


Abstract
The ability to efficiently generate targeted point mutations in the chromosome without the need for antibiotics, or other means of selection, is a powerful strategy for genome engineering. Although oligonucleotide-mediated recombineering (ssDNA recombineering) has been utilized in Escherichia coli for over a decade, the successful adaptation of ssDNA recombineering to Gram-positive bacteria has not been reported. Here we describe the development and application of ssDNA recombineering in lactic acid bacteria. Mutations were incorporated in the chromosome of Lactobacillus reuteri and Lactococcus lactis without selection at frequencies ranging between 0.4% and 19%. Whole genome sequence analysis showed that ssDNA recombineering is specific and not hypermutagenic. To highlight the utility of ssDNA recombineering we reduced the intrinsic vancomymycin resistance of L. reuteri >100-fold. By creating a single amino acid change in the d-Ala-d-Ala ligase enzyme we reduced the minimum inhibitory concentration for vancomycin from >256 to 1.5 µg/ml, well below the clinically relevant minimum inhibitory concentration. Recombineering thus allows high efficiency mutagenesis in lactobacilli and lactococci, and may be used to further enhance beneficial properties and safety of strains used in medicine and industry. We expect that this work will serve as a blueprint for the adaptation of ssDNA recombineering to other Gram-positive bacteria.
INTRODUCTION
Lactococci and lactobacilli are lactic acid bacteria (LAB) that are commonly used in industry for the fermentation of food and beverage products. Also, numerous strains are of interest to both the industrial and medical communities as they elicit health-promoting properties to the host [for a review see (1,2)]. As many LAB are generally recognized as safe, lactobacilli and lactococci have been genetically engineered to serve as protein delivery vehicles (3–5), or have been modified for optimized production of metabolic products (6–10). In the United States and Western Europe the use of LAB strains in industry and medicine represents a multi-billion dollar market that is predicted to expand in the coming years.
Recombineering was developed in Escherichia coli and refers to ‘Engineering recombinant DNA molecules by in vivo homologous recombination’ (11). Initial applications of this technology relied on the expression of three lambda (λ) phage-derived proteins: Beta, Gam and Exo, collectively named the λ-Red system (12,13). Beta is a single-stranded DNA (ssDNA)-binding protein, Gam suppresses host nucleases, and Exo is a 5′–3′ exonuclease. Double-stranded DNA with short flanking sequences homologous to the target site is introduced into cells expressing the λ-Red proteins and single-stranded overhangs are generated by Exo. These overhangs are subsequently bound by Bet, followed by a homologous recombination event at the target site.
More recent applications of recombineering involve single-stranded oligonucleotides as a substrate rather than double-stranded DNA. In this case only expression of Bet, or the functional homolog RecT of the Rac prophage, is required to incorporate the ssDNA into the genome (11). In E. coli, ssDNA recombineering is most efficient with oligonucleotides that are 40–70 bp (11,14), and there is a lagging-strand bias in both E. coli and Mycobacterium (11,15). In E. coli the efficiency of recombineering is impacted by the methyl-directed mismatch repair system (MMR) (16,17). With proper oligonucleotide design the MMR may be avoided in E. coli by using oligonucleotides yielding C•C mismatches, multiple adjacent mismatches, multiple mismatches at consecutive wobble base positions (11,14,18,19), or the use of chemically modified bases (17) yielding a recombinant genotype in 20–50% of the population (16,17). These levels are high enough to identify mutations without the need for antibiotic selection, whereas in Mycobacterium ssDNA recombineering levels only allowed identification of mutants within the pool of cells that were able to take up plasmid DNA (20). In ssDNA recombineering it has been proposed that the oligonucleotide is incorporated via an annealing and replication-dependent mechanism (11,21) rather than by homologous recombination. The ssDNA recombineering has been demonstrated in Gram-negative bacteria (11,22–24), and in Mycobacterium (20).
In this study we describe the development of targeted ssDNA recombineering in Lactobacillus reuteri ATCC PTA 6475, a human isolate which is a candidate probiotic with anti-inflammatory properties (25,26) without the need for antibiotic selection (Figure 1). Also, we extended this technology to Lactococcus lactis which is widely used in the dairy industry for fermentation processes and has been applied as a biotherapeutic delivery vehicle (3,27). RecT-mediated recombineering is not hypermutagenic, and may serve as a powerful and novel tool for strain improvement through directed evolution by genetic engineering in LAB. The use of recombineering in the development of LAB as biotherapeutics is discussed.
Figure 1.

Overview of recombineering in L. reuteri. (i) Electrocompetent cells in which RecT is expressed are transformed with a recombineering oligonucleotide. Transformation efficiencies of ∼ ≥105cfu/µg DNA are required for high recombineering efficiencies. Wavy black lines with a red dot represent recombineering oligonucleotides with multiple non-complementary bases; the green circle represents a bacterial cell; pJP042 is the sakacin-based expression vector that contains recT; the black double helix represents chromosomal DNA; expressed RecT proteins are denoted in the cell. (ii) An oligonucleotide identical to the lagging strand contains multiple non-complementary bases that avoid the mismatch repair system resulting in increased recombineering efficiency. (iii) Viable cells are recovered on antibiotic-free plates and recombinants are detected by a mismatch amplification mutation assay-PCR (MAMA-PCR). Two oligonucleotides (blue) will yield a 1-kb fragment, whereas a third oligonucleotide (red) will only be extended by the polymerase when the mutations are incorporated in the chromosome yielding a second amplicon of 500 bp. As the recombineering oligonucleotide only targets one strand during DNA replication the colonies will be of mixed genotype. The red dot on the chromosome indicates that the mutations are incorporated. (iv) Single colony purification is performed to separate the wild-type genotype from the mutant genotype. (v) MAMA-PCR is repeated as described in section iii to identify a pure genotype mutant. A 1:1 ratio of wild-type and mutant genotypes is suggestive that during replication a single chromosome is being replicated per cell. (vi) The recombineering plasmid pJP042 can now be cured from the mutant strain by passaging bacteria without antibiotic selection to yield a plasmid-free derivative (vii).
MATERIALS AND METHODS
Bacterial strains, plasmids and media
Bacterial strains and plasmids used in this study are listed in Supplementary Table S1. Lactobacilli and their derivatives were cultured under anaerobic conditions at 37°C in deMan Rogosa Sharpe (MRS) medium (Difco, BD BioSciences). Lactococcus and its derivatives were cultured static at 30°C in M17-broth (Difco, BD BioSciences) that was supplemented with glucose to a final concentration of 0.5% (w/v). Electrocompetent cells of lactobacilli and Lactococcus were prepared as described before (28–30). If required, antibiotics were added as follows: 5 µg/ml erythromycin or chloramphenicol for lactobacilli and Lactococcus, 25 and 50 µg/ml rifampicin for lactobacilli and lactococci, respectively.
Reagents and enzymes
Restriction enzymes and T4 DNA ligases were purchased from New England Biolabs (NEB). PCR amplicons for cloning purposes were generated by KOD DNA hot start polymerase (Novagen), and PCR reactions for screening purposes were performed with Taq DNA polymerase (Denville Scientific). Pellet Paint Precipitant (Novagen) was used to precipitate DNA prior to restriction digestion, ligation and transformation. All oligonucleotides used in this study were purchased from Integrated DNA Technologies.
