


Abstract
During the origin of life, the biological information of nucleic acid polymers must have increased to encode functional molecules (the RNA world). Ribozymes tend to be compositionally unbiased, as is the vast majority of possible sequence space. However, ribonucleotides vary greatly in synthetic yield, reactivity and degradation rate, and their non-enzymatic polymerization results in compositionally biased sequences. While natural selection could lead to complex sequences, molecules with some activity are required to begin this process. Was the emergence of compositionally diverse sequences a matter of chance, or could prebiotically plausible reactions counter chemical biases to increase the probability of finding a ribozyme? Our in silico simulations using a two-letter alphabet show that template-directed ligation and high concatenation rates counter compositional bias and shift the pool toward longer sequences, permitting greater exploration of sequence space and stable folding. We verified experimentally that unbiased DNA sequences are more efficient templates for ligation, thus increasing the compositional diversity of the pool. Our work suggests that prebiotically plausible chemical mechanisms of nucleic acid polymerization and ligation could predispose toward a diverse pool of longer, potentially structured molecules. Such mechanisms could have set the stage for the appearance of functional activity very early in the emergence of life.
INTRODUCTION
The biology of modern organisms is based on RNA, DNA and proteins, but this biochemistry was probably preceded by a stage in which RNA molecules acted both as chemical catalysts and carriers of genetic information. Evidence for this early stage of life (the ‘RNA world’) includes the similarity of ancient chemical cofactors to certain ribonucleotides and the discovery that the catalytic core of the ribosome is composed of RNA (1–5). Possible pathways for the prebiotically plausible synthesis of the components of RNA and the polymerization of ribonucleotides have been reported by several groups (3,6–8). While natural selection could enhance low catalytic activity, the very earliest ribozymes must have arisen through chemical processes (3). Understanding the details of this initial emergence is a deep conceptual puzzle (9).
The first ribozymes must have emerged from pools of short sequences that were low in diversity and information content, but it is unclear how the complexity of these pools could be increased (10,11). These sequence pools would have been limited for at least two reasons. First, monomers would have different abundances because they are synthesized and degraded by different pathways. For example, a concentrated eutectic phase solution of ammonium cyanide yields significantly more adenine than guanine, uracil and cytosine (roughly 10× or more) (6). Degradation affects the nucleobases differently, with cytosine being particularly susceptible to spontaneous deamination (12). Indeed, the abundances of nucleobases detected in meteorites also vary by one or more orders of magnitude (13–16). Second, the rate at which different monomers are polymerized can vary by an order of magnitude (6,17–20). In one study of montmorillonite-catalyzed RNA synthesis, this bias led to a large reduction in diversity, as only 3 out of 32 possible pentamer sequences were formed in detectable amounts from a mixture of activated A and C monomers (21). These biases reduce the diversity of the sequences generated, restricting exploration of sequence space and thus reducing the probability of generating a sequence with biological function. While compositional biases might increase the probability of generating functional RNA (22–24), the magnitude of the biases associated with prebiotically plausible polymerization is still substantially larger than potentially favorable biases. Interestingly, ribozymes with limited compositional diversity have been made by a combination of rational design and in vitro evolution on restricted alphabets, but the resulting ribozymes exhibited decreased catalytic efficiency (the three-letter alphabet gave a 2500-fold decrease in kcat relative to the four-letter alphabet; the two-letter alphabet gave a further 10-fold decrease in kcat as well as a large decrease in total product conversion due to ribozyme misfolding) (25,26). While fine-tuning the conditions—e.g. by adjusting monomer ratios to counteract reduced reactivity or limiting UV irradiation to attain an appropriate monomer ratio (7,27,28)—could potentially overcome these biases, such conditions would be unlikely early on. Therefore, we sought more general mechanisms to counter compositional bias in nucleic acid pools undergoing prebiotically plausible reactions. Our experiments using DNA and simulations of binary sequences demonstrate that template-directed ligation is one such mechanism. Our RNA folding simulations suggest that compositionally diverse sequences are more likely to fold into stable structures compared with the substantially biased sequences that would be derived from template-independent processes. Greater stability is one of the factors promoting greater functional activity in RNA aptamers (29,30). In addition, our simulations indicate that template-directed ligation would shift the pool toward longer sequences, another important factor for activity (31,32). Our results suggest that a broad exploration of interesting sequence space was possible in prebiotic sequence pools despite initial chemical biases.
MATERIALS AND METHODS
Simulations
Two types of simulation were performed (stochastic and deterministic). Both simulations are limited for computational reasons. The stochastic simulation keeps track of each monomer or oligomer and implements a specific reaction at each time step. In practice, the simulation cannot keep track of an infinite number of reactants and possible reactions, so the system is limited by the total number of monomers considered. This mimics a protocell containing a relatively small number of monomers (e.g. 400, in which case the longest possible sequence would be 400 monomers). The stochastic simulation can generate sequences of ribozyme length. In contrast, the deterministic simulation keeps track of all possible species and reactions among them simultaneously, mimicking a very large, well-mixed system. In practice, the deterministic simulation cannot keep track of an infinite number of species, so the system must be truncated at a certain maximum length (e.g. 12).
The stochastic simulation was based on the Gillespie algorithm (33). Each simulation began with a pool of monomers. The total number of monomers in the system was typically limited to 400. During each iteration, an exponential waiting time was generated before a single reaction (concatenation, template-directed ligation or hydrolysis) occurred according to the relative rates of all possible reactions. Concatenation could occur between any two monomers or polymers. Realistic bias was introduced in the simulation as either a concatenation rate that depended on the identity of the 5′ monomer (e.g. reactivity ratio of 19:1), or as a difference in the initial number of monomers of each type (e.g. abundance ratio of 9:1). Template-directed ligation was possible if a 6-mer segment of one sequence (the template) was complementary to the trimer at the 3′-end of one substrate and the trimer at the 5′-end of the second substrate. Any sequence of length 6 or greater was a potential template; any sequence of length 3 or greater was a potential substrate. Circularization reactions were not considered. Hydrolysis of phosphodiester bonds occurred at a constant rate per bond. Characteristics of the system (average length and Ck) were measured at exponentially increasing time steps (i.e. 0, 1, 2, 4, 8, etc), and steady state was considered to be achieved when the characteristics at consecutive time steps deviated by <0.1%. SEs for these measurements were calculated from multiple simulation runs. The deterministic simulation used the same reactions as the stochastic simulation and kept track of the abundances of all possible sequences as the system evolved. The system was truncated at a maximum polymer length (typically 12) for computational tractability (i.e. polymers of the maximum length could not undergo further concatenation or ligation). Average Ck, average length and diversity were computed using steady-state abundances. The simulations were performed on the Odyssey Cluster of the FAS Research Computing Group at Harvard University. See Supplementary Data for simulation details.
Experiment: degenerate oligonucleotides for template-directed ligation of a heterogeneous sequence pool of DNA (four bases)
In heterogeneous pool reactions, all oligonucleotides were composed of A,C,G,T. Degenerate DNA oligonucleotides were obtained from Keck Oligo Synthesis Resource (Yale University, New Haven, CT, USA). Octamers and templates (40-mer) were synthesized as 5′-NNNN… using the facility's standard procedure for equimolar, degenerate oligonucleotides. Oligos were purified by reverse-phase cartridge. Octamers were phosphorylated as noted below, using non-radiolabeled ATP.
