

Abstract
Numerous studies support the role for mutations in the phosphatase and tensin homologue (PTEN) tumor suppressor gene and unopposed estrogen stimulation in the pathogenesis of uterine endometrioid carcinoma. However, the relation between PTEN signaling and estrogen/estrogen receptor in endometrial tumorigenesis remains unresolved. We used genetically engineered mice as a model to address this relation. Mice with a single deleted Pten allele (Pten+/−) spontaneously develop complex atypical hyperplasia and ∼20% develop endometrial cancer. To determine the effect of removing endogenous estrogen, we performed oophorectomies on Pten+/− mice. Although there was a reduction in the number and severity of hyperplastic lesions, the endometrial phenotype persisted, suggesting that Pten mutation, independent of estrogen, can initiate the development of complex atypical hyperplasia. To recapitulate the situation in women with unopposed estrogen, we implanted 17β-estradiol pellets in adult female Pten heterozygous mice, resulting in increased carcinoma incidence. Because studies have shown that estrogen largely acts on the endometrium via estrogen receptor ERα, we generated Pten+/−ERα−/− mice. Strikingly, 88.9% of Pten+/−ERα−/− mice developed endometrial hyperplasia/carcinoma. Furthermore, Pten+/−ERα−/− mice showed a higher incidence of in situ and invasive carcinoma, suggesting that endometrial tumorigenesis can progress in the absence of ERα. Thus, the relation between Pten alterations and estrogen signaling in the development of endometrial carcinoma is complex; the results presented herein have important implications for the treatment of endometrial hyperplasia and carcinoma in women.
Endometrial cancer is the most common malignancy of the female genital tract, and, like most cancers, it is a complex disease comprising a number of different types of carcinoma. Clinicopathologic, epidemiologic, and genetic studies have supported a dualistic model of endometrial carcinoma categorized as type I and type II. Type I carcinoma is the most common, and, although there are a number of different histologic subtypes, it is usually of endometrioid histology. Previous studies have shown that women with uterine endometrioid carcinoma (UEC) often have increased circulating levels of estrogen and low levels of progesterone, a situation that results in unopposed estrogen stimulation of the endometrium. Furthermore, UEC is usually preceded by complex atypical hyperplasia (CAH), which is also thought to be a result of unopposed estrogen stimulation. Conversely, type II carcinoma is not associated with unopposed estrogen stimulation, arises in postmenopausal women, and is predominately of serous histology.1 It has been found that the most common genetic alteration in UEC is mutation of the phosphatase and tensin homologue (PTEN) tumor suppressor gene, an alteration that is uncommon in uterine serous carcinoma.2,3 PTEN mutations have also been identified in CAH at approximately the same frequency as in UEC.4,5 Thus, both aberrant PTEN function and unopposed estrogen stimulation are thought to play a role in the pathogenesis of UEC, suggesting a possible relation between signaling pathways downstream of estrogen and PTEN. However, little is known about the connection between estrogen and PTEN signaling in the development of endometrial hyperplasia and UEC.
It is currently thought that the effects of estrogen on the endometrium are mediated primarily via the estrogen receptor ERα.6 ERα is a member of a superfamily of nuclear receptors that act as transcription factors through estrogen-independent and estrogen-dependent activation domains.7 Although the endometrial epithelium expresses ERα,8 studies have suggested that the mitogenic effects of estrogen on the epithelium occur through its interaction with ERα in the endometrial stromal cells.9–11 This leads to elaboration of growth factors from the stromal cells that stimulate the epithelium by binding the cognate receptors expressed on the surface of the epithelial cells.12 Growth factors, for example, insulin-like growth factor and epidermal growth factor, are known to regulate a diverse number of cellular processes, including cell proliferation, differentiation, motility, and invasion.
PTEN functions primarily as a lipid phosphatase to regulate the phosphatidyl inositol kinase (PI3K)/AKT pathway.13 The PI3K/AKT pathway is activated by growth factors, such as those elicited by the endometrial stromal cells in response to estrogen.1 This results in the phosphorylation of phosphatidylinositol-4,5-phosphate (PIP2) to generate phosphatidlyinositol-3,4,5-phosphate (PIP3), previously reported to occur in uterine tissue in response to estrogen and involving ER.14 Increased levels of PIP3 lead to phosphorylation of AKT, which in turn leads to the phosphorylation of a large number of proteins that regulate cell proliferation, survival, and growth. One of the actions of PTEN is to dephosphorylate PIP3 to PIP2; thus, loss of PTEN function results in unchecked activation of the pathway, leading to increased levels of phosphorylated AKT.15 Several in vitro and in vivo studies have suggested cross talk between PI3K/AKT and estrogen signaling. ERα can bind to the regulatory subunit of PI3K in the absence or presence of estradiol in epithelial cells and subsequently activate PI3K/AKT2.16,17 By contrast, PI3K and AKT can phosphorylate and activate ERα in the absence of estrogen, resulting in its increased ability to activate the transcription of several target genes.18,19 These findings suggest that loss of PTEN may function, at least in part, through activation of ERα. In addition, nongenomic actions of estrogen via a surface receptor have been well documented,20–22 and this may be another mechanism by which PTEN and estrogen signaling intersect.
Rodent models have been invaluable to the study of estrogen's role in endometrial tumorigenesis. Treatment of certain mouse strains with diethylstilbestrol (DES) in the neonatal period results in the development of endometrial carcinoma with histologic features similar to UEC.23 It has been shown that 100% of Pten+/− female mice develop complex atypical hyperplasia with ∼20% developing endometrial carcinoma by 1 year of age.24 Thus, in mice, as in humans, excess estrogen and alterations of Pten appear to be causative factors in the development of endometrial hyperplasia and carcinoma. To better understand the relation of Pten and estrogen/estrogen receptor in the development of endometrial hyperplasia and carcinoma, we used genetically engineered mouse models with alterations in Pten and ERα.
Materials and Methods
Generation of Mouse Strains
Pten+/− mice in a mixed C57BL6/129SvJ (B6/129) background have been described previously.24 To generate Pten+/− genotype in a pure C57BL6 background, the mice were backcrossed to wild-type C57BL6 females for 10 generations. Pten+/− CD-1 mice were generated similarly by breeding the Pten+/− mixed background mice to wild-type CD-1 females. ERα heterozygous mice, on a C57BL6 background, were generated at the National Institutes of Environmental Health Sciences (Research Triangle Park, NC) under an approved animal study protocol.6 The Pten+/− mixed strain mice were crossed with ERα+/− mice to generate Pten+/−;ERα−/− mice and the appropriate littermate controls. Animals were genotyped with the use of multiplex PCR on genomic DNA prepared from tail samples.6,24
Treatment of Mice with Hormones
To determine the role of estrogen in the development of endometrial neoplasia, nine virgin Pten+/− and wild-type B6/129 female mice underwent oophorectomy at 3 weeks of age. At 32 weeks after oophorectomy, the mice were euthanized, and uteri were harvested for further analysis.
For investigating the effect of excess estrogen, we used two approaches. For approach 1, we followed an established protocol of treating neonatal mice with DES.23 Pten+/− mice in the mixed background were treated with DES (2 μg/day per pup) by subcutaneous injections from day 1 through day 5 of age. Oil-injected animals served as controls. The animals were sacrificed 24 and 32 weeks after treatment. The uterine weights were measured and were submitted for histochemical analysis.