Construction of RecT expression vectors: pJP005 and pJP042
Oligonucleotides for cloning are listed in Supplementary Table S2. The RecT expression vectors (pJP005 and pJP042) were constructed as follows: pNZ8048 was digested with NcoI and KpnI, followed by Pellet Paint precipitation (Novagen). Genomic DNA of L. reuteri ATCC PTA 6475 (Wizard genomic DNA purification kit, Promega) was used as template for PCR amplification of the recT1 gene with oligonucleotide pair oJP022–oJP023, and the amplicon was digested and precipitated as described for pNZ8048. Purified DNA was quantified using Nanodrop-1000 (Thermo Scientific), and ligations were performed using a 1:1 (vector:insert) molar ratio using conditions specified by the manufacturer (NEB). The ligation mixture was transformed in L. lactis NZ9000, and Cm-resistant colonies were subjected to colony PCR with oligonucleotides that flank the multiple cloning site (oJP024–oJP025). Insertion was verified by restriction digest analysis and the integrity of the sequence was confirmed by sequence analysis. The resultant construct was named pJP005.
pSIP411 (a kind gift from Lars Axelsson, Nofima, Norway) is known to yield inducible and titratable gene expression in L. reuteri ATCC PTA 6475 (Lars Axelsson, personal communication) and was therefore used for this work. The backbone of pSIP411 was amplified with oligonucleotide pair oJP367–oJP368, followed by Pellet Paint precipitation, and digestion with NcoI and XbaI. Using oligonucleotide pair oJP369–oJP370 recT1 was amplified as described above, and the amplicon was purified followed by digestion with NcoI and XbaI. Ligation was performed as described for the construction of pJP005, and the ligation mix was transformed in L. lactis MG1363. Em-resistant colonies were screened by colony PCR for insertion of recT using oligonucleotide pair oJP415–oJP416. Integrity of the resultant clone, hereafter referred to as pJP042, was done in an identical manner as described for pJP005.
Preparation of cells for recombineering
All recombineering experiments with lactobacilli harboring pJP042 were performed with the following conditions: induction at OD600 > 0.55 and <0.65 for 20 min with 10 ng/ml induction peptide (MAGNSSNFIHKIKQIFTHR; Peptide2.0 Inc), followed by 5 min incubation on ice and preparation of competent cells as described above. All electroporations described were performed with a BioRad Genepulser and 2-mm electroporation cuvettes (BioRad).
Lactococcus lactis NZ9000 harboring pJP005 was induced for 30 min with 0.1% (v/v) filter-sterilized nisin at OD600 > 0.2 and < 0.3, followed by preparation of competent cells. Nisin was derived from an overnight culture of L. lactis NZ9700, and aliquots were stored at −20°C for up to 6 months.
Optimization of recombineering in L. reuteri
All oligonucleotides for recombineering are listed in Supplementary Table S2. Oligonucleotides for recombineering purposes were ordered at 100 nmol scale, desalted, and without any purifications. All recombineering oligonucleotides are complementary to the leading strand, and the non-complementary bases for all recombineering oligonucleotides are located centrally. With exception of the experiment to assess the effect of the olignucleotide concentration on the recombineering efficiency, we used 100 µg of oligonucleotide. Electrocompetent cells harboring pJP042 were prepared as described above, and for all experiments a volume of 5 µl (oligonucleotide or water) was electroporated. One microlitre of MRS was added, and bacteria were recovered for 2.5 h followed by serial dilution plating on MRS agar plates and MRS agar plates containing 25 µg/ml rifampicin. The number of rifampicin-resistant colonies is expressed per 109 bacteria.
To assess the efficiency of recombineering in relation to a single or multiple mismatches we targeted the rpoB gene. Based on work in E. coli (31) we predicted the amino acid change H488R would yield a rifampicin-resistant phenotype. Oligonucleotides had a single mismatch (oJP133), or multiple mismatches (oJP311 and oJP577). No multiple mismatches could be designed which, when incorporated into a new DNA strand, would be silent mutations. As it is unknown whether an amino acid change adjacent to H488 affects the rifampicin-resistant phenotype, we identified experimentally which amino acid change would still yield a rifampicin-resistant phenotype, yielding oJP311 and oJP577.
Other optimization studies, including the effect of oligonucleotide concentration or multiple mutations in the wobble base in relation to the recombineering efficiency, were performed in an identical manner as described above.
Non-selected recombineering in L. reuteri
Competent cells of L. reuteri harboring pJP042 were transformed with 100 µg oligonucleotide oJP577 and after recovery serial dilutions were plated on MRS plates. One hundred colonies were replica plated on MRS agar plates and MRS agar plates containing 25 µg/ml rifampicin. Rifampicin-resistant colonies were subjected to PCR (using oligonucleotides oJP097–oJP098) followed by sequence analysis to confirm incorporation of the recombineering oligonucleotide.
For genes other than rpoB, a selection of genes was chosen that targeted different regions of the chromosome of L. reuteri. For each gene a 90-mer oligonucleotide was designed with multiple consecutive mismatches. With exception of oJP475 and oJP810, recombineering oligonucleotides were designed such that an in-frame stop codon and a restriction endonuclease recognition site (either EcoRI, BamHI or HindIII) were created upon incorporation. For all genes 100 µg recombineering oligonucleotide was transformed and after recovery serial dilutions were spread on MRS plates without antibiotic selection. From the pool of transformants approximately 300 colonies were subjected to screening. We applied two screening strategies. The first strategy involves identification of mutations by restriction digest when the mutation being incorporated generated a new restriction site. PCR amplification of a 1-kb fragment in which the target site is located centrally was performed. Amplicons were subsequently subjected to restriction digest analysis with the appropriate enzyme. A partial or completely digested amplicon was indicative of incorporation of the recombineering oligonucleotide. The rationale for the partial digest is that one strand is targeted by oligonucleotide recombineering, i.e. the PCR product can be derived from mixed genotypes yielding a mix of amplicons. Alternatively, a fully digested amplicon can be expected if the mixed genotypes separated during recovery. The second method is a mismatch amplification mutation analysis–PCR (MAMA–PCR) (32,33), and relies on the use of an oligonucleotide of which the 3′-end is complementary to the recombinant sequence. Upon identification of a recombinant genotype by either method, cells were streaked on plate to separate the genotypes followed by screening to identify a pure genotype. The oligonucleotide sequences related to the targets listed in Figure 3b are available upon request.
Figure 3.
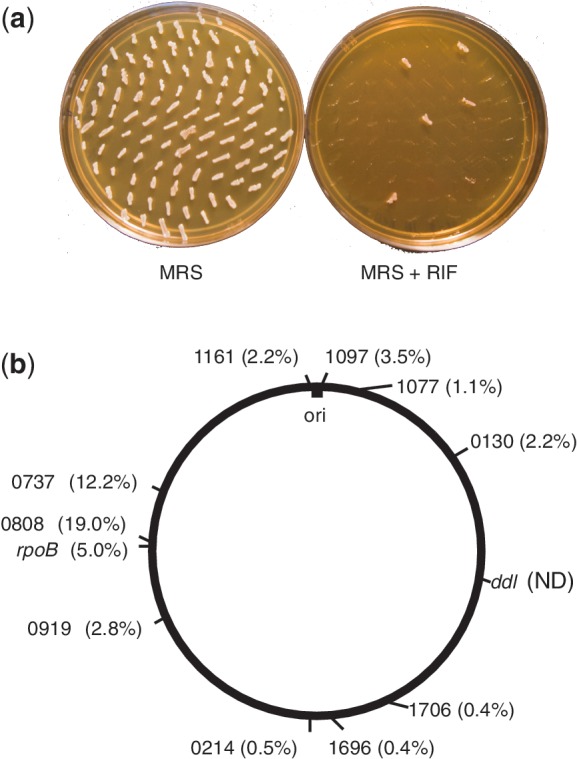
Non-selected recombineering in L. reuteri. (a) One hundred colonies derived from transformation with 100 µg oJP577, which upon incorporation yields a rifampicin-resistant phenotype (RpoB H488R), were patched onto MRS agar without antibiotic selection (left—MRS), and onto MRS agar containing rifampicin (right—MRS + RIF). (b) Overview of a selection of genes mutated by non-selected recombineering in the L. reuteri ATCC PTA 6475 chromosome. The locations of the mutated genes on the L. reuteri ATCC PTA 6475 chromosome are indicated based on the closed genome of the closely related strain L. reuteri F275 (NCBI accession number NC_010609). The numbers displayed represent the last four digits of the locus tag (HMPREF0535_XXXX) for each of the genes targeted and the recombineering frequency shown in parentheses. The rpoB gene (DNA directed RNA polymerase; locus tag HMPREF0536_0828) and the ddl gene (d-Ala-d-Ala ligase, locus tag HMPREF0536_1572) are listed using their gene names based on experimental evidence. ND: not determined; ori: origin of replication.