Experiment: template-directed chemical ligation reactions (four bases)
Reagents were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise specified. Synthetic degenerate oligonucleotides (templates of length 40; 5′-phosphorylated substrates of length 8) were mixed and ligated using cyanogen bromide following a previously published procedure (34). Reactions contained 1–2 µM DNA template and 16 µM DNA octamers. DNA sequences were mixed with buffer [0.23 M 2-(N-morpholino)ethanesulfonic acid, pH 7.4] and 19 mM MgCl2, in 4.5 µl of aqueous solution, heated to 95° for 3 min and annealed by cooling on the benchtop for 15 min. The solution was placed on ice for 5 min and 0.5 µl CNBr (5 M in acetonitrile) was added (final 0.5 M). After 1 min, the reactions were quenched by 100 µl ethanol, ethanol precipitated for >1 h at −20° and centrifuged at 12 000g at 4° for 30 min. Supernatant was removed and the pellets were dried on the benchtop overnight. The pellets were resuspended in 50 µl of water and aliquots were mixed with loading buffer for electrophoresis through a 24% urea-polyacrylamide gel following a standard protocol.
Experiment: next-generation sequencing of templates and products of template-directed ligation (four bases)
Ligation products of length 16 were purified by urea–PAGE. Products were dephosphorylated with alkaline phosphatase following the manufacturer's protocol (all enzymes were obtained from New England Biolabs, Ipswich, MA, USA, unless otherwise specified), phenol–chloroform extracted and ethanol precipitated. Products, templates and octamers were resuspended and ligated to a barcoded 3′ adapter sequence for Illumina sequencing using T4 RNA ligase. The barcoded 3′ adapter sequences were (lowercase = RNA):
TAG1: 5′-ucgTGTCGTATGCCGTCTTCTGCTTGTddC,
TAG2: 5′-ucgCATCGTATGCCGTCTTCTGCTTGTddC,
3-TAG1: 5′-ucgTACTCGTATGCCGTCTTCTGCTTGTddC,
3-TAG2: 5′-ucgACATCGTATGCCGTCTTCTGCTTGTddC,
3-TAG3: 5′-ucgCTATCGTATGCCGTCTTCTGCTTGTddC.
Products were gel purified by urea–PAGE and phosphorylated by PNK following the manufacturer's protocol, phenol–chloroform extracted and ethanol precipitated. Phosphorylated products were then ligated to the 5′ adapter sequence for Illumina sequencing using T4 RNA ligase. The 5′ adapter sequence was: 5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACaguccgacgauc. Products were gel purified by urea–PAGE and reverse transcribed with SuperScript III RT (Invitrogen, Carlsbad, CA, USA). cDNA products were gel purified on urea–PAGE and directly sequenced on an Illumina (Solexa) Genome Analyzer II. Polymerase chain reaction was not performed in order to avoid bias due to differential PCR amplification. As controls, template and substrate DNA were also prepared for sequencing following the same procedure.
Experiment: sequence analysis (four bases)
Sequence reads of the appropriate length containing the appropriately barcoded 3′ adapter sequence were extracted. The compositional diversity of each sequence was calculated for subsequence length k = 3. To eliminate effects due to the total length of the sequences, length 16 was chosen for analysis. Products were already of this length. For templates (length 40), C3 was calculated for all 16-mers contained in the template and the average was used as the C3 of the template. For octamers (length 8), simulated 16-mer ‘products’ were generated in silico by randomly joining octamer sequences obtained experimentally, and the C3 of these simulated random products was calculated.
Experiment: template-directed ligation with a single template (two bases)
Each reaction contained 1–2 µM DNA template and 16 µM random radiolabeled DNA octamers (Eurofins MWG Operon, Huntsville, AL, USA and Sigma-Aldrich, St Louis, MO, USA). In single-template reactions, the template oligonucleotides were composed of C and T while the octamers were composed of A and G. The templates used in the reactions of Figure 3b were:
5′-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT,
5′-CTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCT,
5′-TCCTTCCTTCCTTCCTTCCTTCCTTCCTTCCT,
5′-CCTCTTCTCCTCTTCTCCTCTTCTCCTCTTCT,
5′-CTCTTCTTTTCTCCTCTTCTTTTCTCCTCCCC.
Figure 3.

Experimental relationship between compositional diversity and template-directed ligation. (a) Fraction of DNA sequences having high compositional diversity (C3 > 0.95; analysis length = 16) after template-directed ligation in a heterogeneous pool of degenerate oligonucleotides including four bases. A greater fraction of reaction products (yellow) have high C3 relative to the templates (red = average C3 of 16-mers contained in sequenced 40-mer templates) and substrates (orange = C3 of 16-mers from in silico non-templated, random concatenation of experimentally sequenced octamers). The fraction of high C3 sequences in a uniform random pool is shown in green. Error bars are SDs from replicate sequencing experiments. (b) Polyacrylamide gel showing higher molecular weight products of template-directed ligation for different single templates from a binary alphabet. Molecular weight markers are given in the left lane. ‘Ø’ indicates a reaction without template added. Template C3 increases from left (C3 = 0) to right (C3 = 0.97; see ‘Materials and Methods’ section for list of sequences).
This set of templates utilizes a relatively small subset (25) of the theoretically possible octamers. These 25 octamers were mixed in equimolar ratio, phosphorylated and radiolabeled by T4 polynucleotide kinase (New England Biolabs, Ipswich, MA, USA) and added to the reaction, which was performed as described earlier. Band intensities were measured using a Typhoon TRIO Variable Mode Imager (Piscataway, NJ, USA). See Supplementary Data for more details.
DEFINITIONS AND MODEL
Measuring compositional bias for a given sequence
To measure the compositional bias, we use a definition inspired by grammar complexity and the Shannon entropy (35–37). For a sequence s of length L, we define Hk(s) by
where i is the index of a unique string of length k (k < L; i = 1 to 2k for a binary string) and pi is the frequency of the ith k-mer within s. If k = 1, Hk is just the Shannon entropy. The Hk for various k describe the compositional bias of a sequence, and the Hk for k > 1 reveal information that H1 by itself does not. Consider this hypothetical scenario: ‘0’ tends to follow ‘1’ and vice versa, such that most sequences are alternating (e.g. ‘01010101…’). In this case, H1 would be high even though the sequence is not very diverse. However, H2 is low, reflecting the lack of diversity on the ‘2-mer’ scale, and so on for higher k.
The Hk have the advantage of being straightforward to compute and relevant for RNA folding, and measuring the diversity of potential template subsequences (see below). Like any simple measure, however, they suffer from some drawbacks. First, Hk for a fixed k can suggest high internal sequence diversity when the sequence is actually highly regular. Some well-known measures of complexity [e.g. the number of states in the smallest finite state machine accepting the sequence (38), or algorithmic complexity, the length of the shortest program generating the sequence (39,40)] do not suffer from this limitation, but they are intractable to compute and do not incorporate information about the scale at which the underlying chemical processes are operating. Second, the calculation of Hk for k > 1 involves non-independent, overlapping sequences. This would be appropriate for measuring the diversity of potential ligation sites. However, one might imagine a scenario with much competition for binding to the same template (high concentrations of similar sequences); in that case, overlapping sites might interfere with one another and perhaps an alternative measure of diversity would be more appropriate. Nevertheless, we find Hk to have heuristic value for the analysis described below.