For approach 2, to more closely simulate the situation of estrogen exposure in women with endometrial carcinoma, we treated CD-1 Pten+/− and wild-type females with time-release estrogen pellets. Mice underwent oophorectomy at 12 weeks of age, allowing the uteri to become fully mature, and the pellets were implanted immediately after oophorectomy. Seven CD-1 Pten+/− mice were implanted subcutaneously once with 90-day time-release pellets that contained 0.72 mg of 17β-estradiol (Innovative Research of America, Sarasota, FL) according to the manufacturer's instruction. CD-1 Pten+/− (n = 7) mice implanted with placebo pellets (Innovative Research of America) at the time of oophorectomy served as controls. In addition, 11 CD-1 Pten+/+ mice were also implanted with estrogen pellets. Furthermore, CD-1 Pten+/− mice (n = 5) were treated with estrogen pellets twice, once at 12 weeks (immediately after oophorectomy) and the second time after another 12 weeks. CD-1 Pten+/+ mice (n = 5) treated with estrogen pellets twice and CD-1 Pten+/− mice (n = 5) treated with placebo pellets twice were used as control animals. The pellets resulted in serum levels of 200 to 250 pg/mL, which are approximately 10 times higher than endogenous circulating levels. The animals were sacrificed at indicated times after treatment, and their uteri were harvested.
IHC Analysis
The uteri harvested after treatment from all animals were processed for H&E staining as well as immunohistochemical (IHC) analysis. Tissues were formalin-fixed, dehydrated through an alcohol series, and subsequently embedded in paraffin. Five-micron sections were stained with H&E. For IHC, after deparaffinization and hydration through alcohol grades, slides were subjected to antigen retrieval by boiling in 10 mmol/L sodium citrate. Antibodies used were anti-Ki-67 (RM-9106; Lab Vision, Fremont, CA), anti-phospho-GSK3β S9 (9323; Cell Signaling, Beverly, MA), anti-ERα (SC-542; Santa Cruz Biotechnologies, Santa Cruz, CA), anti-cyclin D1 (RM-9104; Lab Vision, Fremont, CA). IHC staining was performed with Elite Vectastain ABC Kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Progesterone receptor (PR) IHC was performed with the Mouse-on-Mouse (M.O.M.) staining kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Anti-PR antibody (Immunotech, Marseille, France) was used at 1:50 dilution for 45 minutes at room temperature.
The number of positive epithelial nuclei for Ki-67was counted per 1000 nuclei and expressed as percentage values. Scoring of cyclin D1, p-GSK3β, and ERα and PR IHC were done to quantify the expression in lesions and normal epithelium. The localization was determined to be either nuclear or cytoplasmic. First, staining intensities were ranked from 1 to 4, corresponding to increasing intensity with 1 being the least and 4 the strongest. Then the percentage of positive cells were estimated by scoring at least five fields at magnification ×100 and ranked from 0 to 4 (0 = 0%, 1 = 1% to 25%, 2 = 26% to 50%, 3 = 51% to 75%, and 4 = 74% to 100%). Final scores were calculated by multiplying intensity rank numbers with the percentage of rank numbers and were verified in a single-blinded fashion by two pathologists (L.H.E. and J.S.Y.C.).
Cell Culture
Parental and PTEN-deleted Hec1A cells were obtained from the laboratory of Dr. Todd Waldman at the Lombardi Comprehensive Cancer Center of Georgetown University (Washington, DC). Cells were maintained in McCoy's 5A medium supplemented with 10% charcoal-stripped fetal bovine serum. Cell proliferation was measured with the XTT (sodium 2,3,-bis(2-methoxy-4-nitro-5-sulfophenyl)−5-[(phenylamino)-carbonyl]-2H-tetrazolium inner salt) assay. For XTT assays, 4000 cells were plated in 96-well plates, and the medium was changed to phenol red-free Dulbecco's modified Eagle's medium/F12 supplemented with 15 mmol/L HEPES and either low (1%) or high (10%) charcoal-stripped fetal bovine serum the next day. To determine proliferation, XTT solution was added to wells for each time point, and absorbance was measured at 492 nm with 595 nm as the reference wavelength every alternate day for 12 days. For the rest of the wells, medium was changed every other day. Wells with media only were used as controls. The absorbance values were converted to percentage of cell growth with respect to values on day 1. For effect of serum starvation and restimulation, cells were plated in 6-well dishes in McCoy's 5A medium at ∼50% to 60% confluence and changed to serum-free and phenol red-free McCoy's 5A medium the next day. The cells were serum starved for 24 hours, restimulated with 10% serum for indicated time periods, and harvested for immunoblot analysis.
Immunoblot Analysis
For immunoblot analysis, cells stimulated with either 1% or 10% serum for indicated time points were lysed in 250 μL/well of a 6-well dish in ice-cold 1× radioimmunoprecipitation assay buffer with protease and phosphatase inhibitors. Cells were passed through a 27-gauge needle and clarified by centrifugation at 13,000 rpm for 15 minutes at 4°C. All manipulations were performed on ice. Lysates were resolved on 10% SDS-PAGE, transferred to polyvinylidene difluoride membrane, and blocked in 5% nonfat dried milk dissolved in 1× Tris-buffered saline with 0.1% Tween-20 (TBS-T). Membranes were probed with anti-Pten (9559), anti-phospho-Akt S473 (9271), anti-Akt (9272), anti-phospho-Erk1/2 (9101), anti-Erk (4695; from Cell Signaling), anti-cyclin D1 (RM-9104; Lab Vision), anti-cyclin D3 (SC-182; Santa Cruz Biotechnologies), and anti-Actin (A5060; Sigma-Aldrich, St. Louis, MO) antibodies. All antibodies were used at a dilution of 1:1000, and membranes were incubated overnight at 4°C with primary antibody, washed liberally at room temperature in TBS-T, and incubated for 1 hour with horseradish peroxidase-conjugated secondary antibody. After liberal washing in TBS-T, blots were incubated in Western Lightning Plus ECL reagent (PerkinElmer, Waltham, MA) and visualized by exposure to electrochemiluminescence film (Eastman Kodak Co., Rochester, NY).
Statistical Analysis
IHC scores were analyzed with the nonparametric Mann–Whitney test with P < 0.05. The percentage of positive Ki-67 cells, immunoblot quantitations, and data in the tables were analyzed with unpaired t-test (two-tailed) with P < 0.05. All analyses were performed with GraphPad Prism version 5.0 for Mac (GraphPad Software, San Diego, CA).
Results
Development of CAH in Pten+/− Mice Is Estrogen Independent
We first explored whether lack of Pten, excess estrogen, or both are initiating events for the development of atypical hyperplasia. Immature (3-week-old) Pten+/− and wild-type mice on the B6/129 background underwent oophorectomy and were sacrificed at 32 weeks of age, the age at which 100% of the Pten+/− females develop endometrial hyperplasia.24 All of the mice that had undergone oophorectomy exhibited an ∼75% reduction in uterine weights, as expected because of the absence of estrogen. CAH did not develop in wild-type mice that had undergone oophorectomy or intact wild-type mice (Figure 1, A and C). Histologic analysis found the presence of CAH in all (9 of 9) intact Pten+/− uteri (Figure 1B). Interestingly, 8 of 9 Pten+/− mice that had undergone oophorectomy also developed CAH (Figure 1D). Although the lesions were qualitatively similar, the size and number of lesions were smaller than those in the intact mice. The results have been summarized in Table 1. These observations suggest that Pten mutation, in the absence of estrogen, is sufficient for development of CAH as shown previously.25
Figure 1.

H&E stained sections of Pten+/+ mice (A), Pten+/− mice (B), Pten+/+ mice that had undergone oophorectomy (Ovx) (C), and Pten+/− mice that had undergone oophorectomy (D). CAH develops in the absence of estrogen. Original magnification, ×20.
Table 1.