Recombineering in other LAB
We isolated natural rifampicin-resistant mutants for Lactobacillus plantarum BAA-793, Lactobacillus gasseri ATCC 33323, and L. lactis NZ9000, and determined the sequence of rpoB by sequence analysis. For lactobacilli, 109 cells were plated on MRS containing 25 µg/ml rifampicin whereas for Lactococcus 109 cells were plated on GM17 harboring 50 µg/ml rifampicin. Five rifampicin-resistant colonies of each strain were subjected to PCR using oligonucleotide pair oJP451–oJP458, oJP497–oJP505 and oJP528–oJP536 for L. plantarum, L. gasseri and L. lactis, respectively, and the sequence of the amplicons was determined and analyzed. Mutations that were identified in the rpoB genes were a G-T transversion at position 1438 (H480Y) for L. plantarum, a C–T transition at position 1468 (H490Y) for L. gasseri, and a C-A transversion at position 1456 (H486N) for L. lactis. Next we designed recombineering oligonucleotides targeting the rpoB gene for L. plantarum (oJP492), L. gasseri (oJP579) and L. lactis (oJP547 and oJP563). For L. gasseri we designed an oligonucleotide that, when incorporated, yields the amino acid changes T489S and H490R, and we determined experimentally that these changes yield a rifampicin-resistant phenotype. Competent cells for recombineering purposes were prepared as described above, followed by transformation of 100 µg of the corresponding oligonucleotides. The number of rifampicin-resistant colonies were determined and expressed per 109 viable cells.
For construction of a double mutant by recombineering in L. lactis NZ9000, competent cells expressing RecT were transformed with 50 µg oJP563 (to yield a rifampicin-resistant phenotype) and 50 µg oJP584 which targets malG, and transformants were recovered on GM17 agar plates harboring 50 µg/ml rifampicin. A total of approximately 300 rifampicin-resistant colonies were screened by MAMA-PCR using oligonucleotides oJP575, oJP576 and oJP598. Two percent of the screened colonies yielded two amplicons, indicative for co-incorporation of oJP584. A streak plate was made to separate the genotypes, and subsequently 10 colonies were screened with the above oligonucleotides to identify a pure genotype yielding strain RPRB4004.
Mutating the active site of Ddl
Active site residues of the Ddl protein of E. coli had previously been identified and in vitro analysis with recombinant protein showed that a single amino acid change (Y216F) could convert a dipeptide ligase to a depsipeptide ligase (34). To identify the corresponding active site of Ddl from L. reuteri ATCC PTA 6475 we first obtained the predicted 3D structure using the SWISS-MODEL server (35). Next we used Chimera software (36) to perform a structural overlay of Ddl proteins derived from L. reuteri and E. coli (37) to determine the predicted active site residue of Ddl L. reuteri that corresponds to the Y216 of E. coli, and this was predicted to be F258 for Ddl-L. reuteri. E. coli crystal structures (PDB ID: 1IOV for wild-type, and 1IOW for Ddl-Y126F) were obtained from the RCSB protein data bank.
An 80-mer oligonucleotide was designed (oJP810) which, upon incorporation, yields seven base changes which resulted in a single amino acid change (F258Y). Electrocompetent cells expressing RecT1 were transformed with 100 µg oJP810. We anticipated a slower growth phenotype for Ddl (F258Y) compared to the wild-type strain. Overgrowth by wild-type cells in a mixed population makes it difficult to identify of a mutant genotype by colony screening. We therefore recovered the cells for 4 h instead of 2.5 h to allow separation of the genotypes, and plated the cells on MRS agar plates. After three days incubation, seven small colonies were subjected to PCR with oligo pair oJP697–oJP698 to yield a 500-bp product (Supplementary Figure S1), followed by ApoI restriction digest analysis according to the manufacturer specified conditions. As incorporation of oJP810 alters a single ApoI recognition site in the amplicon it is expected that an undigested PCR product is indicative for a mutant genotype (Supplementary Figure S1). Digested amplicons which yielded fragments corresponding to 300 and 200 bp are indicative for a wild-type genotype. The resultant mutant strain was named RPRB3003.
Vancomycin susceptibility testing
The susceptibility to vancomycin was assessed with Etest strips (BioMérieux), and the minimum inhibitory concentration (MIC) was expressed as micrograms per millilitre. The range of vancomycin concentrations tested on the strip was 0.016–256 µg/ml. The assay was performed as previously described (38).
Whole genome sequence analysis
We found that introduction of pSIP411 or its derivatives in L. reuteri ATCC PTA 6475 introduces a null-mutation in locus HMPREF0536_1706 in a sub-population of the transformants (unpublished results, Laura Ortiz, JPVP and RB). Prior to whole genome sequence analysis strains were selected in which no mutation was present in this locus. After identification of the mutant genotype, strains were cured from pJP042 by growing the strains for one passage without antibiotic selection followed by plating on non-selective MRS agar plates. By patch plating we identified erythromycin-sensitive colonies that are considered cured from pJP042. All strains with exception of RPRB1696 were subjected by Illumina genome sequencing by the Research Technology Support Facility (RTSF) at Michigan State University. Genomic DNA from L. reuteri and L. lactis was isolated with the Wizard Genomic DNA kit (Promega). For whole genome sequencing, genomic DNA samples were further processed with the Illumina Genomic Sample Prep kit according to the manufacturer's recommendations. Paired-end 75 base runs were performed on the Illumina GAIIx sequencer(39). Strain RPRB1696 was subjected to whole genome sequencing using a Roche 454 GS Junior sequencer. Samples were prepared according to the manufacturer's recommendations. Reads were mapped to the closed L. reuteri F275 genome sequence yielding for the Illumina data on average 139 ± 14-fold depth and 99.986 ± 0.01% genome coverage. The 454 sequencing resulted in a 36-fold depth and 99.446% genome coverage. Fold depth and coverage percentages were determined using the utility genomeCoverageBed within the BEDtools suite(40).
SNP analysis was performed with the software packages ‘From Reads to Results (R2R)’(41), MAQ (Sourceforge) and BWA(42) using standard settings. Identified SNPs in each of the recombineered strains were confirmed by PCR amplification of the mutated region followed by sequence analysis. The presence of larger INDELs and inversions was assessed by MAQ, BWA and Pindel(43).
RESULTS
Establishing recombineering in L. reuteri ATCC PTA 6475
Although there are several genetic tools available for LAB (44–47) there is no system to generate targeted point mutations in the chromosomes of members of the phylum Firmicutes without the need for antibiotic selection. Therefore we wanted to establish ssDNA recombineering in L. reuteri that may serve as a model for other lactobacilli and lactococci. Using E. coli recT as a query we searched the draft genome sequence of L. reuteri ATCC PTA 6475 and identified two recT homologs, recT1 (locus tag HMPREF0536_0455) and recT2 (locus tag HMPREF0536_0582).