The maximum possible Hk (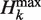 ) grows with total sequence length L. To focus on composition rather than length, we define the relative compositional diversity Ck of a sequence to be the ratio Hk(s)/
) grows with total sequence length L. To focus on composition rather than length, we define the relative compositional diversity Ck of a sequence to be the ratio Hk(s)/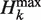 (L). In addition, we only compare Ck for sequences of the same length. Ck(s) measures the heterogeneity of strings of length k within sequence s. Ck(s) is zero when all subsequences are identical, and Ck(s) is one when all possible subsequences appear equally often in s (Table 1). For our purpose, if k is chosen to be too small, the compositional bias of longer k-mers is not captured, but if k is too large, Ck cannot distinguish well among different sequences. We generally used k = 3 except when analyzing long sequences (50-mer), when we used k = 4.
(L). In addition, we only compare Ck for sequences of the same length. Ck(s) measures the heterogeneity of strings of length k within sequence s. Ck(s) is zero when all subsequences are identical, and Ck(s) is one when all possible subsequences appear equally often in s (Table 1). For our purpose, if k is chosen to be too small, the compositional bias of longer k-mers is not captured, but if k is too large, Ck cannot distinguish well among different sequences. We generally used k = 3 except when analyzing long sequences (50-mer), when we used k = 4.
Table 1.
Example RNA sequences, compositional diversity and folding energy predicted by Viennafold (41)
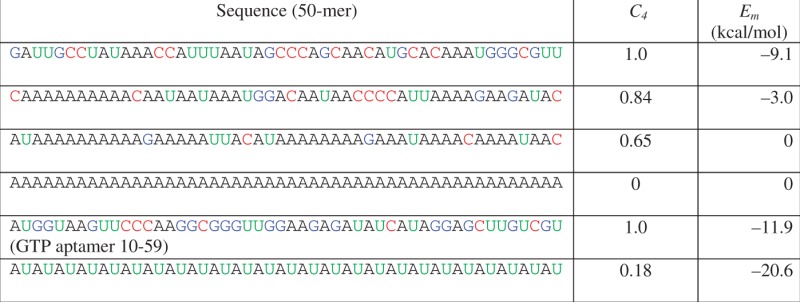 |
In rows 2 and 3, adenine is 5- or 10-fold more abundant than the other bases, respectively, representing a realistic bias in composition (6). The fifth row contains the sequence of a known aptamer (42); other sequences were computer-generated. The sixth row illustrates an unusual sequence with low C4 and low folding energy.
We find Ck to be a useful measure of the diversity of k-mers within sequence s, because values of Ck close to 1 would be desirable in the RNA world for at least two reasons. First, high Ck characterizes the vast majority of sequence space, because the number of different sequences corresponding to a particular composition is greater if the composition is more uniform (43). The total number of possible unique sequences varies approximately exponentially with Ck (Figure 1a). Biases in monomer composition and reactivity would decrease the average Ck and thus restrict the exploration of sequence space. For example, a 10-fold bias in composition decreases the average Ck from 0.94 to 0.43, which represents a severe restriction in sequence space given the exponential dependence (Supplementary Data). While sequence space might contain many potentially structured molecules (44), any search through sequence space for which the average Ck is low would under-represent or omit a large fraction of possible sequences. Therefore, high average Ck is desirable for finding rare, functional molecules.
Figure 1.

Compositional diversity, sequence space and predicted RNA folding energy. (a) Most of sequence space is of high compositional diversity. Histogram of C4 for RNA sequences, computed from random sampling of 109 sequences of length 50 (black dots) in silico. The complete histogram for all possible sequences of shorter length is computable and is similar to that of the random sample of 50-mers (length 10 = blue, 12 = pink, 14 = green, 17 = orange). (b) Compositional diversity (C4) and predicted minimum folding energy (Em) for known ribozymes (length 40–60; see Supplementary Data) (45) are shown as blue dots with mean and SD (blue lines). (c) C4 versus Em (black dots) predicted by Viennafold (41) for 2.5 × 106 RNA sequences of length 50. To minimize effects from GC-content, we restricted the in silico sampling to sequences whose GC content is 40–60%. To avoid sampling artifacts, sequences were assigned to five bins according to C4, and an equal number of unique sequences were analyzed in each bin. The bin averages are shown as the red line (see Supplementary Data for values and SDs).
Second, Ck appears to be correlated with RNA folding energy. Intuitively, an internally diverse sequence would present more independent opportunities for finding complementary regions compared to a repetitive sequence of the same length. A simple toy model for hairpin formation verifies this effect (Supplementary Data). Known ribozymes have nearly maximal C4 (average C4 of ribozyme sequence families of length 40–60 is 0.970 ± 0.013; Figure 1b, Supplementary Data) (45). On the other hand, some repetitive sequences have both low Ck and low folding energy [e.g. (AU)n; Table 1]. It is conceivable that high Ck in ribozymes could be merely a reflection of the fact that many were isolated from pools of nearly random sequences, which have high average Ck. To understand the relationship between Ck and folding, we computed the minimum folding energy (Em) for a large number of random RNA sequences (Figure 1c and Table 1). The fact that most random sequences have high Ck would cause a spurious correlation between Ck and Em simply because a greater range of Em is sampled by more sequences at high Ck. To determine the correlation between Ck and Em independently of this effect, we binned the RNA sequences according to C4 and analyzed an equal number of unique sequences in five bins centered between 0.6 and 1. We also limited our analysis to sequences with GC content of 40–60%, to avoid potential effects due to GC content alone. The folding energies are correlated with C4, and collectively the Ck explain 61% of the variance in Em according to principal components regression analysis (Supplementary Data). There is a notable paucity of energetically stable, low C4 sequences.
While stable folding is believed to be a prerequisite for function (46), Ck is not a perfect predictor of function. For example, sequence libraries containing deliberate repetitive patterns (alternating purine/pyrimidine) would have lower Ck on an average, but they perform at least as well as random libraries during SELEX because the design favors hairpin formation (46). Also, some rare structures might only be formed if the composition is biased. For example, sequences depleted in U and enriched in G are more likely to form stable structures, presumably because small regions of base-pairing are stabilized by this composition (22). Computational studies suggest that structures with long loop regions are favored by enrichment for A and C, and the optimal composition depends on the desired motif and structure (24). Loop regions in ribosomal RNAs tend to be A-rich, while G, C and U tend to comprise the stems (47). RNA folding simulations suggest that folding could be improved by a small compositional bias (nucleotide frequencies within 2-fold of each other) (23). It should be noted that the relationship of sequence, structure and function is complex and not yet fully understood, and well-folded structures are not always functional; mutations that preserve ribozyme fold might still destroy activity. Nevertheless, to the extent that structure may be important for function, in general Ck is a measure of compositional diversity and structural potential for a given sequence.
Measuring diversity of a pool
To quantify the diversity among different sequences in a pool, we also measure the population-level entropy D of a pool of molecules:
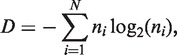 |
where i is an index for unique sequences, N is the total number of unique sequences in the pool and ni is the fraction of sequences in the pool that consist of copies of the ith sequence. D is zero if all molecules are identical and D is maximal when molecules are distributed uniformly through sequence space. In contrast to Ck (a property of each sequence), D is a property of the entire pool.