Endometrial Lesions in Mice That Have Undergone Oophorectomy
| Age (weeks) | n | No. (%) of mice with lesions | No. of lesions per mouse (mean ± SD) | Size of lesion (mm2) | Range of size (mm2) | |
|---|---|---|---|---|---|---|
| Pten+/+ | 32 | 4 | 0 | NA | NA | NA |
| Pten+/+;Ovx | 32 | 9 | 0 | NA | NA | NA |
| Pten+/− | 32 | 9 | 9 (100) | 18.56 ± 8.57⁎ | 0.22 ± 0.12 | 0.04−0.75 |
| Pten+/−;Ovx | 32 | 9 | 8 (90) | 8.20 ± 10.27⁎ | 0.13 ± 0.13 | 0.01−0.96 |
NA, not applicable; Ovx, oophorectomy.
P = 0.03, difference was statistically significant.
In addition, we treated neonatal mice with DES according to established protocols and sacrificed the mice at 24 and 32 weeks (see Supplemental Figure S1 at http://ajp.amjpathol.org). The mice treated with DES showed no significant difference in the uterine weights than mice treated with oil. Of the DES-treated Pten+/+ mice, only 33% to 36% developed mild endometrial hyperplasia (simple hyperplasia). By contrast, all Pten+/− mice treated with oil alone or DES developed CAH (Table 2). Interestingly, Pten+/− mice treated with DES and sacrificed after 32 weeks showed fewer foci of CAH than did Pten+/− mice treated with oil alone. Similar to previous reports26 we also observed marked fibrosis of the endometrial stroma in DES-treated mice (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Table 2.
Endometrial Lesions in DES-treated Mice
| Genotype | Age (weeks) | Treatment | n | No.(%) of mice with lesions | No. of lesions per mouse (mean ± SD) | Size of lesion (mm2) | Range of size (mm2) |
|---|---|---|---|---|---|---|---|
| +/− | 24 | DES | 11 | 8 (73) | 2.45 ± 2.77 | 0.11 ± 0.08 | 0.04−0.3 |
| +/− | 24 | Oil | 9 | 8 (88.9) | 9.78 ± 5.91 | 0.19 ± 0.16 | 0.04−0.64 |
| +/− | 32 | DES | 10 | 8 (80) | 6.70 ± 7.06⁎ | 0.14 ± 0.12 | 0.03−0.48 |
| +/− | 32 | Oil | 9 | 9 (100) | 18.56 ± 8.57⁎ | 0.22 ± 0.12 | 0.04−0.75 |
| +/+ | 24 | DES | 9 | 3 (33) | 1.22 ± 1.99 | 0.03 ± 0.01 | 0.02−0.04 |
| +/+ | 32 | DES | 11 | 4 (36) | 1.18 ± 1.83 | 0.11 ± 0.14 | 0.04−0.48 |
P = 0.0072, difference was statistically significant.
We next determined proliferation indices of uterine epithelia in the Pten+/− mice that had undergone oophorectomy, intact Pten+/− mice, and wild-type mice. Ki-67 immunostaining was used as a marker for proliferation, and positive cells were expressed as a percentage of the total counted in the normal epithelium and in the CAH. Oophorectomy significantly reduced the Ki-67-positive cells in the normal epithelium of Pten+/+ (Figure 2, A and B) and Pten+/− (Figure 2, C and D) mice. These observations suggest that lack of estrogen has a similar effect on normal epithelium irrespective of Pten haploinsufficiency. The CAH in the intact mice also had significantly more Ki-67-positive cells than the mice that had undergone oophorectomy (Figure 2, C–E). These findings provide a likely mechanism for the greater number and increased size of the CAH foci seen in the intact Pten+/− mice.
Figure 2.

Ki-67 immunostaining on uterine sections of intact Pten+/+ mice (A), Pten+/+ mice that had undergone oophorectomy (Ovx) (B), intact Pten+/− mice (C), and Pten+/− mice that undergone oophorectomy (D). Original magnification, ×40. (E) Quantitation of Ki-67-positive cells. Oophorectomy reduced Ki-67-positive cells in the normal epithelium of Pten+/+ mice. Statistical analysis was performed with the unpaired t-test. *P values ranging between 0.01 and 0.05 were considered significant; ***P values < 0.001 were considered highly significant.
We also performed immunostaining on the sections using antibodies to cyclin D1 (Figure 3). Cyclin D1 was localized to the nucleus in luminal and glandular epithelia, as well as CAH. The slides were scored for staining intensities as previously described. Overall, there was no significant difference between cyclin D1-positive cells in the normal epithelium of Pten+/+ uteri as compared to Pten+/− mice (Figure 3, A, C, and E). Absence of estrogen (oophorectomy) significantly reduced cyclin D1-positive cells in the normal epithelium of Pten+/+ and Pten+/− mice (Figure 3, B, D, and E). In intact Pten+/− mice, cyclin D1-positive cells in CAH were significantly reduced compared with the normal epithelium (Figure 3, E and F), showing down-regulation of cyclin D1 in association with Pten loss in the setting of physiological estrogen levels. However, in the absence of estrogen, no difference was observed in the levels of cyclin D1 between CAH and normal epithelium. In addition, cyclin D1-positive cells in CAH of Pten+/− mice or Pten+/− mice that had undergone oophorectomy (Figure 3F) did not differ significantly, despite the differences in proliferative indices as measured by Ki-67 staining. The proliferative response to estrogen in the normal luminal and glandular epithelium likely involves cyclin D1 in contrast to CAH in which cyclin D1 is down-regulated in the presence or absence of estrogen. This suggests that Pten loss is associated with a decrease in cyclin D1 levels.
Figure 3.

Cyclin D1 immunostaining of intact Pten+/+ mice (A), Pten+/+ mice that had undergone oophorectomy (Ovx) (B), intact Pten+/− mice (C), and Pten+/− mice that had undergone oophorectomy (D). Original magnification, ×20. (E) Quantitation of cyclin D1-positive cells. (F) Loss of Pten reduced cyclin D1 levels in CAH lesions compared with the normal epithelium. Statistical analysis was performed with the Mann–Whitney test. *P values between 0.01 and 0.05 were considered significant; **P values between 0.001 and 0.01 were considered very significant.
Inactivation of GSK3β Is an Estrogen-Independent Downstream Event in the Pten Pathway
Numerous studies have shown that Pten exerts its tumor suppressive function through a variety of mechanisms. The most common mechanism is preventing activation of Akt by inhibiting phosphorylation on Ser473. In the endometrium, all CAH display the characteristic membrane staining of p-Akt, further supporting activation of Akt to be a consistent signaling event in endometrial tumorigenesis downstream of Pten loss.24 GSK3β is considered a critical downstream element of the PI3K/Akt pathway. Phospho-Akt inhibits GSK3β action by phosphorylating Ser9, leading to progression of cells into S phase.27–31 We therefore investigated the expression of p-GSK3β in CAH by immunostaining (Figure 4). Approximately 90% of CAHs in Pten+/− mice that had undergone oophorectomy (Figure 4B) showed increased expression of phosphorylated GSK3β compared with CAH in the intact Pten+/− mice (Figure 4A), indicating that GSK3β inactivation, initiated by Pten loss, occurs most robustly in the absence of estrogen when cyclin D1 levels are down (Mann–Whitney test, P = 0.005; Figure 4C). Despite increased levels of p-GSK3β, proliferation in CAH of Pten+/− mice that had undergone oophorectomy was reduced compared with intact mice.
Figure 4.

p-GSK3β(S9) immunostaining in CAH of intact Pten+/− mice (A) and Pten+/− mice that had undergone oophorectomy (B). Original magnification, ×20. Phosphorylation at Ser 9 was significantly up-regulated in the mice that had undergone oophorectomy (C). Statistical analysis was performed with the Mann–Whitney test. **P = 0.005.