To assess the ability of the L. reuteri RecT proteins to support recombineering we placed recT1 under the control of an inducible promoter using the sakacin-based expression vector pSIP411(48) to yield plasmid pJP042. A simple method to quantitatively assess the efficacy of recombineering is to mutate a site in the RNA polymerase gene (rpoB) that results in an amino acid change yielding a rifampicin-resistant phenotype. We designed an 80-mer oligonucleotide (oJP133) targeting the lagging strand of DNA synthesis which has a single centrally located non-complementary base that results in a C•A mismatch (Figure 2a). Incorporation of oJP133 generates the amino acid change H488R in RpoB that confers rifampicin resistance in L. reuteri. After optimization of RecT1 expression conditions (data not shown), transformation of 1 µg oJP133 yielded approximately a 10-fold increase in rifampicin-resistant colonies compared to background levels (Figure 2b). Expression of RecT2 yielded similar recombineering efficiencies (data not shown), and we used RecT1 for all future recombineering experiments in L. reuteri.
Figure 2.

Establishing and optimizing recombineering in L. reuteri. (a) The dsDNA sequence of the targeted rpoB region of L. reuteri is shown aligned with amino acids specified by each codon. On the left the leading and lagging strand are indicated and below the oligonucleotides are listed which result in different mismatches, all resulting in a rifampicin-resistant phenotype. The corresponding amino acid changes are listed on the right. (b) Titrations of the amount of oligonucleotides oJP133 (Ο) and oJP577 (Δ) were performed with 1, 5, 25 and 100 µg of oligonucleotide. Rifampicin resistant colonies derived from the control transformation are represented as a bar graph. Data are expressed as rifampicin-resistant cfu per 109 cells. Data shown are the averages of three independent experiments and error bars represent standard deviation. (c) Assessment of recombineering efficiencies with 100 µg oJP133, oJP311, oJP813 and oJP577 in the wild-type strain (black bars), and in the mismatch repair deficient strains MutS1− (light grey bars) and MutL− (dark grey bars). Data shown are the averages of three independent experiments and error bars represent standard deviation.
High concentration of ssDNA required for efficient recombineering
In E. coli recombineering levels are saturated when using ∼115 ng of a 70 base oligonucleotide(14,49). To assess whether recombineering in L. reuteri is saturated with 1 µg, we transformed oJP133 over a two-log range with a maximum of 100 µg oligonucleotide (Figure 2b). There is a linear correlation between the number of recombinants that were obtained and the amount of oligonucleotide transformed up to 100 µg (R2 = 0.98; Figure 2b). At 100 µg of oligonucleotide we observed a 1000-fold increase in recombineering efficiency over background levels. No increase in recombinants could be obtained by using >100 µg of oligonucleotide as this significantly reduced cell viability and recombineering efficiency. Co-transformation of carrier DNA or the use of oligonucleotides with phosphorothiorate linkages to prevent host nuclease activity did not increase the recombineering efficiency in L. reuteri (data not shown).
Increased recombineering efficiency using mutations that avoid mismatch repair
To assess if mismatch repair can be avoided in L. reuteri in a similar way as described for E. coli (see introduction), we designed an oligonucleotide (oJP311) with two non-complementary bases, one of which yields a C•C mismatch that is, at least in E. coli, not efficiently recognized by the MMR (Figure 2a). oJP311 also showed a linear correlation (R2 = 0.99) for the increase in rifampicin-resistant colonies compared to the oligonucleotide concentration that was transformed (Figure 2b). The presence of an adjacent non-complementary base that yields a C•C mismatch did increase the recombineering efficiency, yielding on average 3 ± 1-fold more recombinants when compared to oJP133 (Figure 2b).
To assess whether the resultant C•C mismatch completely avoids mismatch repair in L. reuteri we inactivated two key players in MMR, mutS and mutL (see ‘Materials and Methods’ section for details), followed by testing the recombineering efficiencies with 100 µg of the above oligonucleotides. The MMR is only completely avoided when an oligonucleotide yields a similar number of recombinants in MutS− and MutL− strains compared to the wild-type. The level of recombinants obtained with oJP133 and oJP311 is on average 75-fold and 20-fold, respectively, lower in the wild-type background compared to MutS− or MutL− strains, demonstrating that in L. reuteri mismatch repair is not completely avoided with a single C•C mismatch (Figure 2c). We also find that, unlike in E. coli, an oligonucleotide yielding multiple mismatches at wobble positions (oJP813, Figure 2a) flanking the mutation yielding H488R in RpoB only partially avoided mismatch repair (Figure 2c).
To investigate whether the MMR can be completely avoided with an oligonucleotide with multiple adjacent mismatches we designed an oligonucleotide (oJP577) with five consecutive mismatches (Figure 2a). oJP577 yielded over a 100-fold increase in recombineering levels compared to recombineering levels obtained with an oligonucleotide with a single non-complementary base yielding a C•A mismatch. Next we assessed the recombineering levels of oJP577 in the MMR-deficient strains, showing that five adjacent mismatches yields similar levels of recombinants, showing that it completely avoids the MMR in L. reuteri (Figure 2c). Notably this level of recombineering yielded a 100 000-fold increase in recombineering compared to background levels, which indicated we had sufficient recombineering frequency to generate mutations without the need for selection.
Isolation of mutants without phenotypic selection in L. reuteri
We use the term ‘non-selected recombineering’ for the identification of mutants within a pool of cells without antibiotic or other phenotypic selection. Non-selected targeted recombineering is a powerful time-efficient approach that allows, when avoiding the MMR, construction of strain variants that are comparable to strains obtained through directed evolution. To identify if we could perform non-selected recombineering in L. reuteri we transformed oJP577, the most efficient oligonucleotide in this study, and plated the transformations on medium without rifampicin selection. One hundred colonies were picked from antibiotic-free plates and replica-plated on plates containing rifampicin. We found 5% of the transformants had incorporated the mutation into their genome (Figure 3a). These results show that targeted non-selected mutations can be made in L. reuteri.
To show that the non-selected recombineering is not dependent on the location of rpoB, we targeted ten additional genes in L. reuteri that are found at different locations in the chromosome (Figure 3b). For each of these genes basepair changes were made without selection with recombineering rates that ranged from 0.4% to 19%. This demonstrates that non-selected recombineering can be performed throughout the genome. Currently we are investigating the basis for variation in recombineering efficiency at different loci.
Extending recombineering to other LAB
Current technologies do not allow the construction of targeted point-mutations in a high-throughput manner in L. lactis, a species with industrial and medical relevance for its application in the dairy industry in fermentation processes and its application as a biotherapeutic delivery vehicle. As pJP042 is not functional in L. lactis MG1363 (data not shown), we switched to the well-established nisin-inducible expression system for the expression of RecT1 [for a review, see (50)]. We used L. lactis NZ9000 as the expression host for pJP005, a derivative of pNZ8048 in which the L. reuteri recT1 gene was cloned under the control of the nisin-inducible promoter PnisA. We designed oligonucleotides with one and four non-complementary bases that will yield a rifampicin resistant phenotype (Figure 4a). In L. lactis recombineering is more efficient with an oligonucleotide (oJP563) with multiple adjacent non-complementary bases as 50-fold more recombinants were obtained compared to a single non-complementary base that yields a T•C mismatch (Figure 4b). As observed in L. reuteri we find in L. lactis that recombineering efficiencies obtained with oJP563 are high enough for non-selected recombineering, yielding a 1% efficiency (data not shown).
Figure 4.

Establishing recombineering in other LAB. (a) The dsDNA sequence of the targeted rpoB region of L. lactis is shown aligned with the encoded amino acids for each codon. On the left are the leading and lagging strand indicated and below oligonucleotides are listed yielding different mismatches, all resulting in a rifampicin-resistant phenotype. The corresponding amino acid changes are listed on the right. (b) Recombineering efficiencies obtained in L. lactis transformed with 100 µg oJP547 or oJP563 and the level of natural rifampicin resistant L. lactis colonies per 109 cells (control). (c) Comparison of recombineering levels between L. reuteri (oJP577), L. lactis (oJP563), L. gasseri (oJP579) and L. plantarum (oJP492). All data shown are averages from three independent experiments and error bars represent standard deviation.