Simulations of prebiotically plausible reactions
To understand the effect of prebiotically plausible chemical reactions on compositional diversity, we first simulated a population of binary sequences undergoing three reactions: concatenation, template-directed ligation and hydrolysis. During concatenation, the 5′-end of one monomer (or polymer) reacts with the 3′-end of another monomer (or polymer) with rate constant kcon. This process first polymerizes monomers into oligomers, and later joins monomers and oligomers in template-independent reactions. Template-directed ligation can occur when two oligomers anneal adjacent to one another on a template sequence, leading to the ligation of the two oligomers with rate constant klig (48). Template-directed ligation appears to be a general phenomenon, occurring with peptides, small molecules and nucleic acids (49–51), and it can greatly accelerate bond formation (52–54). Interestingly, template-directed ligation of oligonucleotides appears to be relatively unbiased compared to monomer polymerization, permitting the incorporation of nucleotides that are effectively unreactive as monomers (55). For the purpose of modeling, we assume that three or more adjacent ‘Watson–Crick base-pairs’ (i.e. 0's pairing with 1's in our two-letter model) are required for annealing (34,54,56,57). Finally, hydrolysis of phosphodiester bonds, an important process for RNA molecules, occurs in our model at a constant rate per bond (kh).
Model parameters
The parameters of our simulations are the two dimensionless ratios: rcon = kconc0/kh, where c0 is the concentration of monomers, which gives the relative strength of concatenation and hydrolysis; and rlig = kligc0/kcon, which gives the relative strength of template-directed ligation and concatenation. In experiments, c0 is usually in the millimolar range, rcon is roughly 1–100, and rlig is between 103 and 107 (see Supplementary Data), so we use these parameters in our simulations. For computational tractability, we use a two-base system in the modeling for the purpose of building intuition. A two-base system has been proposed as a progenitor of the four-base system (12,58), and a ribozyme can be composed of only two bases (25). However, because a four-base system would be more realistic and it has been argued that this alphabet size is optimal (59), we also investigated the four-base system to the extent that it was computationally tractable. Regardless, a four-base system is used in our DNA-based experiments testing the predictions of the simulations.
Based on these chemical reactions, we implemented a stochastic simulation of a small reactor (e.g. a protocell) and a deterministic simulation mimicking a very large reactor. The stochastic simulations were initiated with ∼400 monomers (corresponding to a concentration of ∼10 mM in a protocell ∼50–100 nm in diameter). We recorded the average Ck, D and the length distribution after the reactors reached steady state (Supplementary Data). While the stochastic results are most relevant to prebiotic protocells, we used the deterministic results to understand diversity for computational reasons. The deterministic simulations are generalizations of a previously described ‘prelife’ framework (Supplementary Data) (60–62). We examined both possible sources of bias: (i) biased reactivity and (ii) biased initial monomer abundance. Based on the reactivity and abundance differences of the literature cited earlier, the bias examined in each case was roughly one order of magnitude.
RESULTS
Simulation: template-directed ligation causes a shift toward longer sequences
In the absence of template-directed ligation, both sources of bias resulted in an exponential relationship between length and abundance at steady state, with the scaling determined by the ratio rcon. We give an analytical proof of this relationship, which has been seen in other models of polymerization (63), in Supplementary Data. While increasing concatenation would also create longer products due to greater bond formation, even high concatenation rates would still give an exponentially decreasing distribution of lengths. In contrast, template-directed ligation skewed the distribution qualitatively toward longer lengths (Figure 2a and Supplementary Data), resulting in a substantial excess of long sequences compared to an exponential distribution. The skew may occur because this process uses somewhat long substrates (greater than or equal to three bases) to make longer products in relatively few steps. Since ribozymes and aptamers typically have a length of 30 bases or greater (45), template-directed ligation could improve the chance of obtaining functional molecules simply by increasing the number of long polymers. For example, as rlig increased from 0 to 106 (rcon = 10), the mass fraction of ribozyme-length sequences (>30 bases) increased from 0% (numerically undetectable) to >5%. A similar trend is seen using a four-base simulation (Supplementary Data).
Figure 2.

Template-directed ligation increases average length and compositional diversity in silico. (a) Length distribution of binary sequences with or without template-directed ligation [rlig = 0 (red) or 106 (blue); rcon = 10 in both cases]. Length is the number of bases per molecule. (b) Compositional diversity C3 at several rcon values, with or without template-directed ligation, when monomer reactivity is biased [19-fold difference between kcon; rlig = 0 (red) or 106 (blue); length = 15].
Simulation: template-directed ligation increases compositional diversity
Template-directed ligation increased the average compositional diversity C3 beyond that achieved by concatenation alone when analyzing product sequences of the same length. When reactivities were biased, C3 depended on the rates of both concatenation and template-directed ligation. Without template-directed ligation, a system with higher rcon had higher average C3 (Figure 2b). This effect appears to be a consequence of mass action, as the less reactive monomer is increasingly incorporated into polymers when concatenation is fast relative to hydrolysis. A simplified analytical model demonstrates this effect (Supplementary Data). However, at a given rcon, template-directed ligation further increased average C3 (Figure 2b), particularly at low concatenation rates. A four-base system appears to give similar results, with the caveat that our analysis was limited by computational tractability (Supplementary Data). Since template-directed ligation, like concatenation, increased the amount of bond formation relative to hydrolysis, one possible explanation might again be mass action. Therefore, we also measured C3 as a function of the total rate of bond-forming events (concatenation and template-directed ligation). At the same rate of bond formation, template-directed ligation still increased the C3 of the sequences, such that the majority of the increase of C3 with rlig was not simply the result of increased bond formation (Supplementary Data). Another possible mechanism for this increase is that template-directed ligation is a relatively unbiased mode of ligation compared to concatenation. To illustrate this point, we performed simulations in which we artificially relaxed the requirement for complementary base-pairing in template-directed ligation, which we call ‘relaxed-ligation’. Relaxed-ligation retains the effect of introducing unbiased reactions while eliminating the more subtle effects stemming from the information content of the reacting sequences. Relaxed-ligation produces average C3 that are similar to comparable template-directed ligation simulations (Supplementary Data). This result suggests that the unbiased nature of template-directed ligation is an important explanation for the increase of C3 with template-directed ligation.
In simulations where monomer abundance was biased, concatenation alone resulted in relatively low average C3 (∼0.4), independent of rcon. The effect of template-directed ligation was complicated but it tended to increase compositional diversity (Supplementary Data). This may be due to increased incorporation of the less abundant monomer through complementary base pairing in addition to the effects detailed below. Overall, both sets of simulations suggested that template-directed ligation caused a relative increase of compositionally diverse sequences.
Simulation: diversity of the pool
While Ck measures internal heterogeneity in a sequence and D measures population-wide diversity, we found that these measures were highly correlated in our simulations across a range of parameters (Supplementary Data). Therefore, increased average compositional diversity within a sequence implied increased diversity among molecules in the pool.