Prolonged Estrogen Exposure in Pten+/− Mice Accelerates Progression to Invasion
To avoid the use of mixed background B6-129 mice for studying hormonal effects, we generated congenic C57BL6 Pten+/− mice by back-crossing the mixed B6-129 Pten+/− mice with C57BL6 mice for 10 generations (N10). The N10 generation, however, completely lost the endometrial phenotype (see Supplemental Figure S2, A and B, at http://ajp.amjpathol.org), which was restored in the F1 hybrids generated by the cross between Pten+/− N10 C57BL6 and wild-type 129SvJ mice. The reason for loss of phenotype is unknown, but, as this experiment definitely found, it is due to strain differences. Therefore, we generated Pten+/− mice in the CD-1 background because this outbred strain had been used to study the effect of neonatal exposure to DES. In this strain, neonatal DES treatment resulted in endometrial adenocarcinomas in 90% of the mice by 18 months of age.23,32 CD-1 Pten+/− mice develop CAH similar to the mixed C57BL6/129SvJ strain, both qualitatively and quantitatively. Adult CD-1 Pten+/− females that had undergone oophorectomy were implanted with either estrogen pellets or placebos once or twice as described in Materials and Methods and were sacrificed after 12 or 24 weeks of estradiol exposure.
As expected, only the Pten+/− mice developed CAH or carcinoma (Table 3 and Figure 5). Wild-type mice, even those treated with estrogen for 24 weeks, developed dilated complex hyperplasia without cytologic atypia (Figure 5A). The Pten+/− mice, treated with placebo pellets (Figure 5B) or estrogen pellets for 12 weeks (Figure 5C), developed CAH with no significant difference between the number of lesions or the size (Table 3). However, three of four Pten+/− mice (75%) treated with estradiol for 24 weeks (Figure 5D) developed myoinvasive carcinomas, whereas the placebo controls for this group developed only CAH. Of note, the number of lesions in the Pten+/− mice did not differ significantly between those treated with estradiol for 24 weeks and the corresponding placebo controls (Table 3). Thus, prolonged exposure to supraphysiological levels of estrogen increased the incidence of invasive endometrial carcinoma in the background of Pten heterozygosity. Of note, we have never observed carcinoma at 36 weeks of age in intact Pten+/− mice not treated with estrogen.
Table 3.
Endometrial Lesions in Estradiol-Treated Pten Heterozygous Mice That Had Undergone Oophorectomy
| Genotype | Treatment (weeks) | n | No. (%) of mice with disease | No. (%) of mice with CAH | Mice with invasion | No. of CAH per mouse (mean ± SD) | Size (mm2) of CAH (mean ± SD) | Range of size (mm2) |
|---|---|---|---|---|---|---|---|---|
| Pten+/−(E) | 24 | 5 | 5/5 (100) | 5 | 3 | 19.8 ± 3.86‡ | 0.20 ± 0.17 | 0.04–1.2 |
| Pten+/−(P) | 24 | 5 | 5/5 (100) | 5 | 0 | 24.4 ± 9.29‡ | 0.14 ± 0.11 | 0.04–0.8 |
| Wild type (E) | 24 | 5⁎ | 0 | 0 | 0 | |||
| Pten+/−(E) | 12 | 7 | 7/7 (100)† | 6 | 0 | 6.86 ± 4.30§ | 0.11 ± 0.09 | 0.04–0.25 |
| Pten+/−(P) | 12 | 7 | 7/7 (100) | 7 | 0 | 8.4 ± 5.27§ | 0.12 ± 0.09 | 0.04–0.4 |
| Wild type (E) | 12 | 6⁎ | 0 | 0 | 0 |
E, estradiol pellets; P, placebo pellets.
Mice had dilated complex hyperplasia without atypia.
One mouse showed hyperplasia without atypia.
P > 0.05, difference was not statistically significant.
P > 0.05, difference was not statistically significant. These numbers were not compared with those in ‡.
Figure 5.

H&E staining of uteri of CD-1 Pten+/+ mice that had undergone oophorectomy (A) treated with estradiol pellets for 24 weeks. Pten+/− mice (B) treated with placebo pellets for 24 weeks, estradiol pellets for 12 weeks (C) and 24 weeks (D). Prolonged and excess estradiol exposure increased the incidence of invasive carcinoma (arrows in D). E, estradiol; P, placebo pellets. ERα immunostaining of Pten+/− mice treated with placebo pellets (E) or estradiol pellets for 24 weeks (F). Quantitation of nuclear ERα IHC (G). Invasive carcinoma was associated with reduced nuclear ERα (Mann–Whitney test, *P = 0.02), as seen in the insets. Original magnification: ×20 (A–F)l ×60 (insets of E and F).
Uterine sections of CD-1 Pten+/− mice were further analyzed for expression of ERα and p-GSK3β by IHC. In this mouse model as well, we observed increased phosphorylation of GSK3β in CAH of the placebo-treated group compared with the hormone-treated animals (Mann–Whitney test, P = 0.016; data not shown). Analysis of ERα IHC found that ERα expression was predominantly nuclear. When CAH arising in the placebo-treated animals (Figure 5E) was compared with the carcinomas in the estradiol-treated animals (Figure 5F), the nuclear ERα expression was significantly reduced in the carcinomas (Figure 5G). Treatment with estradiol for 24 weeks did not significantly affect the expression of ERα in the normal epithelium compared with the placebo group (Figure 5G).
Endometrial Tumorigenesis in Pten+/− Mice Occurs in the Absence of ERα
ERα is the predominant estrogen receptor in the uterus, substantiated by the phenotypes of the ERα versus ERβ knockout mice.8 Therefore, to dissect the role of ERα in endometrial tumorigenesis, we crossed the Pten+/− mice in the B6-129 mixed background to the ERα+/− mice (C57/BL6 background) to generate Pten+/−;ERα−/− mice. Wild-type (Pten+/+;ERα+/+) uteri were histologically normal (Figure 6A). At 32 weeks (the age at which 100% of Pten+/− mice develop CAH), the uteri of the Pten+/+;ERα−/− (Figure 6B) and Pten+/−;ERα−/− (Figure 6D) mice were hypoplastic, as would be expected because of the absence of ERα. Despite being hypoplastic, uteri of 8 (of 9) Pten+/−;ERα−/− mice had CAH and/or carcinoma (Figure 6D). Further, 4 of the 8 mice with lesions exhibited either in situ carcinoma or carcinoma with myometrial invasion (Table 4). In contrast, all of the age-matched Pten+/−;ERα+/− (Figure 6C) and Pten+/−;ERα+/+ mice developed only CAH. Interestingly, as in the CD-1 mice, the incidence and size of the CAH foci in Pten+/−;ERα+/+ and Pten+/−;ERα+/− mice did not differ significantly (Table 4), but the incidence of carcinoma was significantly increased only in the Pten+/−;ERα−/− mice.
Figure 6.

H&E stained sections of uteri in Pten+/+;ERα+/+ (A), Pten+/+;ERα−/− (B), Pten+/−;ERα+/− (C), and Pten+/−;ERα−/− (D) mice. Carcinoma develops in the absence of ERα and invades into the myometrium. Original magnification, ×20.
Table 4.
Endometrial Lesions in Pten+/−ER−/− Mice and Littermates
| Genotype | Age (weeks) | n | Mice with lesions | Mice with in situ CA | Mice with invasion | Lesions per mouse (mean ± SD) | Size of lesion (mm2) |
|---|---|---|---|---|---|---|---|
| Pten+/+ER+/+ | 32 | 2 | 0 | 0 | 0 | ||
| Pten+/+ER−/− | 32 | 3 | 0 | 0 | 0 | ||
| Pten+/−ER+/+ | 32 | 10 | 10/10 | 0 | 0 | 18.56 ± 8.57 | 0.22 ± 0.12⁎ |
| Pten+/−ER+/− | 32 | 7 | 7/7 | 0 | 0 | 5.5 ± 0.71 | 0.13 ± 0.08⁎ |
| Pten+/−ER−/− | 32 | 9 | 8/9 | 2/8 | 2/8 | 9.57 ± 6.78 | 0.51 ± 0.84 |
P = 0.104, difference was not statistically significant.
