAP induced liver injury in the present study. Notable differences between the present study using MMP-9-deficient mice and a previous study using a pharmacological inhibitor of MMP-9 include their use of DMSO as the solvent for the MMP inhibitor, a compound known to affect APAP metabolism and glutathione depletion.28 The fast before APAP administration was more extensive in that study (24 hours, compared with 15 hours in the present study), and both the dose and the route of APAP administration differed between the two studies. Our studies with MMP-9−⧸− mice strongly suggest that the protection observed previously cannot be attributed to inhibition of MMP-9.
In the clinic, patients who present with APAP overdose arrive with varying degrees of liver injury and at various stages of injury progression. Our data suggest that a PA-plasmin pathway participates in the early progression of APAP-induced liver injury in a mouse model. Inhibition of this pathway could provide a therapeutic benefit to patients during the early progression phase of APAP toxicity. However, the fibrinolytic system and MMPs have also been shown to participate in tissue regeneration and wound healing processes. Deficiency in plasminogen has been shown to derange the wound healing process, including in chronic liver injury.29 A previous study showed that impaired liver regeneration in plasminogen-deficient mice was not affected by a combined deficiency of fibrinogen,30 suggesting that plasminogen contributes to liver repair in a fibrinolysis-independent mechanism in that model. Although we found that fibrinogen deficiency does not affect acute APAP hepatotoxicity development, we cannot exclude a role for fibrinogen in the resolution of APAP-induced liver injury.
Similar questions arise about the potential for differential roles of plasminogen activators and MMPs during the development and recovery phases of APAP-induced liver injury. Accelerated injury resolution has been described in PAI-1−⧸− mice in a dermal injury model.31 Of importance, uPA is known to activate hepatocyte growth factor, a critical cytokine that promotes liver regeneration.32 Bajt et al6 reported that increased liver injury 24 hours after APAP administration in PAI-1−⧸− mice was accompanied by a decrease in hepatic PCNA protein expression, suggesting that these mice have delayed liver regeneration. In that same study, however, PAI-1−⧸− mice with dramatically increased liver injury recovered alongside wild-type mice by 48 hours, suggesting that those mice had enhanced liver regeneration in the recovery phase of APAP.6 Further experiments are needed to determine the role or roles of fibrinogen, fibrin, and fibrinolytic enzymes in the resolution and repair of APAP-induced liver injury.
In summary, we have shown through genetic and pharmacological interventions that fibrin(ogen) are not involved in the development of APAP hepatotoxicity in mice. Furthermore, we found that both genetic (PAI-1−⧸−) and pharmacological (TNK) interventions that increase plasminogen activator activity also increase APAP-induced liver injury and hemorrhage. Of importance, inhibition of plasminogen activation with tranexamic acid and complete plasminogen deficiency reduced liver injury in APAP-treated mice. Taken together, our findings indicate that the plasminogen activators and plasmin contribute to APAP-induced liver injury development in a fibrin(ogen)-independent manner. Inhibition of the PA-plasmin pathway may serve as a potential therapeutic target to limit acute liver injury development in patients presenting with APAP overdose.
Acknowledgments
The authors thank Ruipeng Wang for outstanding technical assistance.
Footnotes
Supported by grants from the NIH (R01-DK087886 to R.A.R. and J.P.L. and R01-ES017537 to J.P.L.). The histology core facility at the University of Kansas is supported by an NIH Center of Biomedical Research Excellence grant (P20 RR021940). B.P.S. was supported in part by an NIH-National Institute of Environmental Health Sciences training grant (T32-ES007079) and by the University of Kansas Medical Center Biomedical Research Training Program. M.J.F. was supported by grants from the NIH (R01-AR056990 and P30-AR47363). Tenecteplase was provided by Genentech (San Francisco, CA).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.02.011.
Supplementary data
PAI-1 deficiency enhances hepatic hemorrhage in APAP-treated mice. Fasted wild-type or PAI-1−⧸− mice were treated with APAP (300 mg/kg); samples were collected at 24 hours. Photomicrographs are representative of H&E-stained liver sections from wild-type (A) and PAI-1−⧸− (B) mice. Original magnification, ×100.