Experimental: compositional diversity in template-directed ligation of DNA
To experimentally test our main prediction that template-directed ligation increases the average compositional diversity of a heterogeneous pool of sequences containing all 4 nt, we performed template-directed ligation in a pool of sequences made by degenerate DNA oligonucleotide synthesis with all four bases (A, C, G, T). The experiments described here were performed with DNA (not RNA). We chose to use DNA because reactivity differences during synthesis are well known (64). Slight differences in phosphoramidite reactivity result in small biases in the composition of a degenerate pool, analogous to the larger biases resulting from reactivity differences in prebiotic syntheses. Since the reactivity biases in phosphoramidite synthesis are relatively small, this experiment is a stringent test of whether template-directed ligation can increase average Ck. We studied whether the bias would be countered by template-directed ligation. Degenerate templates (length 40; four bases) and octamer substrates (four bases) were used to perform ligation. The size difference between the templates, substrates and expected ligation products permitted later gel purification of the products. Bond formation was catalyzed by cyanogen bromide after an annealing step (34) and the products of ligation were isolated by gel purification. Templates, octamers and products were sequenced using the Illumina platform (Supplementary Data). We measured the proportion of sequences that had C3 close to that of ribozymes (C3 of 0.95 or greater) and found that the ligation products were significantly shifted toward higher C3 compared to the templates (Figure 3a and Supplementary Data). Ligation products also had higher C3 than sequences predicted from random concatenation of the sequenced octamers, indicating that template-directed ligation could increase average C3 beyond unbiased concatenation by favoring compositionally diverse templates. The proportion of ligation products of high C3 was similar to that of a uniform random pool, indicating that template-directed ligation quantitatively countered the initial bias of synthesis (Figure 3a). The shift was not due to artifacts from comparing samples of different length, experimental bias during sequencing or a shift in GC content (Supplementary Data). We attempted to ascertain whether sequence elements from a recently described RNA replicase (65) could be found at greater frequency in the pool of template-directed ligation products compared with random concatenation of the octamers; no difference was apparent by this test (Supplementary Data), but the importance of this finding is tempered by our incomplete understanding of the sequence elements supporting ribozyme function.
The error of our measurements of compositional diversity in ligation reactants and products could be calculated in two ways: (i) SD among experiments, as shown in Figure 3a or (ii) SD from bootstrapping subsamples. The error (i) reflects the deviation between experiments. The error (ii) reflects the sampling error of sequencing. The sampling error is similar in magnitude to the error between experiments (Supplementary Data). Also, a possible source of differences between templates, octamers and ligation products is bias introduced by RNA ligase during preparation for deep sequencing. This bias is most pronounced at the 5′- and 3′-ends of the sequence reads (66). To confirm that the differences in measured Ck among these samples were not due to artifacts from bias at the ends of the sequences, we randomized the first and last base of each sequence read (i.e. replaced the 3′ and 5′ bases with a randomly chosen base: A,C,G,T). The Ck of this end-randomized set of sequences were calculated. We found that end-randomization did not affect the conclusion that ligation products had significantly higher Ck than the templates and octamers (Supplementary Data).
These results confirmed the prediction that template-directed ligation increases the compositional diversity of a heterogeneous pool of DNA sequences comprising four bases. One possible mechanism for this increase could be that internally diverse sequences were better templates. We can calculate the ratio R of the probabilities of template-directed ligation occurring on a high Ck (phigh) versus low Ck (plow) template:
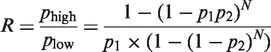 |
where p1 is the probability of the template annealing to two adjacent fragments and p2 is the probability of bond formation. Both p1 and p2 are <1 in practical situations. The ratio R is therefore always >1 (Supplementary Data), meaning that high Ck sequences are more likely to be templates. Therefore, templates with high Ck should be more likely to propagate their sequence information.
To test this experimentally, we studied the dependence of ligation efficiency on C3 for different templates. A set of binary DNA templates (two bases: C,T; 32-mer) of varying C3 was designed such that any 8-mer subsequence within the templates was 1 of 25 known octamer sequences (two bases: A,G), allowing the use of a defined set of substrates. Although this base composition is not a good mimic of a prebiotic reaction, it was chosen to minimize intramolecular secondary structure in order to focus on the effect of sequence heterogeneity. The templates were mixed with an excess of radiolabeled 5′-phosphorylated binary DNA octamers (A,G; Supplementary Data). All sequences had a GC content of 47–50% except for the template with C3 = 0. Ligation reactions were analyzed by polyacrylamide gel electrophoresis to visualize higher molecular weight products. We found a positive relationship between C3 and amount of products formed (Figure 3b). A similar trend was observed using random degenerate octamers and longer templates (Supplementary Data). This suggests that ligation efficiency on heterogeneous templates is one mechanism by which Ck increases in the products of template-directed ligation.
DISCUSSION
Our simulations and experiments suggest that compositional diversity in a pool of nucleic acids could have emerged early on, despite biases in monomer abundance and reactivity. The increase due to template-directed ligation may have at least two causes. First, template-directed ligation is relatively unbiased compared to concatenation, so it would counter the intrinsic bias of the system. Second, ligation may happen more efficiently on a compositionally diverse template, because internal sequence correlations in a low Ck template reduce the number of independent possibilities for ligation. Essentially, a compositionally diverse template could utilize a greater fraction of the substrate pool while the subsequences within a repetitive template would compete with each other for substrates.
While it may be possible to imagine scenarios where different biases cancel one another to produce complex pools, such finely tuned rates are unlikely in real systems. Based on our results using two-letter simulations and template-dependent DNA ligation, we can suggest the following prebiotic scenario (Figure 4). Initially the sequence pool would consist of short, highly compositionally biased sequences resulting from differences in reactivity among the nucleotides. However, once these sequences become long enough to serve as templates (length ≥ 6), the general mechanism of template-directed ligation would favor propagation of internally diverse sequences. A small increase in diversity would correspond to an exponentially large increase in the fraction of sequence space that would be explored. One should note that the optimal compositional diversity for forming secondary structures may be less than the maximum possible. For example, compositional bias toward GC-rich sequences may be a reasonable criterion for identifying non-coding RNAs in the genome due to the effect on folding stability (67), although formal structure itself does not appear to be a good criterion (perhaps because the compositional diversity of a genome tends to be fairly high, giving a relatively large probability of forming structures). Nevertheless, it is unclear whether the bias from prebiotic processes would be in the correct direction to favor structures, and it would still be desirable to increase diversity over the substantial ∼10-fold initial compositional bias estimated for prebiotic reactions. As the compositional bias disappeared, well-folded sequences could emerge. In addition, template-directed ligation would rapidly stitch together short sequences to produce a qualitative shift toward long sequences. The combined effects on diversity and length would enable the generation of ribozymes. These first inefficient ribozymes could then ‘jump-start’ the evolution of sequences with greater function and complexity through natural selection, especially within a spatially restricted context (44,63,68).
Figure 4.

Proposed prebiotic scenario. Monomers first concatenate into compositionally biased short oligomers. When the oligomers are long enough to act as templates, template-directed ligation produces relatively long, compositionally diverse sequences. These sequences can fold into stable structures, some of which may be catalytically active, leading to the RNA world.