Next, we performed Ki-67 immunostaining on uterine sections of mice of all genotypes (Figure 7, A–D), and the percentage of positive Ki-67 cells in CAH and/or carcinoma and normal epithelium were determined (Figure 8A). As expected, the normal epithelium in Pten+/+;ERα−/− (Figure 7B) and Pten+/−;ERα−/− (Figure 7D) mice showed reduced proliferation compared with the Pten+/+;ERα+/+ (Figure 7A) and Pten+/−;ERα+/− (Figure 7C) genotypes. The CAH and carcinoma in the Pten+/−;ERα−/− mice (7.2 ± 4.6) had reduced Ki-67 staining compared with the CAH in Pten+/−;ERα+/+ (46.25 ± 9.6) or Pten+/−;ERα+/− (38 ± 7.2) mice (Figure 8A).
Figure 7.

Immunostaining with Ki-67 (A–D) and cyclin D1 (E–H) on uteri from Pten+/+;ERα+/+ (A and E), Pten+/+;ERα−/− (B and F), Pten+/−;ERα+/− (C and G), and Pten+/−;ERα−/− (D and H) mice. Invasive carcinomas in the Pten+/−;ERα−/− mice exhibit reduced number of Ki-67-positive cells but increased staining of cyclin D1. Original magnification, ×20.
Figure 8.
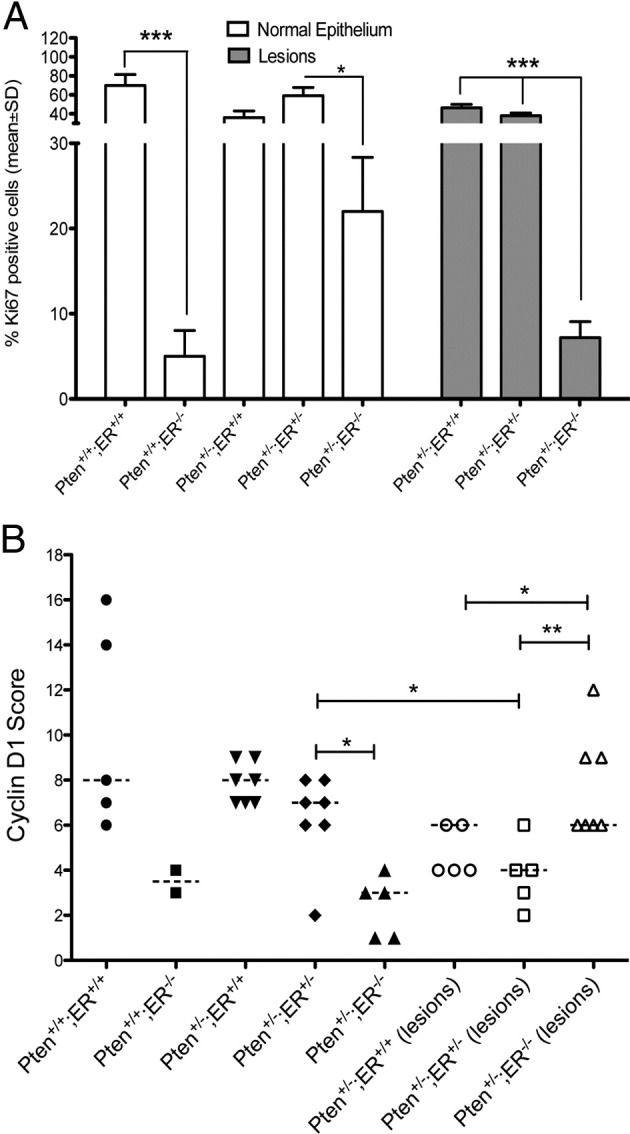
Quantitation of Ki-67 (A) and Cyclin D1 (B) immunostaining depicted in Figure 7. Statistical analysis for that percentage of positive Ki-67 cells was performed with the unpaired t-test. *P values ranging between 0.01 and 0.05 were considered significant; ***P values < 0.001 were considered highly significant. For cyclin D1, Mann–Whitney test was used. *P values ranging between 0.01 and 0.05 were considered significant; **P values ranging between 0.001 and 0.01 were considered very significant.
Expression of cyclin D1 was analyzed by immunostaining (Figure 7, E–H), and the slides were scored for staining intensities (Figure 8B). Loss of ERα reduced the number of cyclin D1-positive cells in the normal epithelium in both the Pten+/+ and Pten+/− uteri (Figure 7, F and H) compared with the wild-type mice (Figure 7E). CAH in Pten+/−;ERα+/− (Figure 7G) exhibited reduced cyclin D1-positive cells compared with that in the normal epithelium (Figure 8B), similar to results with the Pten+/− mice that had undergone oophorectomy, supporting the notion that Pten loss is associated with down-regulation of cyclin D1. Pten+/−;ERα−/− mice (Figure 7H) exhibited a different pattern. The normal epithelium had reduced levels of cyclin D1-positive cells compared with Pten+/−;ERα+/− and Pten+/−;ERα+/+ mice (Figure 8B). However, paradoxically the invasive carcinomas expressed more cyclin D1-positive cells compared with CAH in Pten+/−;ERα+/− and Pten+/−;ERα+/+ mice despite reduced Ki-67 staining (Figure 8B).
We analyzed the expression of progesterone receptor in the uteri of Pten+/−;ERα−/− mice by IHC (see Supplemental Figure S3, A and B, at http://ajp.amjpathol.org). PR expression was predominantly nuclear in the epithelium and stroma. CAH and carcinomas were also positive for PR. We did not observe significant differences in the expression of PR between the normal epithelium and CAH or between CAH and invasive carcinoma in all of the genotypes. Even the highly invasive lesions observed in the Pten+/−;ERα−/− mice expressed PR at similar levels.
Analysis of Pathways Using Hec1A Cells with Somatic Deletion of PTEN
To analyze the interaction between estrogen-ERα signaling and Pten in vitro, we used the human endometrial carcinoma cell line Hec1A, which expresses wild-type PTEN as well as ERα (Figure 9). We obtained two isogenic clones (clone 16 and clone 22), which differ from the parental cell line only for genomic deletion of PTEN. These clones exhibit increased phosphorylation of AKT (Figure 9A). Surprisingly, cyclin D1 levels in clones 16 and 22 were always reduced compared with the parental cells under normal cell culture conditions (Figure 9A). Clones 16 and 22 have similar expression profiles of p-AKT and cyclin D1; therefore, only clone 16 was used in subsequent experiments.
Figure 9.

Analysis of parental and PTEN-deleted (clones 16 and 22) Hec1A cells by immunoblot analysis (A). Deletion of PTEN activated AKT in clones 16 and 22. Both clones 16 and 22 expressed reduced cyclin D1 levels under normal growth conditions. Growth of parental cells was affected by high (10%) or low (1%) serum, whereas serum had no effect on growth of clone 16 (C). All time points for the proliferation assay were done in triplicate, and the values represent results from three independent experiments. Immunoblot analysis of PTEN, p-AKT, p-ERK, and cyclin D1 in whole-cell lysates of serum-starved cells after adding serum for indicated time periods (B) and represent the results obtained from three independent experiments. Addition of 10% serum increased cyclin D1 levels in the parental cells by at least threefold to fourfold as early as 30 minutes (D).