We also extended recombineering to two human isolates of Lactobacillus whose genomes are publicly available: L. plantarum BAA 793 (51) and L. gasseri ATCC 33323 (52). We used pJP042 in combination with 100 µg oligonucleotide that targets rpoB to yield the amino acid changes D480Y and T489S + H490Y in L. plantarum and L. gasseri, respectively. We show, without any further optimization in these strains, that recombineering in L. gasseri yields ∼104 rifampicin-resistant colonies with an oligonucleotide containing five adjacent mismatches (Figure 4c). We were only able to design an oligonucleoitde with a single mismatch in L. plantarum (oJP492) that yields a rifampicin-resistant phenotype, yielding approximately 103 rifampicin-resistant colonies. Although recombineering levels in both strains are lower compared to L. reuteri and L. lactis, we anticipate that optimization of electroporation conditions, testing RecT from different hosts, optimizing expression levels of RecT, combined with oligonucleotide design will further increase the level of recombineering in these strains.
ssDNA recombineering did not yield untargeted genome changes in L. reuteri and L. lactis
A major concern of recombineering, which so far has not been adequately addressed in any organism, is the specificity of ssDNA recombineering and the possibility of obtaining additional untargeted mutations and genome rearrangements. In E. coli prolonged expression of the λ-Red genes yielded a hypermutagenic phenotype based on the increase of natural rifampicin-resistant colonies (53). To assess whether our approach is mutagenic we sequenced the complete genome of three L. reuteri strains: the wild-type control strain, a strain in which RecT was expressed from pJP042 but not exposed to ssDNA (RPRB0000), and a strain which has undergone a single recombineering event using 100 µg of oligonucleotide o1696 that targets a putative 6-aminohexanoate-cyclic-dimer hydrolase (locus tag HMPREF0536_1696; RPRB1696). In RPRB0000, a single base insertion was identified in the gene encoding dextransucrase (locus tag HMPREF0536_1850) resulting in a frame-shift with a consequent stop codon downstream. In RPRB1696 we confirmed the correct mutation in locus tag HMPREF0536_1696 specified by o1696. In addition we identified a single non-targeted point-mutation in a gene encoding a putative transcription regulation protein (locus tag HMPREF0536_1614), a G–T transversion in the third base of the start codon.
We also sequenced the genome of a L. lactis strain that has undergone a double recombineering event in which rpoB (locus tag LLNZ_10235) and malG (locus tag LLNZ_03840) were mutated (RPRB4004) simultaneously. Compared to the control strain (RPRB4000) no point mutations or other genome alterations were identified other than the mutations introduced by the oligonucleotides. Taken together these data indicate ssDNA recombineering is specific and not hypermutagenic in the sequenced L. reuteri and L. lactis isolates that have undergone a recombineering event.
Using recombineering to create a vancomycin-sensitive L. reuteri strain
To emphasize the utility of recombineering we converted L. reuteri from vancomycin resistant to vancomycin sensitive by mutating the active site of Ddl directly in the chromosome. Vancomycin inhibits the peptidoglycan biosynthesis by binding to muramylpentapeptides that terminate in d-alanyl-d-alanine (d-Ala-d-Ala) and blocks the addition of these precursors into the peptidoglycan backbone (54,55). Many lactobacilli incorporate d-alanyl-d-lactate (d-Ala-d-Lac) into their muramylpentapeptides, and this is the basis for their vancomycin resistance as peptides that terminate in d-Ala-d-Lac are bound 1000-fold less efficiently by vancomycin compared to peptides terminating in d-Ala-d-Ala (56,57). The enzyme responsible for the addition of d-Ala or d-lac is d-Ala-d-Ala ligase (Ddl). In E. coli Ddl is a dipeptide ligase as it catalyzes the formation of d-Ala-d-Ala whereas in L. mesenteorides, and probably in many lactobacilli, Ddl is a depsipeptide ligase as it catalyzes the formation of d-Ala-d-Lac. Superimposing the structures of Ddl-E. coli and Ddl-L. reuteri show that both proteins are structurally related (Figure 5a). In vitro analyses in E. coli and L. mesenteroides have previously identified active site residues in Ddl that upon mutation could switch from a dipeptide ligase to a depsipeptide ligase and vice versa(34,58). In Ddl-E. coli the residues Y216–E15–S150 form a hydrogen-bonding triad (Figure 5b) that is linked with the specificity of the enzyme although the exact mechanism for the shift in specificity is unknown (34). By replacing the tyrosine at position 216 with a phenylalanine (Y216F) in Ddl-E. coli there is no hydrogen bond formed between F216–E15, and as a consequence Ddl-Y216F has gained depsipeptide activity (Figure 5b) (34). To translate this to L. reuteri, we identified the predicted active site residues in Ddl- L. reuteri by superimposing Ddl-L. reuteri with Ddl-E. coli (Fig 5c). The L. reuteri triad is predicted to be F258–E17–S186, and mutating phenylalanine at position 258 to a tyrosine (F258Y) would, based on the E. coli model, yield a hydrogen-bond between Y258-E17 and change subsequently the enzyme activity from a depsipeptide ligase to a dipeptide ligase. As a result the cells render sensitivity to vancomycin.
Figure 5.

Converting vancomycin-resistant L. reuteri to vancomycin-sensitive by a single amino acid change. (a) The determined 3D structure of the d-ala-d-ala ligase (Ddl) protein of E.coli (PDB ID 1IOV; blue) and the modeled protein structure of Ddl of L. reuteri (green) were superimposed to locate the putative active site residues in Ddl of L. reuteri which, in E. coli, form a hydrogen-bonded network (Y216–E15–S150; boxed region). (b) Superimposing the determined structures of the Ddl proteins of the E. coli wild-type (blue) and a mutant (PDB ID 1IOW; grey) show that replacing a tyrosine at position 216 with a phenylalanine (Y216F) does not form a hydrogen-bond between F216–E15, and changes the enzymatic activity from a dipeptide ligase to a depsipeptide ligase. (c) We predicted the active site residues of Ddl-L. reuteri by superimposing the wild-type E. coli Ddl protein (blue) with the predicted protein structure of L. reuteri (green), and residues F258–E17–S186 in Ddl-L. reuteri correspond to the E. coli triad Y216–E15–S15, respectively. According to the E. coli model changing the phenylalanine at position 258 to a tyrosine (F258Y) in Ddl-L. reuteri would establish a hydrogen-bond between F258–E17 and may therefore yield dipeptide ligase activity which subsequently would result in a vancomycin-sensitive phenotype. (d) The dsDNA sequence of the ddl region of L. reuteri is shown aligned with the encoded amino acids for each triplet. Italic sequence represents the ApoI restriction site that is mutated by incorporation of oJP810. On the left are the leading and lagging strand indicated, and below is oJP810 showing the different mismatches with the resulting amino acid change (F258Y) on the right. (e) Susceptibility of L. reuteri wild-type (left) and the mutant RPRB3003 (right) to vancomycin using an Etest assay. The MIC is determined by identifying where bacterial growth intersects the Etest strip.
We designed an oligonucleotide that mutates two bases in the F258 codon of ddl along with bases in wobble positions in flanking codons to generate the single amino acid change F258Y (Figure 5d). The base changes disrupt an ApoI restriction endonuclease site and therefore we screened colonies by PCR amplification of the target region followed by ApoI restriction digest analysis. An undigested amplicon was obtained (Supplementary Figure S1) and we confirmed by sequence analysis that the mutation was successfully incorporated into the ddl gene. The resulting strain RPRB3003 showed increased sensitivity to vancomycin (MIC = 1.5 µg/ml) compared to the wild-type (MIC >256 µg/ml; Figure 5e). This demonstrates that non-selected ssDNA recombineering may be applied to strains used as biotherapeutic delivery vehicles to remove undesirable characteristics, such as unwanted antibiotic resistance.