Although it has been previously hypothesized that abstract measures of primary sequence information are unrelated to RNA function (11,69), we found a correlation between folding energy and Ck in silico. This suggests that internal heterogeneity, which is calculable from primary sequence alone, is an interesting measurement in addition to functional information or genomic complexity (which require knowledge of functional activity or fitness, respectively) (36,69). An important caveat regarding this relationship is that the minimum free energy of a sequence is only one of several features that would be important for functional activity. Other features would also be desirable [e.g. a large energy gap between the most stable fold and misfolded structures, or low structural ‘plasticity’ (70)]. Many desirable features are poorly understood. Interestingly, our simulations results suggest that, although template-independent processes result in exponential length distributions, template-directed ligation would skew the distribution qualitatively toward long sequences. A minimum length appears to be required to find certain activities. This was demonstrated by a series of selections for isoleucine aptamers that differed only by the length of the random region; no aptamers were isolated at the shortest length (16 bases) (32). Therefore, template-directed ligation may increase the probability of finding functional molecules by increasing the frequency of long sequences. Our results also highlight the importance of templating as a special property of nucleic acids during the origin of life: in addition to enabling the faithful replication of information, the ability to template could have promoted a search of functionally rich regions of sequence space.
Our modeling, while adequate for generating a hypothesis that could be experimentally tested, could be made more realistic in several ways. The alphabet size could be increased to include four bases, although this modification is computationally expensive because longer sequences would be needed to differentiate among the more varied compositions. Secondary structure could be included, which might interfere with templating and protect against degradation (71). The fact that our DNA experiments (including a four-letter alphabet and the possibility of secondary structure) show that template-directed ligation increases compositional diversity suggests that the overall result is not greatly influenced by the simplifications in the modeling. The rate of non-templated concatenation might depend on the length of the reactants, and circularization reactions might select against certain lengths. Interestingly, models of prebiotic polymerization of monomers that neglect the concatenation of oligomers (i.e. strong dependence of rate on substrate length) also yield exponential length distributions at equilibrium (61–63,72), suggesting that the qualitative distribution is not changed by the inclusion of oligomer concatenation. More experimental investigation would be required to understand the possible dependencies. Advances in the modeling based on such realistic features would be worthwhile for a more detailed prediction of the outcome of prebiotically plausible reactions. Our experiments used DNA because biases during synthesis are well known and we expect DNA to resemble RNA with respect to annealing of oligonucleotides. While the details of formation of secondary structure differ for DNA and RNA, both types of molecule can fold into catalytically active structures (73). Nevertheless, an investigation of compositional diversity using RNA would be more realistic as a model of the RNA world.
It is generally assumed that the emergence of ribozymes was the result of natural selection for replication in a pool of RNA sequences (3,44), but the pathway for generating the very first ribozymes is unknown. The probability of finding functional sequences also depends on the desired activity. Functions that can be performed by short motifs, such as aminoacylation or self-cleavage (24,31,74), could potentially be found even in highly biased sequence pools since the probability of finding the motif is relatively large. However, more sophisticated functions that appear to require longer motifs, such as an RNA polymerase (27,65), would be more likely to arise in a sequence pool if the prebiotic compositional bias were at least somewhat mitigated. It has been suggested that physical ordering effects alone could not produce functional molecules (11). Here, we have shown how long, compositionally diverse and well-folded sequences might be produced as a consequence of prebiotically plausible chemical mechanisms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–24 and Supplementary References [75–86].
FUNDING
Human Frontiers Science Program (postdoctoral fellowships to J.D. and R.X.); Harvard University (I.A.C., Bauer Fellow); NIH grant GM068763 to the Center for Modular Biology at Harvard; NSF/NIH Joint Program in Mathematical Biology (NIH grant R01GM078986 to M.A.N.); John Templeton Foundation (M.A.N.); Bill and Melinda Gates Foundation (Grand Challenges grant 37874 to M.A.N.) and J. Epstein (M.A.N.). Funding for open access charge: NIH (grant GM068763).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Niles Lehman, Jeffrey Bada, Jim Collins, Allan Drummond, Suckjoon Jun, Arthur Lander, David Liu, Andrew Murray, Eugene Shakhnovich and Jack Szostak for comments.
REFERENCES
- 1.Crick FH. The origin of the genetic code. J. Mol. Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 2.Orgel LE. Evolution of the genetic apparatus. J. Mol. Biol. 1968;38:381–393. doi: 10.1016/0022-2836(68)90393-8. [DOI] [PubMed] [Google Scholar]
- 3.Orgel LE. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004;39:99–123. doi: 10.1080/10409230490460765. [DOI] [PubMed] [Google Scholar]
- 4.Woese CR, Dugre DH, Dugre SA, Kondo M, Saxinger WC. On the fundamental nature and evolution of the genetic code. Cold Spring Harb. Symp. Quant. Biol. 1966;31:723–736. doi: 10.1101/sqb.1966.031.01.093. [DOI] [PubMed] [Google Scholar]
- 5.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 6.Miyakawa S, Cleaves HJ, Miller SL. The cold origin of life: B. Implications based on pyrimidines and purines produced from frozen ammonium cyanide solutions. Orig. Life Evol. Biosph. 2002;32:209–218. doi: 10.1023/a:1019514022822. [DOI] [PubMed] [Google Scholar]
- 7.Powner MW, Gerland B, Sutherland JD. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature. 2009;459:239–242. doi: 10.1038/nature08013. [DOI] [PubMed] [Google Scholar]
- 8.Rajamani S, Vlassov A, Benner S, Coombs A, Olasagasti F, Deamer D. Lipid-assisted synthesis of RNA-like polymers from mononucleotides. Orig. Life Evol. Biosph. 2008;38:57–74. doi: 10.1007/s11084-007-9113-2. [DOI] [PubMed] [Google Scholar]
- 9.Davies P. The origin of life.. II: how did it begin? Sci. Prog. 2001;84:17–29. doi: 10.3184/003685001783239096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joyce GF. Nonenzymatic template-directed synthesis of informational macromolecules. Cold Spring Harb. Symp. Quant. Biol. 1987;52:41–51. doi: 10.1101/sqb.1987.052.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Abel DL, Trevors JT. Three subsets of sequence complexity and their relevance to biopolymeric information. Theor. Biol. Med. Model. 2005;2:29. doi: 10.1186/1742-4682-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy M, Miller SL. The stability of the RNA bases: implications for the origin of life. Proc. Natl Acad. Sci. USA. 1998;95:7933–7938. doi: 10.1073/pnas.95.14.7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Glavin,D. and Bada,J. (2004) 35th Lunar and Planetary Science Conference. League City, TX, pp. 1022.