As a first step, we compared the growth characteristics of parental and PTEN-deleted clone 16 with the use of the XTT assay (Figure 9C). The parental Hec1A cells grew faster than clone 16 in conditions of high serum. Conditions of low serum did not affect the growth of clone 16 but significantly reduced the growth of the parental cells. We next analyzed the response of the cell lines to serum starvation followed by restimulation with 10% fetal bovine serum by immunoblot analysis of downstream signaling molecules in the PI3K/Akt/Pten pathway (Figure 9B). Clone 16 and parental cells differed in their response to serum addition. In the absence of serum, both cell types expressed low levels of cyclin D1. Addition of serum increased the levels of cyclin D1 protein within 15 minutes in the parental cells, and they remained high thereafter (Figure 9D). However, in clone 16, the addition of serum had no effect on cyclin D1, and the levels remained low even 24 hours after the addition of serum. No effect was observed on p-ERK levels in either of the cell lines. All together, these observations suggest that deletion of PTEN renders the cells unresponsive to serum, which may be due, at least in part, to constitutive down-regulation of cyclin D1. In addition, the reduced cyclin D1 expression correlates with observations made in the mouse models in which Pten-negative lesions generally expressed low levels of cyclin D1.
Discussion
Although we have learned a great deal about the relation of genetics and hormones from epidemiologic and molecular studies in humans, genetic mouse models allow direct manipulation of circulating estradiol levels in the setting of relevant genetic changes. In the present study, we used genetically engineered mouse models to investigate the complex interaction between PTEN and estrogen signaling in the pathogenesis of endometrial carcinoma.
We first showed that CAH developed in 90% of Pten+/− mice that had undergone oophorectomy, although the foci were reduced in number and size compared with controls. These findings suggest that in the setting of a Pten mutation physiological estrogen levels may contribute to the development of hyperplasia. Mice that had undergone oophorectomy also lack progesterone, which may affect proliferation of endometrial epithelium; however, a previous study of Pten mutant mice suggested that treatment with progestin did not significantly reduce endometrial hyperplasia.25
Given the association of unopposed estrogen stimulation and endometrial carcinoma, we exposed Pten+/− mice to exogenous estrogen. The presence of prolonged excess estradiol resulted in a significant increase in invasive carcinoma, despite similar numbers and size of CAH foci, suggesting that prolonged exposure to high levels of unopposed estrogen promotes progression of CAH to carcinoma in the setting of Pten loss. It was concluded from recent epidemiologic studies that PTEN mutations were not associated with the development of endometrial carcinoma.33 Our findings provide further support for Pten loss as an important cause of endometrial hyperplasia with additional factors, including unopposed estrogen, promoting the progression of hyperplasia to carcinoma. The mechanisms by which unopposed estrogen causes this progression are not known at this time and require further study. However, it is clear that additional genetic alterations are important. In primary human tumor tissues, we have previously reported that PTEN mutations are common in both hyperplasia and carcinoma, whereas PIK3CA mutations are limited largely to carcinomas,34 suggesting a role for PI3K activation in the invasive phenotype. In addition, it is possible that reduction in ERα expression, as seen here in the mouse model, may play a role in the progression of the disease.
ERα and ERβ are the most well-characterized nuclear receptors for estrogen. In addition, a significant body of literature reports that estrogen signaling occurs via membrane-bound receptors.22,35,36 In the uterus ERα is the predominant receptor as shown by the phenotypes of ERα and ERβ knockout mice.6,37 In addition, the proliferative response to growth factors in the uterus is mediated via the full-length ERα.38 Because it had been shown that p-Akt results in ligand-independent phosphorylation of ERα and that ERα can activate the PI3K pathway, we sought to determine the role of ERα in the development of endometrial carcinoma in a Pten+/−;ERα−/− mouse model. Despite being severely hypoplastic, uteri of Pten+/−;ERα−/− mice exhibited endometrial hyperplasia/carcinoma. In fact, absence of ERα led to an increased incidence of in situ and invasive carcinoma. When we analyzed the uteri for the percentage of Ki-67-positive cells, the invasive carcinomas had a significantly lower proliferation index than did CAH in the Pten+/−;ERα+/− and Pten+/−;ERα+/+ mice.
Although epithelial cells express estrogen receptor, it is believed that estrogen-induced epithelial proliferation is driven by soluble paracrine factors produced by the stroma. Given the extensive cross talk in vivo between the stromal and epithelial cells, it is likely that lack of ERα in the stroma might also contribute to endometrial tumorigenesis in Pten+/−;ERα−/− mice. A study that described stroma-specific deletion of adenomatous polyposis coli tumor suppressor gene in mice led to endometrial cancer.39 Adenomatous polyposis coli deletion affected estrogen responsiveness of the stroma, which was because of reduced ERα expression. Normal epithelial PR expression is also down-regulated by estrogen, acting via stromal ERα receptors.40 Interestingly, the invasive carcinomas in Pten+/−;ERα−/− uteri did not exhibit reduced PR expression, likely because of ERα-negative stroma. Although there are differences between stromal-epithelial interactions in rodent models and humans, potentially important information can be provided by these mouse models for subsequent validation in human tissue and cells. To this end, we plan on generating epithelial cell-specific Pten−/−;ERα−/− mice by breeding PtenloxP/loxP mice with previously described exon 3-floxed αER knockout mice.41 In a previous study, ICI treatment to reduce the levels of ERα resulted in a loss of the endometrial hyperplasia/carcinoma phenotype.19 That study was one of the reasons the present genetic approach was undertaken. There are a number of possible explanations for the different results of the two studies, the most relevant being that treatment with ICI only reduced the levels of ERα but did not eliminate them entirely. Furthermore, the strains of mice and the timing of the ERα loss are different.
Cyclin D1 is normally up-regulated in estrogen-induced epithelial cell proliferation in the uterus31,42,43 and is overexpressed in several cancers, including UEC. Despite the presence of considerable proliferation, cyclin D1-positive cells in CAH of intact and oophorectomized Pten+/− uteri were significantly lower than the normal epithelium. Another intriguing observation was the increased phosphorylation of GSK3β in CAH of Pten+/− mice that had undergone oophorectomy. Phosphorylation at serine 9 by Akt inactivates GSK3β, allowing cells to progress to S phase, resulting in proliferation in response to growth factor signaling,44,45 mediated in part by stabilization of cyclin D1. Estrogen signaling (via membrane-associated ERs) can also activate the PI3K pathway and lead to phosphorylation of GSK3β. Given that CAH is negative for Pten expression, our observations suggest that biallelic inactivation of Pten, in the absence of estrogen, activates alternate proliferation pathways in epithelial cells via inactivation of GSK3β, which do not involve cyclin D1. Further support for altered response of cells because of Pten deletion was provided by the observation that PTEN-deleted human endometrial carcinoma cell line Hec1A was insensitive to the presence of serum for growth characteristics and modulation of cell cycle proteins compared with the parental cell line. Interestingly, similar to the mouse models presented here, cyclin D1 expression was significantly reduced in the clone, possibly causing the refractoriness to serum. However, invasive carcinomas also had significantly higher cyclin D1-positive cells, suggesting that, in the progression to invasive endometrial disease, cyclin D1 may be involved in other processes besides entry into the G1 phase of the cell cycle. Given the observation that cyclin D1 is overexpressed in many other tumor types and that it cannot induce cellular transformation by itself, several alternate functions for cyclin D1 other than the classical G0 to G1 transition in the cell cycle have been suggested.46 Among the several functions, its ability to influence transcription by interaction with steroid receptors and the transcription machinery47,48 might be highly relevant in the context of endometrial carcinoma.