References
- 1.Chun L.J., Tong M.J., Busuttil R.W., Hiatt J.R. Acetaminophen hepatotoxicity and acute liver failure. J Clin Gastroenterol. 2009;43:342–349. doi: 10.1097/MCG.0b013e31818a3854. [DOI] [PubMed] [Google Scholar]
- 2.Ganey P.E., Luyendyk J.P., Newport S.W., Eagle T.M., Maddox J.F., Mackman N., Roth R.A. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology. 2007;46:1177–1186. doi: 10.1002/hep.21779. [DOI] [PubMed] [Google Scholar]
- 3.Gazzard B.G., Henderson J.M., Williams R. Early changes in coagulation following a paracetamol overdose and a controlled trial of fresh frozen plasma therapy. Gut. 1975;16:617–620. doi: 10.1136/gut.16.8.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 2005;25:2273–2281. doi: 10.1161/01.ATV.0000183884.06371.52. [DOI] [PubMed] [Google Scholar]
- 5.Schaller J., Gerber S.S. The plasmin-antiplasmin system: structural and functional aspects. Cell Mol Life Sci. 2011;68:785–801. doi: 10.1007/s00018-010-0566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bajt M.L., Yan H.M., Farhood A., Jaeschke H. Plasminogen activator inhibitor-1 limits liver injury and facilitates regeneration after acetaminophen overdose. Toxicol Sci. 2008;104:419–427. doi: 10.1093/toxsci/kfn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reilly T.P., Bourdi M., Brady J.N., Pise-Masison C.A., Radonovich M.F., George J.W., Pohl L.R. Expression profiling of acetaminophen liver toxicity in mice using microarray technology. Biochem Biophys Res Commun. 2001;282:321–328. doi: 10.1006/bbrc.2001.4576. [Erratum in Biochem Biophys Res Commun 2001;283:536] [DOI] [PubMed] [Google Scholar]
- 8.Legrand C., Polette M., Tournier J.M., de Bentzmann S., Huet E., Monteau M., Birembaut P. uPA/plasmin system-mediated MMP-9 activation is implicated in bronchial epithelial cell migration. Exp Cell Res. 2001;264:326–336. doi: 10.1006/excr.2000.5125. [DOI] [PubMed] [Google Scholar]
- 9.Gong Y., Hart E., Shchurin A., Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest. 2008;118:3012–3024. doi: 10.1172/JCI32750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazzieri R., Masiero L., Zanetta L., Monea S., Onisto M., Garbisa S., Mignatti P. Control of type IV collagenase activity by components of the urokinase-plasmin system: a regulatory mechanism with cell-bound reactants. embo j. 1997;16:2319–2332. doi: 10.1093/emboj/16.9.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monea S., Lehti K., Keski-Oja J., Mignatti P. Plasmin activates pro-matrix metalloproteinase-2 with a membrane-type 1 matrix metalloproteinase-dependent mechanism. J Cell Physiol. 2002;192:160–170. doi: 10.1002/jcp.10126. [DOI] [PubMed] [Google Scholar]
- 12.Takano A., Hirata A., Inomata Y., Kawaji T., Nakagawa K., Nagata S., Tanihara H. Intravitreal plasmin injection activates endogenous matrix metalloproteinase-2 in rabbit and human vitreous. Am J Ophthalmol. 2005;140:654–660. doi: 10.1016/j.ajo.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 13.Ito Y., Abril E.R., Bethea N.W., McCuskey R.S. Inhibition of matrix metalloproteinases minimizes hepatic microvascular injury in response to acetaminophen in mice. Toxicol Sci. 2005;83:190–196. doi: 10.1093/toxsci/kfh291. [DOI] [PubMed] [Google Scholar]
- 14.Luyendyk J.P., Mackman N., Sullivan B.P. Role of fibrinogen and protease-activated receptors in acute xenobiotic-induced cholestatic liver injury. Toxicol Sci. 2011;119:233–243. doi: 10.1093/toxsci/kfq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luyendyk J.P., Cantor G.H., Kirchhofer D., Mackman N., Copple B.L., Wang R. Tissue factor-dependent coagulation contributes to alpha-naphthylisothiocyanate-induced cholestatic liver injury in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G840–G849. doi: 10.1152/ajpgi.90639.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewart M.R., Hatton M., Basford J.M., Dodgson K.S. The proteolytic action of arvin on human fibrinogen. Biochem J. 1969;115:17P. doi: 10.1042/bj1150017p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suh T.T., Holmbäck K., Jensen N.J., Daugherty C.C., Small K., Simon D.I., Potter S., Degen J.L. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- 18.Verstraete M. Third-generation thrombolytic drugs. Am J Med. 2000;109:52–58. doi: 10.1016/s0002-9343(00)00380-6. [DOI] [PubMed] [Google Scholar]