- 14.Shimoyama A, Hagishita S, Harada K. Search for nucleic-acid bases in carbonaceous chondrites from Antarctica. Geochem. J. 1990;24:343–348. [Google Scholar]
- 15.Stoks PG, Schwartz AW. Uracil in carbonaceous meteorites. Nature. 1979;282:709–710. [Google Scholar]
- 16.Stoks PG, Schwartz AW. Nitrogen-heterocyclic compounds in meteorites - significance and mechanisms of formation. Geochim. Cosmochim. Acta. 1981;45:563–569. [Google Scholar]
- 17.Kawamura K, Ferris JP. Clay catalysis of oligonucleotide formation: kinetics of the reaction of the 5′-phosphorimidazolides of nucleotides with the non-basic heterocycles uracil and hypoxanthine. Orig. Life Evol. Biosph. 1999;29:563–591. doi: 10.1023/a:1006648524187. [DOI] [PubMed] [Google Scholar]
- 18.Sawai H, Orgel LE. Letter: oligonucleotide synthesis catalyzed by the Zn-2+ ion. J. Am. Chem. Soc. 1975;97:3532–3533. doi: 10.1021/ja00845a050. [DOI] [PubMed] [Google Scholar]
- 19.Rajamani S, Ichida JK, Antal T, Treco DA, Leu K, Nowak MA, Szostak JW, Chen IA. Effect of stalling after mismatches on the error catastrophe in nonenzymatic nucleic acid replication. J. Am. Chem. Soc. 2010;132:5880–5885. doi: 10.1021/ja100780p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ertem G, Hazen RM, Dworkin JP. Sequence analysis of trimer isomers formed by montmorillonite catalysis in the reaction of binary monomer mixtures. Astrobiology. 2007;7:715–722. doi: 10.1089/ast.2007.0138. [DOI] [PubMed] [Google Scholar]
- 21.Miyakawa S, Ferris JP. Sequence- and regioselectivity in the montmorillonite-catalyzed synthesis of RNA. J. Am. Chem. Soc. 2003;125:8202–8208. doi: 10.1021/ja034328e. [DOI] [PubMed] [Google Scholar]
- 22.Stich M, Briones C, Manrubia SC. On the structural repertoire of pools of short, random RNA sequences. J. Theor. Biol. 2008;252:750–763. doi: 10.1016/j.jtbi.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 23.Kennedy R, Lladser ME, Wu Z, Zhang C, Yarus M, De Sterck H, Knight R. Natural and artificial RNAs occupy the same restricted region of sequence space. RNA. 2010;16:280–289. doi: 10.1261/rna.1923210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knight R, De Sterck H, Markel R, Smit S, Oshmyansky A, Yarus M. Abundance of correctly folded RNA motifs in sequence space, calculated on computational grids. Nucleic Acids Res. 2005;33:5924–5935. doi: 10.1093/nar/gki886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reader JS, Joyce GF. A ribozyme composed of only two different nucleotides. Nature. 2002;420:841–844. doi: 10.1038/nature01185. [DOI] [PubMed] [Google Scholar]
- 26.Rogers J, Joyce GF. A ribozyme that lacks cytidine. Nature. 1999;402:323–325. doi: 10.1038/46335. [DOI] [PubMed] [Google Scholar]
- 27.Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP. RNA-catalyzed RNA polymerization: accurate and general RNA-templated primer extension. Science. 2001;292:1319–1325. doi: 10.1126/science.1060786. [DOI] [PubMed] [Google Scholar]
- 28.Muller UF. Re-creating an RNA world. Cell. Mol. Life Sci. 2006;63:1278–1293. doi: 10.1007/s00018-006-6047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carothers JM, Oestreich SC, Szostak JW. Aptamers selected for higher-affinity binding are not more specific for the target ligand. J. Am. Chem. Soc. 2006;128:7929–7937. doi: 10.1021/ja060952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carothers JM, Davis JH, Chou JJ, Szostak JW. Solution structure of an informationally complex high-affinity RNA aptamer to GTP. RNA. 2006;12:567–579. doi: 10.1261/rna.2251306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knight R, Yarus M. Finding specific RNA motifs: function in a zeptomole world? RNA. 2003;9:218–230. doi: 10.1261/rna.2138803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Legiewicz M, Lozupone C, Knight R, Yarus M. Size, constant sequences, and optimal selection. RNA. 2005;11:1701–1709. doi: 10.1261/rna.2161305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gillespie D. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977;81:2340–2361. [Google Scholar]
- 34.James KD, Ellington AD. Surprising fidelity of template-directed chemical ligation of oligonucleotides. Chem. Biol. 1997;4:595–605. doi: 10.1016/s1074-5521(97)90245-3. [DOI] [PubMed] [Google Scholar]
- 35.Shannon C. A mathematical theory of communication. Bell Syst. Tech. J. 1948;27:379–423. [Google Scholar]
- 36.Adami C, Ofria C, Collier TC. Evolution of biological complexity. Proc. Natl Acad. Sci. USA. 2000;97:4463–4468. doi: 10.1073/pnas.97.9.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehman N, Donne MD, West M, Dewey TG. The genotypic landscape during in vitro evolution of a catalytic RNA: implications for phenotypic buffering. J. Mol. Evol. 2000;50:481–490. doi: 10.1007/s002390010051. [DOI] [PubMed] [Google Scholar]
- 38. Sipser,M. (2005) Introduction to the Theory of Computation, 2nd edn. Thomson Course Technology, Boston.
- 39.Chaitin GJ. Theory of program size formally identical to information-theory. J. ACM. 1975;22:329–340. [Google Scholar]
- 40.Chaitin GJ. Algorithmic information-theory. IBM J. Res. Dev. 1977;21:350–359. [Google Scholar]
- 41.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carothers JM, Oestreich SC, Davis JH, Szostak JW. Informational complexity and functional activity of RNA structures. J. Am. Chem. Soc. 2004;126:5130–5137. doi: 10.1021/ja031504a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stich M, Manrubia SC. Motif frequency and evolutionary search times in RNA populations. J. Theor. Biol. 2011;280:117–126. doi: 10.1016/j.jtbi.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Briones C, Stich M, Manrubia SC. The dawn of the RNA World: toward functional complexity through ligation of random RNA oligomers. RNA. 2009;15:743–749. doi: 10.1261/rna.1488609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JF, Hesselberth JR, Meyers LA, Ellington AD. Aptamer database. Nucleic Acids Research. 2004;32:D95–100. doi: 10.1093/nar/gkh094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruff KM, Snyder TM, Liu DR. Enhanced functional potential of nucleic acid aptamer libraries patterned to increase secondary structure. J. Am. Chem. Soc. 2010;132:9453–9464. doi: 10.1021/ja103023m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gutell RR, Cannone JJ, Shang Z, Du Y, Serra MJ. A story: unpaired adenosine bases in ribosomal RNAs. J. Mol. Biol. 2000;304:335–354. doi: 10.1006/jmbi.2000.4172. [DOI] [PubMed] [Google Scholar]
- 48.Li X, Liu DR. DNA-templated organic synthesis: nature's strategy for controlling chemical reactivity applied to synthetic molecules. Angew. Chem. Int. Ed. Engl. 2004;43:4848–4870. doi: 10.1002/anie.200400656. [DOI] [PubMed] [Google Scholar]
- 49.Lee DH, Granja JR, Martinez JA, Severin K, Ghadiri MR. A self-replicating peptide. Nature. 1996;382:525–528. doi: 10.1038/382525a0. [DOI] [PubMed] [Google Scholar]
- 50.Naylor R, Gilham PT. Studies on some interactions and reactions of oligonucleotides in aqueous solution. Biochemistry. 1966;5:2722–2728. doi: 10.1021/bi00872a032. [DOI] [PubMed] [Google Scholar]