DISCUSSION
We have adapted the ssDNA recombineering technology to efficiently construct non-selected targeted mutations on the chromosomes of L. reuteri and L. lactis and show proof-of-concept in two other human-derived probiotic lactobacilli. In optimizing this work we have uncovered important parameters that should facilitate adaptation of non-selected recombineering to other Firmicutes. Keys to efficient recombineering in LAB were (i) high ssDNA concentration (up to 100 µg), (ii) optimal electroporation conditions and (iii) oligonucleotide design that will avoid the MMR system. Efficient recombineering required 100 µg of oligonucleotide, nearly a 1000-fold excess over the amount of oligonucleotide needed for optimal recombineering in E. coli (14,49). We suspect the high amount of oligonucleotide required is due to the inability of ssDNA to enter the cells, presumably because of the thick peptidoglycan layer found in Gram-positive bacteria. Extensive degradation of ssDNA by host nucleases, which can be overcome in E. coli by co-transformation of carrier DNA or by using phosphorothioate linkages in the oligonucleotide, is not a major factor in L. reuteri. In contrast to E. coli, the MMR in L. reuteri can only be avoided with an oligonucleotide harbouring multiple consecutive non-complementary bases. C•C mismatches and multiple mutations at wobble base positions can increase recombineering efficiency, but are still 30-fold lower than oligonucleotides containing five consecutive mismatches. We do not understand why L. reuteri's MMR system appears to be more stringent than MMR of E. coli. Finally, without optimization we achieved successful ssDNA recombineering in the human-derived strains L. plantarum and L. gasseri, and we envision that systematic optimization will further increase the efficiency of recombineering. Currently the level of recombineering in L. gasseri is high enough to isolate co-selected mutants in a similar way as previously described in Mycobacterium (15). We expect similar levels to be achieved in L. plantarum with an oligonucleotide that avoids the MMR.
When avoiding MMR, the efficiency of recombineering in L. reuteri and L. lactis was high enough to obtain non-selected mutants in a wild-type background. The generation of mutations in the chromosome without the need for selection was previously only available in only E. coli (14). Our demonstrated ability to subtly alter the genomes of LAB by recombineering creates the possibility to generate many molecular alterations that improve upon existing technologies. These include the ability to generate in-frame stop codons, to alter gene expression via point mutations in promoters and ribosome-binding sites, to enhance or change enzyme activity, and to generate an unlimited number of mutations in a single genome. Mutations can be generated in less than a week, and in L. reuteri the plasmid DNA can easily be cured from the cells as after a single passage without antibiotic selection 20% of the population have lost pJP042 (unpublished results, JPvP and RB). Our results showed that ssDNA recombineering in both L. lactis and L. reuteri isolates is specific, efficient, and not hypermutagenic. This opens up the possibility to apply multiplex automated genome engineering (MAGE) (59) and trackable multiplex recombineering (TRMR) (60), elegant systems to perform genome-wide genetic engineering.
There is increasing interest to apply lactococci and lactobacilli as protein delivery vehicles for biotherapeutics and vaccines. The majority of lactobacilli, however, have intrinsic resistance to the antibiotic vancomycin (61,62). In addition to the use of (lactobacilli) probiotics in healthy individuals (63,64), it is anticipated that there will be increased use of lactobacilli as delivery vehicles for therapeutics in patients with diseases and/or reduced immune function. This raises serious safety concerns since numerous cases which associate lactobacilli with bacteremia infections in immunocompromised patients have been documented (65–69). Since vancomycin is the antibiotic most often used to treat Gram-positive bacteraemia infections (70), engineered biotherapeutics that are sensitive to clinically relevant levels of vancomycin would allow efficient eradication should a patient show signs of infection. To highlight the utility of ssDNA recombineering in enhancing strain safety by enzyme modification we altered the enzymatic activity of the Ddl enzyme in L. reuteri. A single amino acid change reduced the MIC to vancomycin by two orders of magnitude to clinically relevant levels (71). The mutation only caused a minor effect on growth rate and growth yield (data not shown) and thus can be further explored as a potential therapeutic delivery system.
Using recombineering it is now possible to generate a strain that contains only a single-point mutation when compared to the starting strain, without the need for selection. This can be achieved by creating a 5-bp mutation in a first round of recombineering and then repairing four of the mismatches back to the wild-type sequence in a second round, resulting in a single nucleotide change. Thus strains created by recombineering may contain a single mutation that is molecularly indistinguishable from mutations that arise via natural selection or directed evolution. We therefore propose that strains created by recombineering will be equally safe as strains in which mutations are selected for in the laboratory and we expect that RecT-mediated genetic engineering will stimulate debate over the use of bacteria modified using recombineering technology in industry and medicine.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–2, Supplementary Figure 1, and Supplementary References [72–74].
FUNDING
Funding for open access charge: RAB by National Institutes of Health (R01AT004326) to James Versalovic; and by a Strategic Partnership Grant by Michigan State University.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Kar Mun Neoh, Jan Niklas Galle, and Christopher Radek for technical assistance, Nikhil Jain for his help on using Chimera software, Lothar Steidler (ActoGeniX NV, Zwijnaarde, Belgium) for critically reading the manuscript, and Todd Klaenhammer for providing L. gasseri ATCC 33323.
REFERENCES
- 1.Lebeer S, Vanderleyden J, de Keersmaecker SCJ. Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat. Rev. Microbiol. 2010;8:171–184. doi: 10.1038/nrmicro2297. [DOI] [PubMed] [Google Scholar]
- 2.Ventura M, O'Flaherty S, Claesson MJ, Turroni F, Klaenhammer TR, van Sinderen D, O'Toole PW. Genome-scale analyses of health-promoting bacteria: probiogenomics. Nat. Rev. Microbiol. 2009;7:61–71. doi: 10.1038/nrmicro2047. [DOI] [PubMed] [Google Scholar]
- 3.Steidler L, Hans W, Schotte L, Neirynck S, Obermeier F, Falk W, Fiers W, Remaut E. Treatment of Murine Colitis by Lactococcus lactis Secreting Interleukin-10. Science. 2000;289:1352–1355. doi: 10.1126/science.289.5483.1352. [DOI] [PubMed] [Google Scholar]
- 4.Krüger C, Hu Y, Pan Q, Marcotte H, Hultberg A, Delwar D, van Dalen PJ, Pouwels PH, Leer RJ, Kelly CG, et al. In situ delivery of passive immunity by lactobacilli producing single-chain antibodies. Nat. Biotechnol. 2002;20:702–706. doi: 10.1038/nbt0702-702. [DOI] [PubMed] [Google Scholar]