These studies highlight the complex interaction between hormones and genetics in the development of UEC. Importantly, however, the mouse studies have shed light on some important issues that are encountered in the treatment of this common cancer in women. For example, the results presented herein provide evidence for the fact that biallelic inactivation of PTEN can result in CAH in the absence of estrogen. This finding may explain why women without clinical evidence of unopposed estrogen stimulation can develop CAH. Furthermore, we also show that unopposed estrogen stimulation in the setting of PTEN mutation may increase the likelihood of progression to carcinoma. These findings may have clinical ramifications for the treatment of PTEN-deficient endometrial hyperplasia and the treatment of Cowden disease. Finally, these studies suggest that the loss of ERα may not simply be a consequence of decreasing tumor differentiation, as is the common interpretation, but may be a mechanism by which tumors become more aggressive.
Footnotes
Supported by grants from the National Cancer Institute, NIH (R01 CA095427 to L.H.E.) and from the National Institute of Environmental Health Sciences Division of Intramural Research (Z01ES70065 to K.S.K.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.03.006.
Supplementary data
Photomicrographs of H&E-stained uterine sections of Pten+/− mice with neonatal treatment of DES or oil. DES-treated mice exhibited stromal fibrosis as indicated by arrows in the inset compared with normal stroma in mice treated with oil. Original magnification: ×20; ×60 (inset).
Loss of uterine phenotype in N10 generation of Pten+/− C57BL6 mice. A: Photomicrographs of H&E-stained uterine section from Pten+/− C57BL6 N10 and F1 hybrid C57BL6/129SvJ strain. CAH was observed only in the F1 hybrids (arrow). B: The number of lesions in Pten+/− mice from the two strains. C57BL6/129SvJ F1 hybrid mice had significantly more lesions compared with the pure C57BL6 strain (P < 0.0001).
Progesterone receptor (PR) immunostaining and scoring. A: Photomicrographs of H&E-stained uterine sections from Pten+/+;ERα−/−, Pten+/−;ERα−/−, Pten+/−;ERα+/−, and Pten+/−;ERα+/+ mice. PR staining was nuclear in normal epithelium and CAH. B: IHC scores for PR staining. No significant difference was observed between nuclear staining of PR in normal epithelium, CAH, and invasive carcinoma.
References
- 1.Di Cristofano A., Ellenson L.H. Endometrial carcinoma. Annu Rev Pathol. 2007;2:57–85. doi: 10.1146/annurev.pathol.2.010506.091905. [DOI] [PubMed] [Google Scholar]
- 2.Risinger J.I., Hayes A.K., Berchuck A., Barrett J.C. PTEN/MMAC1 mutations in endometrial cancers. Cancer Res. 1997;57:4736–4738. [PubMed] [Google Scholar]
- 3.Tashiro H., Blazes M.S., Wu R., Cho K.R., Bose S., Wang S.I., Li J., Parsons R., Ellenson L.H. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res. 1997;57:3935–3940. [PubMed] [Google Scholar]
- 4.Levine R.L., Cargile C.B., Blazes M.S., van Rees B., Kurman R.J., Ellenson L.H. PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res. 1998;58:3254–3258. [PubMed] [Google Scholar]
- 5.Maxwell G.L., Risinger J.I., Gumbs C., Shaw H., Bentley R.C., Barrett J.C., Berchuck A., Futreal P.A. Mutation of the PTEN tumor suppressor gene in endometrial hyperplasias. Cancer Res. 1998;58:2500–2503. [PubMed] [Google Scholar]
- 6.Couse J.F., Korach K.S. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 7.Ellmann S., Sticht H., Thiel F., Beckmann M.W., Strick R., Strissel P.L. Estrogen and progesterone receptors: from molecular structures to clinical targets. Cell Mol Life Sci. 2009;66:2405–2426. doi: 10.1007/s00018-009-0017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Couse J.F., Lindzey J., Grandien K., Gustafsson J.A., Korach K.S. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology. 1997;138:4613–4621. doi: 10.1210/endo.138.11.5496. [DOI] [PubMed] [Google Scholar]
- 9.Donjacour A.A., Cunha G.R. Stromal regulation of epithelial function. Cancer Treat Res. 1991;53:335–364. doi: 10.1007/978-1-4615-3940-7_16. [DOI] [PubMed] [Google Scholar]
- 10.Cooke P.S., Buchanan D.L., Lubahn D.B., Cunha G.R. Mechanism of estrogen action: lessons from the estrogen receptor-alpha knockout mouse. Biol Reprod. 1998;59:470–475. doi: 10.1095/biolreprod59.3.470. [DOI] [PubMed] [Google Scholar]
- 11.Cooke P.S., Buchanan D.L., Young P., Setiawan T., Brody J., Korach K.S., Taylor J., Lubahn D.B., Cunha G.R. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc Natl Acad Sci U S A. 1997;94:6535–6540. doi: 10.1073/pnas.94.12.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunha G.R., Cooke P.S., Kurita T. Role of stromal-epithelial interactions in hormonal responses. Arch Histol Cytol. 2004;67:417–434. doi: 10.1679/aohc.67.417. [DOI] [PubMed] [Google Scholar]
- 13.Salmena L., Carracedo A., Pandolfi P.P. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 14.Ignar-Trowbridge D.M., Hughes A.R., Putney J.W., Jr, McLachlan J.A., Korach K.S. Diethylstilbestrol stimulates persistent phosphatidylinositol lipid turnover by an estrogen receptor-mediated mechanism in immature mouse uterus. Endocrinology. 1991;129:2423–2430. doi: 10.1210/endo-129-5-2423. [DOI] [PubMed] [Google Scholar]
- 15.Cantley L.C. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 16.Sun M., Paciga J.E., Feldman R.I., Yuan Z., Coppola D., Lu Y.Y., Shelley S.A., Nicosia S.V., Cheng J.Q. Phosphatidylinositol-3-OH kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res. 2001;61:5985–5991. [PubMed] [Google Scholar]
- 17.Simoncini T., Hafezi-Moghadam A., Brazil D.P., Ley K., Chin W.W., Liao J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell R.A., Bhat-Nakshatri P., Patel N.M., Constantinidou D., Ali S., Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 19.Vilgelm A., Lian Z., Wang H., Beauparlant S.L., Klein-Szanto A., Ellenson L.H., Di Cristofano A. Akt-mediated phosphorylation and activation of estrogen receptor alpha is required for endometrial neoplastic transformation in Pten+/− mice. Cancer Res. 2006;66:3375–3380. doi: 10.1158/0008-5472.CAN-05-4019. [DOI] [PubMed] [Google Scholar]
- 20.Levin E.R. Nuclear receptor versus plasma membrane oestrogen receptor. Novartis Found Symp. 2000;230:41–50. doi: 10.1002/0470870818.ch5. discussion 50–45. [DOI] [PubMed] [Google Scholar]
- 21.Coleman K.M., Smith C.L. Intracellular signaling pathways: nongenomic actions of estrogens and ligand-independent activation of estrogen receptors. Front Biosci. 2001;6:D1379–D1391. doi: 10.2741/coleman. [DOI] [PubMed] [Google Scholar]
- 22.Moggs J.G., Orphanides G. Estrogen receptors: orchestrators of pleiotropic cellular responses. EMBO Rep. 2001;2:775–781. doi: 10.1093/embo-reports/kve185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newbold R.R., Bullock B.C., McLachlan J.A. Uterine adenocarcinoma in mice following developmental treatment with estrogens: a model for hormonal carcinogenesis. Cancer Res. 1990;50:7677–7681. [PubMed] [Google Scholar]