- 19.Iwamoto M. Plasminogen-plasmin system IX: Specific binding of tranexamic acid to plasmin. Thromb Diath Haemorrh. 1975;33:573–585. [PubMed] [Google Scholar]
- 20.Wang H., Zhang Y., Heuckeroth R.O. PAI-1 deficiency reduces liver fibrosis after bile duct ligation in mice through activation of tPA. FEBS Lett. 2007;581:3098–3104. doi: 10.1016/j.febslet.2007.05.049. [DOI] [PubMed] [Google Scholar]
- 21.von Montfort C., Beier J.I., Kaiser J.P., Guo L., Joshi-Barve S., Pritchard M.T., States J.C., Arteel G.E. PAI-1 plays a protective role in CCl4-induced hepatic fibrosis in mice: role of hepatocyte division. Am J Physiol Gastrointest Liver Physiol. 2010;298:G657–G666. doi: 10.1152/ajpgi.00107.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Copple B.L., Banes A., Ganey P.E., Roth R.A. Endothelial cell injury and fibrin deposition in rat liver after monocrotaline exposure. Toxicol Sci. 2002;65:309–318. doi: 10.1093/toxsci/65.2.309. [DOI] [PubMed] [Google Scholar]
- 23.Neubauer K., Knittel T., Armbrust T., Ramadori G. Accumulation and cellular localization of fibrinogen/fibrin during short-term and long-term rat liver injury. Gastroenterology. 1995;108:1124–1135. doi: 10.1016/0016-5085(95)90211-2. [DOI] [PubMed] [Google Scholar]
- 24.Bailie M.B., Mullaney T.P., Roth R.A. Characterization of acute 4,4′-methylene dianiline hepatotoxicity in the rat. Environ Health Perspect. 1993;101:130–133. doi: 10.1289/ehp.93101130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujiwara K., Ogata I., Ohta Y., Hirata K., Oka Y., Yamada S., Sato Y., Masaki N., Oka H. Intravascular coagulation in acute liver failure in rats and its treatment with antithrombin III. Gut. 1988;29:1103–1108. doi: 10.1136/gut.29.8.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luyendyk J.P., Kassel K.M., Allen K., Guo G.L., Li G., Cantor G.H., Copple B.L. Fibrinogen deficiency increases liver injury and early growth response-1 (Egr-1) expression in a model of chronic xenobiotic-induced cholestasis. Am J Pathol. 2011;178:1117–1125. doi: 10.1016/j.ajpath.2010.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knight T.R., Kurtz A., Bajt M.L., Hinson J.A., Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci. 2001;62:212–220. doi: 10.1093/toxsci/62.2.212. [DOI] [PubMed] [Google Scholar]
- 28.Jaeschke H., McGill M.R., Williams C.D., Ramachandran A. Current issues with acetaminophen hepatotoxicity—a clinically relevant model to test the efficacy of natural products. Life Sci. 2011;88:737–745. doi: 10.1016/j.lfs.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pohl J.F., Melin-Aldana H., Sabla G., Degen J.L., Bezerra J.A. Plasminogen deficiency leads to impaired lobular reorganization and matrix accumulation after chronic liver injury. Am J Pathol. 2001;159:2179–2186. doi: 10.1016/S0002-9440(10)63069-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bezerra J.A., Bugge T.H., Melin-Aldana H., Sabla G., Kombrinck K.W., Witte D.P., Degen J.L. Plasminogen deficiency leads to impaired remodeling after a toxic injury to the liver. Proc Natl Acad Sci USA. 1999;96:15143–15148. doi: 10.1073/pnas.96.26.15143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan J.C., Duszczyszyn D.A., Castellino F.J., Ploplis V.A. Accelerated skin wound healing in plasminogen activator inhibitor-1-deficient mice. Am J Pathol. 2001;159:1681–1688. doi: 10.1016/S0002-9440(10)63015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimizu M., Hara A., Okuno M., Matsuno H., Okada K., Ueshima S., Matsuo O., Niwa M., Akita K., Yamada Y., Yoshimi N., Uematsu T., Kojima S., Friedman S.L., Moriwaki H., Mori H. Mechanism of retarded liver regeneration in plasminogen activator-deficient mice: impaired activation of hepatocyte growth factor after Fas-mediated massive hepatic apoptosis. Hepatology. 2001;33:569–576. doi: 10.1053/jhep.2001.22650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PAI-1 deficiency enhances hepatic hemorrhage in APAP-treated mice. Fasted wild-type or PAI-1−⧸− mice were treated with APAP (300 mg/kg); samples were collected at 24 hours. Photomicrographs are representative of H&E-stained liver sections from wild-type (A) and PAI-1−⧸− (B) mice. Original magnification, ×100.