- 5.Guimarães VD, Innocentin S, Lefèvre F, Azevedo V, Wal JM, Langella P, Chatel J-M. Use of native lactococci as vehicles for delivery of DNA into mammalian epithelial cells. Appl. Environ. Microbiol. 2006;72:7091–7097. doi: 10.1128/AEM.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teusink B, Smid EJ. Modelling strategies for the industrial exploitation of lactic acid bacteria. Nat. Rev. Microbiol. 2006;4:46–56. doi: 10.1038/nrmicro1319. [DOI] [PubMed] [Google Scholar]
- 7.Bron PA, Kleerebezem M. Engineering lactic acid bacteria for increased industrial functionality. Bioeng. Bugs. 2011;2:80–87. doi: 10.4161/bbug.2.2.13910. [DOI] [PubMed] [Google Scholar]
- 8.Sybesma W, Burgess C, Starrenburg M, van Sinderen D, Hugenholtz J. Multivitamin production in Lactococcus lactis using metabolic engineering. Metab. Eng. 2004;6:109–115. doi: 10.1016/j.ymben.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Gaspar P, Neves AR, Ramos A, Gasson MJ, Shearman CA, Santos H. Engineering Lactococcus lactis for production of mannitol: high yields from food-grade strains deficient in lactate dehydrogenase and the mannitol transport system. Appl. Environ. Microbiol. 2004;70:1466–1474. doi: 10.1128/AEM.70.3.1466-1474.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wegkamp A, de Vos WM, Smid EJ. Folate overproduction in Lactobacillus plantarum WCFS1 causes methotrexate resistance. FEMS Microbiol. Lett. 2009;297:261–265. doi: 10.1111/j.1574-6968.2009.01690.x. [DOI] [PubMed] [Google Scholar]
- 11.Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl Acad. Sci. USA. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy KC. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 1998;180:2063–2071. doi: 10.1128/jb.180.8.2063-2071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl Acad. Sci. USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawitzke JA, Costantino N, Li XT, Thomason LC, Bubunenko M, Court C, Court DL. Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J. Mol. Biol. 2011;407:45–59. doi: 10.1016/j.jmb.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Kessel JC, Hatfull GF. Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: characterization of antimycobacterial drug targets. Mol. Microbiol. 2008;67:1094–1107. doi: 10.1111/j.1365-2958.2008.06109.x. [DOI] [PubMed] [Google Scholar]
- 16.Costantino N, Court DL. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc. Natl Acad. Sci. USA. 2003;100:15748–15753. doi: 10.1073/pnas.2434959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang HH, Xu G, Vonner AJ, Church G. Modified bases enable high-efficiency oligonucleotide-mediated allelic replacement via mismatch repair evasion. Nucleic Acids Res. 2011;39:7336–47. doi: 10.1093/nar/gkr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu. Rev. Genet. 2002;36:361–388. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- 19.Swingle B, Markel E, Costantino N, Bubunenko MG, Cartinhour S, Court DL. Oligonucleotide recombination in Gram-negative bacteria. Mol. Microbiol. 2010;75:138–148. doi: 10.1111/j.1365-2958.2009.06976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Kessel JC, Hatfull GF. Recombineering in Mycobacterium tuberculosis. Nat. Meth. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- 21.Huen MS, Li XT, Lu LY, Watt RM, Liu DP, Huang JD. The involvement of replication in single stranded oligonucleotide-mediated gene repair. Nucleic Acids Res. 2006;34:6183–6194. doi: 10.1093/nar/gkl852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerlach RG, Jäckel D, Hölzer SU, Hensel M. Rapid oligonucleotide-based recombineering of the chromosome of Salmonella enterica. Appl. Environ. Microbiol. 2009;75:1575–1580. doi: 10.1128/AEM.02509-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swingle B, Bao Z, Markel E, Chambers A, Cartinhour S. Recombineering using RecTE from Pseudomonas syringae. Appl. Environ. Microbiol. 2010;76:4960–4968. doi: 10.1128/AEM.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grogan DW, Stengel KR. Recombination of synthetic oligonucleotides with prokaryotic chromosomes: substrate requirements of the Escherichia coli/lambdaRed and Sulfolobus acidocaldarius recombination systems. Mol. Microbiol. 2008;69:1255–1265. doi: 10.1111/j.1365-2958.2008.06356.x. [DOI] [PubMed] [Google Scholar]
- 25.Jones SE, Whitehead K, Saulnier D, Thomas CM, Versalovic J, Britton RA. Cyclopropane fatty acid synthase mutants of probiotic human-derived Lactobacillus reuteri are defective in TNF inhibition. Gut Microbes. 2011;2:69–79. doi: 10.4161/gmic.2.2.15282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin YP, Thibodeaux CH, Peña JA, Ferry GD, Versalovic J. Probiotic Lactobacillus reuteri suppress proinflammatory cytokines via c-Jun. Inflamm Bowel Dis. 2008;14:1068–1083. doi: 10.1002/ibd.20448. [DOI] [PubMed] [Google Scholar]
- 27.Foligne B, Dessein R, Marceau M, Poiret S, Chamaillard M, Pot B, Simonet M, Daniel C. Prevention and treatment of colitis with Lactococcus lactis secreting the immunomodulatory Yersinia LcrV protein. Gastroenterology. 2007;133:862–874. doi: 10.1053/j.gastro.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 28.Holo H, Nes IF. High-frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl. Environ. Microbiol. 1989;55:3119–3123. doi: 10.1128/aem.55.12.3119-3123.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahrné S, Molin G, Axelsson L. Transformation of Lactobacillus reuteri with electroporation: studies on the erythromycin resistance plasmid pLUL631. Curr. Microbiol. 1992;24:199–205. [Google Scholar]
- 30.Josson K, Scheirlinck T, Michiels F, Platteeuw C, Stanssens P, Joos H, Dhaese P, Zabeau M, Mahillon J. Characterization of a gram-positive broad-host-range plasmid isolated from Lactobacillus hilgardii. Plasmid. 1989;21:9–20. doi: 10.1016/0147-619x(89)90082-6. [DOI] [PubMed] [Google Scholar]
- 31.Garibyan L, Huang T, Kim M, Wolff E, Nguyen A, Nguyen T, Diep A, Hu K, Iverson A, Yang H, et al. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair (Amst.) 2003;2:593–608. doi: 10.1016/s1568-7864(03)00024-7. [DOI] [PubMed] [Google Scholar]
- 32.Cha RS, Zarbl H, Keohavong P, Thilly WG. Mismatch amplification mutation assay (MAMA): application to the c-H-ras gene. PCR Methods Appl. 1992;2:14–20. doi: 10.1101/gr.2.1.14. [DOI] [PubMed] [Google Scholar]
- 33.Qiang YZ, Qin T, Fu W, Cheng WP, Li YS, Yi G. Use of a rapid mismatch PCR method to detect gyrA and parC mutations in ciprofloxacin-resistant clinical isolates of Escherichia coli. J. Antimicrob. Chemother. 2002;49:549–552. doi: 10.1093/jac/49.3.549. [DOI] [PubMed] [Google Scholar]
- 34.Park IS, Lin CH, Walsh CT. Gain of D-alanyl-D-lactate or D-lactyl-D-alanine synthetase activities in three active-site mutants of the Escherichia coli D-alanyl-D-alanine ligase B. Biochemistry. 1996;35:10464–10471. doi: 10.1021/bi9603128. [DOI] [PubMed] [Google Scholar]
- 35.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Fan C, Park IS, Walsh CT, Knox JR. D-alanine: D-alanine ligase: phosphonate and phosphinate intermediates with wild type and the Y216F mutant. Biochemistry. 1997;36:2531–2538. doi: 10.1021/bi962431t. [DOI] [PubMed] [Google Scholar]
- 38.Egervärn M, Danielsen M, Roos S, Lindmark H, Lindgren S. Antibiotic susceptibility profiles of Lactobacillus reuteri and Lactobacillus fermentum. J. Food Prot. 2007;70:412–418. doi: 10.4315/0362-028x-70.2.412. [DOI] [PubMed] [Google Scholar]