- 51.Tjivikua T, Ballester P, Rebek J. Self-replicating system. J. Am. Chem. Soc. 1990;112:1249–1250. [Google Scholar]
- 52.Kanavarioti A, White DH. Kinetic analysis of the template effect in ribooligoguanylate elongation. Orig. Life Evol. Biosph. 1987;17:333–349. doi: 10.1007/BF02386472. [DOI] [PubMed] [Google Scholar]
- 53.Rohatgi R, Bartel DP, Szostak JW. Nonenzymatic, template-directed ligation of oligoribonucleotides is highly regioselective for the formation of 3′-5′ phosphodiester bonds. J. Am. Chem. Soc. 1996;118:3340–3344. doi: 10.1021/ja9537134. [DOI] [PubMed] [Google Scholar]
- 54.Sievers D, von Kiedrowski G. Self-replication of complementary nucleotide-based oligomers. Nature. 1994;369:221–224. doi: 10.1038/369221a0. [DOI] [PubMed] [Google Scholar]
- 55.Ninio J, Orgel LE. Heteropolynucleotides as templates for non-enzymatic polymerizations. J. Mol. Evol. 1978;12:91–99. doi: 10.1007/BF01733260. [DOI] [PubMed] [Google Scholar]
- 56.Sawai H, Totuka S, Yamamoto K. Helical structure formation between complementary oligonucleotides. Minimum chain length required for the template-directed synthesis of oligonucleotides. Orig. Life Evol. Biosph. 1997;27:525–533. doi: 10.1023/a:1006566212455. [DOI] [PubMed] [Google Scholar]
- 57.Sawai H, Wada M. Nonenzymatic template-directed condensation of short-chained oligouridylates on a poly(A) template. Orig. Life Evol. Biosph. 2000;30:503–511. doi: 10.1023/a:1026583814064. [DOI] [PubMed] [Google Scholar]
- 58.Wachtershauser G. An all-purine precursor of nucleic acids. Proc. Natl Acad. Sci. USA. 1988;85:1134–1135. doi: 10.1073/pnas.85.4.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szathmary E. What is the optimum size for the genetic alphabet? Proc. Natl Acad. Sci. USA. 1992;89:2614–2618. doi: 10.1073/pnas.89.7.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manapat M, Ohtsuki H, Burger R, Nowak MA. Originator dynamics. J Theor. Biol. 2009;256:586–595. doi: 10.1016/j.jtbi.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manapat ML, Chen IA, Nowak MA. The basic reproductive ratio of life. J. Theor. Biol. 2010;263:317–327. doi: 10.1016/j.jtbi.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nowak MA, Ohtsuki H. Prevolutionary dynamics and the origin of evolution. Proc. Natl Acad. Sci. USA. 2008;105:14924–14927. doi: 10.1073/pnas.0806714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu M, Higgs PG. Origin of self-replicating biopolymers: autocatalytic feedback can jump-start the RNA world. J. Mol. Evol. 2009;69:541–554. doi: 10.1007/s00239-009-9276-8. [DOI] [PubMed] [Google Scholar]
- 64.Pollard J, Bell SD, Ellington AD. Design, synthesis, and amplification of DNA pools for construction of combinatorial pools and libraries. Curr. Protoc. Mol. Biol. 2000 doi: 10.1002/0471142727.mb2402s52. Chapter 24, Unit 24.2. [DOI] [PubMed] [Google Scholar]
- 65.Wochner A, Attwater J, Coulson A, Holliger P. Ribozyme-catalyzed transcription of an active ribozyme. Science. 2011;332:209–212. doi: 10.1126/science.1200752. [DOI] [PubMed] [Google Scholar]
- 66.Eun H-M. Enzymology Primer for Recombinant DNA Technology. Academic Press, San Diego; 1996. [Google Scholar]
- 67.Rivas E, Eddy SR. Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics. 2000;16:583–605. doi: 10.1093/bioinformatics/16.7.583. [DOI] [PubMed] [Google Scholar]
- 68.Szabó P, Scheuring I, Czárán T, Szathmary E. In silico simulations reveal that replicators with limited dispersal evolve towards higher efficiency and fidelity. Nature. 2002;420:340–343. doi: 10.1038/nature01187. [DOI] [PubMed] [Google Scholar]
- 69.Hazen RM, Griffin PL, Carothers JM, Szostak JW. Functional information and the emergence of biocomplexity. Proc. Natl Acad. Sci. USA. 2007;104(Suppl. 1):8574–8581. doi: 10.1073/pnas.0701744104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ancel LW, Fontana W. Plasticity, evolvability, and modularity in RNA. J. Exp. Zool. 2000;288:242–283. doi: 10.1002/1097-010x(20001015)288:3<242::aid-jez5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 71.Obermayer B, Krammer H, Braun D, Gerland U. Emergence of information transmission in a prebiotic RNA reactor. Phys. Rev. Lett. 2011;107:018101. doi: 10.1103/PhysRevLett.107.018101. [DOI] [PubMed] [Google Scholar]
- 72.Wu M, Higgs PG. Comparison of the roles of nucleotide synthesis, polymerization, and recombination in the origin of autocatalytic sets of RNAs. Astrobiology. 2011;11:895–906. doi: 10.1089/ast.2011.0679. [DOI] [PubMed] [Google Scholar]
- 73.Klussmann S. The Aptamer Handbook. Weinheim: Wiley-VCH; 2006. [Google Scholar]
- 74.Turk RM, Chumachenko NV, Yarus M. Multiple translational products from a five-nucleotide ribozyme. Proc. Natl Acad. Sci. USA. 2010;107:4585–4589. doi: 10.1073/pnas.0912895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Calderone CT, Liu DR. Nucleic-acid-templated synthesis as a model system for ancient translation. Curr. Opin. Chem. Biol. 2004;8:645–653. doi: 10.1016/j.cbpa.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 76.Ellington AD. Back to the future of nucleic acid self-amplification. Nat. Chem. Biol. 2009;5:200–201. doi: 10.1038/nchembio0409-200. [DOI] [PubMed] [Google Scholar]
- 77.Kozlov IA, De Bouvere B, Van Aerschot A, Herdewijn P, Orgel LE. Efficient transfer of information from hexitol nucleic acids to RNA during nonenzymatic oligomerization. J. Am. Chem. Soc. 1999;121:5856–5859. doi: 10.1021/ja990440u. [DOI] [PubMed] [Google Scholar]
- 78.Lincoln TA, Joyce GF. Self-sustained replication of an RNA enzyme. Science. 2009;323:1229–1232. doi: 10.1126/science.1167856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lohrmann R, Bridson PK, Orgel LE. Efficient metal-ion catalyzed template-directed oligonucleotide synthesis. Science. 1980;208:1464–1465. doi: 10.1126/science.6247762. [DOI] [PubMed] [Google Scholar]
- 80.Luther A, Brandsch R, von Kiedrowski G. Surface-promoted replication and exponential amplification of DNA analogues. Nature. 1998;396:245–248. doi: 10.1038/24343. [DOI] [PubMed] [Google Scholar]
- 81.Mandel J. Use of the singular value decomposition in regression analysis. Am. Stat. 1982;36:15–24. [Google Scholar]
- 82.Rohatgi R, Bartel DP, Szostak JW. Kinetic and mechanistic analysis of nonenzymatic, template-directed oligoribonucleotide ligation. J. Am. Chem. Soc. 1996;118:3332–3339. doi: 10.1021/ja953712b. [DOI] [PubMed] [Google Scholar]
- 83.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2009. [Google Scholar]
- 84.Usher DA, McHale AH. Nonenzymatic joining of oligoadenylates on a polyuridylic acid template. Science. 1976;192:53–54. doi: 10.1126/science.1257755. [DOI] [PubMed] [Google Scholar]
- 85.von Kiedrowski G. A self-replicating hexadeoxynucleotide. Angew. Chem. Int. Ed. Engl. 1986;25:932–935. [Google Scholar]
- 86.Wehrens R, Mevik B-H. 2007. pls: partial least squares regression (PLSR) and principal component regression (PCR). R package version 2.1-0. http://mevik.net/work/software/pls.html (17 March 2010, date last accessed) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.