- 24.Wang H., Douglas W., Lia M., Edelmann W., Kucherlapati R., Podsypanina K., Parsons R., Ellenson L.H. DNA mismatch repair deficiency accelerates endometrial tumorigenesis in Pten heterozygous mice. Am J Pathol. 2002;160:1481–1486. doi: 10.1016/S0002-9440(10)62573-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fyles A., Wood G., Li M., Manoukian A.S., Gowing K., Khokha R., Chapman W., Tsao M.S. Neither ovariectomy nor progestin treatment prevents endometrial neoplasia in pten+/− mice. Gynecol Oncol. 2008;108:395–401. doi: 10.1016/j.ygyno.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 26.Begum M., Tashiro H., Katabuchi H., Suzuki A., Kurman R.J., Okamura H. Neonatal estrogenic exposure suppresses PTEN-related endometrial carcinogenesis in recombinant mice. Lab Invest. 2006;86:286–296. doi: 10.1038/labinvest.3700380. [DOI] [PubMed] [Google Scholar]
- 27.Cross D.A., Alessi D.R., Cohen P., Andjelkovich M., Hemmings B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 28.van Weeren P.C., de Bruyn K.M., de Vries-Smits A.M., van Lint J., Burgering B.M. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation: Characterization of dominant-negative mutant of PKB. J Biol Chem. 1998;273:13150–13156. doi: 10.1074/jbc.273.21.13150. [DOI] [PubMed] [Google Scholar]
- 29.Diehl J.A., Cheng M., Roussel M.F., Sherr C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srivastava A.K., Pandey S.K. Potential mechanism(s) involved in the regulation of glycogen synthesis by insulin. Mol Cell Biochem. 1998;182:135–141. [PubMed] [Google Scholar]
- 31.Chen B., Pan H., Zhu L., Deng Y., Pollard J.W. Progesterone inhibits the estrogen-induced phosphoinositide 3-kinase–>AKT–>GSK-3beta–>cyclin D1–>pRB pathway to block uterine epithelial cell proliferation. Mol Endocrinol. 2005;19:1978–1990. doi: 10.1210/me.2004-0274. [DOI] [PubMed] [Google Scholar]
- 32.Couse J.F., Davis V.L., Hanson R.B., Jefferson W.N., McLachlan J.A., Bullock B.C., Newbold R.R., Korach K.S. Accelerated onset of uterine tumors in transgenic mice with aberrant expression of the estrogen receptor after neonatal exposure to diethylstilbestrol. Mol Carcinog. 1997;19:236–242. doi: 10.1002/(sici)1098-2744(199708)19:4<236::aid-mc4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 33.Lacey J.V., Jr, Mutter G.L., Ronnett B.M., Ioffe O.B., Duggan M.A., Rush B.B., Glass A.G., Richesson D.A., Chatterjee N., Langholz B., Sherman M.E. PTEN expression in endometrial biopsies as a marker of progression to endometrial carcinoma. Cancer Res. 2008;68:6014–6020. doi: 10.1158/0008-5472.CAN-08-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayes M.P., Wang H., Espinal-Witter R., Douglas W., Solomon G.J., Baker S.J., Ellenson L.H. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin Cancer Res. 2006;12:5932–5935. doi: 10.1158/1078-0432.CCR-06-1375. [DOI] [PubMed] [Google Scholar]
- 35.Wehling M., Losel R. Non-genomic steroid hormone effects: membrane or intracellular receptors? J Steroid Biochem Mol Biol. 2006;102:180–183. doi: 10.1016/j.jsbmb.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 36.Kelly M.J., Levin E.R. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab. 2001;12:152–156. doi: 10.1016/s1043-2760(01)00377-0. [DOI] [PubMed] [Google Scholar]
- 37.Wada-Hiraike O., Hiraike H., Okinaga H., Imamov O., Barros R.P., Morani A., Omoto Y., Warner M., Gustafsson J.A. Role of estrogen receptor beta in uterine stroma and epithelium: Insights from estrogen receptor beta-/- mice. Proc Natl Acad Sci U S A. 2006;103:18350–18355. doi: 10.1073/pnas.0608861103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hewitt S.C., Kissling G.E., Fieselman K.E., Jayes F.L., Gerrish K.E., Korach K.S. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J. 2010;24:4660–4667. doi: 10.1096/fj.10-163428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanwar P.S., Zhang L., Roberts D.J., Teixeira J.M. Stromal deletion of the APC tumor suppressor in mice triggers development of endometrial cancer. Cancer Res. 2011;71:1584–1596. doi: 10.1158/0008-5472.CAN-10-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurita T., Lee K.J., Cooke P.S., Taylor J.A., Lubahn D.B., Cunha G.R. Paracrine regulation of epithelial progesterone receptor by estradiol in the mouse female reproductive tract. Biol Reprod. 2000;62:821–830. doi: 10.1093/biolreprod/62.4.821. [DOI] [PubMed] [Google Scholar]
- 41.Winuthayanon W., Hewitt S.C., Orvis G.D., Behringer R.R., Korach K.S. Uterine epithelial estrogen receptor alpha is dispensable for proliferation but essential for complete biological and biochemical responses. Proc Natl Acad Sci U S A. 2010;107:19272–19277. doi: 10.1073/pnas.1013226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altucci L., Addeo R., Cicatiello L., Germano D., Pacilio C., Battista T., Cancemi M., Petrizzi V.B., Bresciani F., Weisz A. Estrogen induces early and timed activation of cyclin-dependent kinases 4, 5, and 6 and increases cyclin messenger ribonucleic acid expression in rat uterus. Endocrinology. 1997;138:978–984. doi: 10.1210/endo.138.3.5002. [DOI] [PubMed] [Google Scholar]
- 43.Tong W., Pollard J.W. Progesterone inhibits estrogen-induced cyclin D1 and cdk4 nuclear translocation, cyclin E- and cyclin A-cdk2 kinase activation, and cell proliferation in uterine epithelial cells in mice. Mol Cell Biol. 1999;19:2251–2264. doi: 10.1128/mcb.19.3.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang J., Slingerland J.M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 45.Katso R., Okkenhaug K., Ahmadi K., White S., Timms J., Waterfield M.D. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 46.Kim J.K., Diehl J.A. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220:292–296. doi: 10.1002/jcp.21791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bienvenu F., Jirawatnotai S., Elias J.E., Meyer C.A., Mizeracka K., Marson A., Frampton G.M., Cole M.F., Odom D.T., Odajima J., Geng Y., Zagozdzon A., Jecrois M., Young R.A., Liu X.S., Cepko C.L., Gygi S.P., Sicinski P. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature. 2010;463:374–378. doi: 10.1038/nature08684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zwijsen R.M., Wientjens E., Klompmaker R., van der Sman J., Bernards R., Michalides R.J. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88:405–415. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Photomicrographs of H&E-stained uterine sections of Pten+/− mice with neonatal treatment of DES or oil. DES-treated mice exhibited stromal fibrosis as indicated by arrows in the inset compared with normal stroma in mice treated with oil. Original magnification: ×20; ×60 (inset).
Loss of uterine phenotype in N10 generation of Pten+/− C57BL6 mice. A: Photomicrographs of H&E-stained uterine section from Pten+/− C57BL6 N10 and F1 hybrid C57BL6/129SvJ strain. CAH was observed only in the F1 hybrids (arrow). B: The number of lesions in Pten+/− mice from the two strains. C57BL6/129SvJ F1 hybrid mice had significantly more lesions compared with the pure C57BL6 strain (P < 0.0001).
Progesterone receptor (PR) immunostaining and scoring. A: Photomicrographs of H&E-stained uterine sections from Pten+/+;ERα−/−, Pten+/−;ERα−/−, Pten+/−;ERα+/−, and Pten+/−;ERα+/+ mice. PR staining was nuclear in normal epithelium and CAH. B: IHC scores for PR staining. No significant difference was observed between nuclear staining of PR in normal epithelium, CAH, and invasive carcinoma.