- 39.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skovgaard O, Bak M, Løbner-Olesen A, Tommerup N. Genome-wide detection of chromosomal rearrangements, indels, and mutations in circular chromosomes by short read sequencing. Genome Res. 2011;21:1388–1393. doi: 10.1101/gr.117416.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Law J, Buist G, Haandrikman A, Kok J, Venema G, Leenhouts K. A system to generate chromosomal mutations in Lactococcus lactis which allows fast analysis of targeted genes. J. Bacteriol. 1995;177:7011–7018. doi: 10.1128/jb.177.24.7011-7018.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goh Y-J, Azcárate-Peril MA, O'Flaherty S, Durmaz E, Valence F, Jardin J, Lortal S, Klaenhammer TR. Development and application of a upp-based counterselective gene replacement system for the study of the S-layer protein SlpX of Lactobacillus acidophilus NCFM. Appl. Environ. Microbiol. 2009;75:3093–3105. doi: 10.1128/AEM.02502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lambert JM, Bongers RS, Kleerebezem M. Cre-lox-based system for multiple gene deletions and selectable-marker removal in Lactobacillus plantarum. Appl. Environ. Microbiol. 2007;73:1126–1135. doi: 10.1128/AEM.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mills DA, Manias DA, McKay LL, Dunny GM. Homing of a group II intron from Lactococcus lactis subsp. lactis ML3. J. Bacteriol. 1997;179:6107–6111. doi: 10.1128/jb.179.19.6107-6111.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sørvig E, Mathiesen G, Naterstad K, Eijsink VG, Axelsson L. High-level, inducible gene expression in Lactobacillus sakei and Lactobacillus plantarum using versatile expression vectors. Microbiology. 2005;151:2439–2449. doi: 10.1099/mic.0.28084-0. [DOI] [PubMed] [Google Scholar]
- 49.Datta S, Costantino N, Zhou X, Court DL. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc. Natl Acad. Sci. USA. 2008;105:1626–1631. doi: 10.1073/pnas.0709089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mierau I, Kleerebezem M. 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl. Microbiol. Biotechnol. 2005;68:705–717. doi: 10.1007/s00253-005-0107-6. [DOI] [PubMed] [Google Scholar]
- 51.Kleerebezem M, Boekhorst J, van Kranenburg R, Molenaar D, Kuipers OP, Leer R, Tarchini R, Peters SA, Sandbrink HM, Fiers MW, et al. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl Acad. Sci. USA. 2003;100:1990–1995. doi: 10.1073/pnas.0337704100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Azcarate-Peril MA, Altermann E, Goh YJ, Tallon R, Sanozky-Dawes RB, Pfeiler EA, O'Flaherty S, Buck BL, Dobson A, Duong T, et al. Analysis of the genome sequence of Lactobacillus gasseri ATCC 33323 reveals the molecular basis of an autochthonous intestinal organism. Appl. Environ. Microbiol. 2008;74:4610–4625. doi: 10.1128/AEM.00054-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy KC, Campellone KG. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 2003;4:11–22. doi: 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 55.Courvalin P. Vancomycin resistance in gram-positive cocci. Clin. Infect. Dis. 2006;42(Suppl 1):S25–S34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 56.Bugg TD, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry. 1991;30:10408–10415. doi: 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- 57.Walsh CT. Vancomycin resistance: decoding the molecular logic. Science. 1993;261:308–309. doi: 10.1126/science.8392747. [DOI] [PubMed] [Google Scholar]
- 58.Park IS, Walsh CT. D-Alanyl-D-lactate and D-alanyl-D-alanine synthesis by D-alanyl-D-alanine ligase from vancomycin-resistant Leuconostoc mesenteroides. Effects of a phenylalanine 261 to tyrosine mutation. J. Biol. Chem. 1997;272:9210–9214. doi: 10.1074/jbc.272.14.9210. [DOI] [PubMed] [Google Scholar]
- 59.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–8. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Warner JR, Reeder PJ, Karimpour-Fard A, Woodruff LBA, Gill RT. Rapid profiling of a microbial genome using mixtures of barcoded oligonucleotides. Nat. Biotechnol. 2010;28:856–862. doi: 10.1038/nbt.1653. [DOI] [PubMed] [Google Scholar]
- 61.Delgado S, Flórez AB, Mayo B. Antibiotic susceptibility of Lactobacillus and Bifidobacterium species from the human gastrointestinal tract. Curr. Microbiol. 2005;50:202–207. doi: 10.1007/s00284-004-4431-3. [DOI] [PubMed] [Google Scholar]
- 62.Bernardeau M, Vernoux JP, Henri-Dubernet S, Guéguen M. Safety assessment of dairy microorganisms: the Lactobacillus genus. Int. J. Food Microbiol. 2008;126:278–285. doi: 10.1016/j.ijfoodmicro.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 63.Savino F, Pelle E, Palumeri E, Oggero R, Miniero R. Lactobacillus reuteri (American Type Culture Collection Strain 55730) versus simethicone in the treatment of infantile colic: a prospective randomized study. Pediatrics. 2007;119:e124–e130. doi: 10.1542/peds.2006-1222. [DOI] [PubMed] [Google Scholar]
- 64.van Baarlen P, Troost F, van der Meer C, Hooiveld G, Boekschoten M, Brummer RJ, Kleerebezem M. Human mucosal in vivo transcriptome responses to three lactobacilli indicate how probiotics may modulate human cellular pathways. Proc. Natl Acad. Sci. USA. 2011;108(Suppl 1):4562–4569. doi: 10.1073/pnas.1000079107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harty DW, Oakey HJ, Patrikakis M, Hume EB, Knox KW. Pathogenic potential of lactobacilli. Int. J. Food Microbiol. 1994;24:179–189. doi: 10.1016/0168-1605(94)90117-1. [DOI] [PubMed] [Google Scholar]
- 66.Cannon JP, Lee TA, Bolanos JT, Danziger LH. Pathogenic relevance of Lactobacillus: a retrospective review of over 200 cases. Eur. J. Clin. Microbiol. Infect. Dis. 2005;24:31–40. doi: 10.1007/s10096-004-1253-y. [DOI] [PubMed] [Google Scholar]
- 67.Svec P, Sevcíková A, Sedlácek I, Bednárová J, Snauwaert C, Lefebvre K, Vandamme P, Vancanneyt M. Identification of lactic acid bacteria isolated from human blood cultures. FEMS Immunol. Med. Microbiol. 2007;49:192–196. doi: 10.1111/j.1574-695X.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 68.Salminen MK, Rautelin H, Tynkkynen S, Poussa T, Saxelin M, Valtonen V, Järvinen A. Lactobacillus bacteremia, species identification, and antimicrobial susceptibility of 85 blood isolates. Clin. Infect. Dis. 2006;42:e35–e44. doi: 10.1086/500214. [DOI] [PubMed] [Google Scholar]
- 69.Husni RN, Gordon SM, Washington JA, Longworth DL. Lactobacillus bacteremia and endocarditis: review of 45 cases. Clin. Infect. Dis. 1997;25:1048–1055. doi: 10.1086/516109. [DOI] [PubMed] [Google Scholar]
- 70.Mermel LA, Allon M, Bouza E, Craven DE, Flynn P, O'Grady NP, Raad II, Rijnders BJA, Sherertz RJ, Warren DK. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 update by the infectious diseases society of America. Clin. Infect. Diseases. 2009;49:1–45. doi: 10.1086/599376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stevens DL. The role of vancomycin in the treatment paradigm. Clin. Infect. Dis. 2006;42(Suppl 1):S51–S57. doi: 10.1086/491714. [DOI] [PubMed] [Google Scholar]
- 72.Kuipers O. Quorum sensing-controlled gene expression in lactic acid bacteria. J. Biotechnol. 1998;64:15–21. [Google Scholar]
- 73.Kuipers OP, Beerthuyzen MM, Siezen RJ, de Vos WM. Characterization of the nisin gene cluster nisABTCIPR of Lactococcus lactis. Requirement of expression of the nisA and nisI genes for development of immunity. Eur. J. Biochem. 1993;216:281–291. doi: 10.1111/j.1432-1033.1993.tb18143.x. [DOI] [PubMed] [Google Scholar]
- 74.de Ruyter P, Kuipers O, Beerthuyzen M, van AlenBoerrigter I, deVos W. Functional analysis of promoters in the nisin gene cluster of Lactococcus lactis. J. Bacteriol. 1996;178:3434–3439. doi: 10.1128/jb.178.12.3434-3439.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.