

Abstract
Lesions displaying a variety of dysplastic changes precede invasive oral and epidermal squamous cell carcinoma (SCC); however, there are no histopathological criteria for either confirming or staging premalignancy. SCCs and dysplasias frequently contain cells that abnormally express the γ2 subunit of laminin-332. We developed cell culture models to investigate γ2 dysregulation. Normal human keratinocytes displayed density-dependent repression of γ2, whereas premalignant keratinocytes and SCC cells overexpressed γ2 and secreted laminin assembly intermediates. Neoplastic cells had hyperactive EGFR/MAPK(ERK) signaling coordinate with overexpressed γ2, and EGFR and MEK inhibitors normalized γ2 expression. Keratinocytes engineered to express HPV16 E6 or activated mutant HRAS, cRAF1, or MEK1 lost density repression of γ2 and shared with neoplastic cells signaling abnormalities downstream of ERK, including increased phosphorylation of S6 and eIF4 translation factors. Notably, qPCR results revealed that γ2 overexpression was not accompanied by increased γ2 mRNA levels, consistent with ERK-dependent, eIF4B-mediated translation initiation of the stem-looped, 5′-untranslated region of γ2 mRNA in neoplastic cells. Inhibitors of MEK, but not of TORC1/2, blocked S6 and eIF4B phosphorylation and γ2 overexpression. Immunostaining of oral dysplasias identified γ2 overexpression occurring within fields of basal cells that had elevated p-S6 levels. These results reveal a causal relationship between ERK-dependent translation factor activation and laminin γ2 dysregulation and identify new markers of preinvasive neoplastic change during progression to SCC.
Squamous cell carcinoma (SCC) is a major cancer that arises from stratified epithelia, including the oral epithelium, epidermis, and the epithelia covering the oropharynx, esophagus, tracheobronchus, bladder, vulva, and cervix. Among attributes shared by the normal basal cells of these epithelia are synthesis of the basement membrane protein laminin-332 (Lam332) (previously known as laminin 5) and control of their cell division by EGF.1,2 Approximately 300,000 new cases of SCC per year occur in the United States, resulting annually in ∼100,000 deaths.3,4 Depending on the tissue of origin, the carcinogenic instigator is chronic exposure to UV radiation (epidermis), tobacco (oral and lung), industrial or environmental carcinogens (lung and bladder), or infection with papillomaviruses HPV16/18 (cervix, some vulva, some oropharynx).5–8 Abnormal hyperactivity of the EGFR/ERK and the PI3K/AKT/mTOR signaling pathways are typical characteristics of advanced SCC.9–11 If not excised at an early stage, SCCs infiltrate deeply and spread rapidly by colonizing regional lymph nodes, such that they can no longer be completely surgically resected. SCCs of all tissue origins have proven refractory to nonsurgical treatment regimens, including small-molecule kinase inhibitors.12–15 Annually in the United States there are 20,000 new cases of oral SCC and 40,000 head and neck SCCs (combined total for oral and oropharynx SCCs). Five-year survival after diagnosis of head and neck SCC is ∼50%, which has remained unchanged for many decades, regardless of treatment protocols.3,16
Visual accessibility of the oral cavity has led to the identification of potential precursor lesions to SCC. Such lesions appear more white, more red, or more scaly than normal epithelium. The incidence of oral lesions is high, at approximately 1% to 3% in adults.17 Some of these lesions eventually disappear, whereas others progress to invasive SCC. It is estimated that, in the United States and Europe, oral dysplasias progress to SCC at a rate of 2% to 6% per year.17–20 Microscopically, many of these lesions show signs of dysplasia, including abnormal cell and nuclear shape and delay of suprabasal differentiation.21,22 There remains much uncertainty about the criteria for a diagnosis of premalignant dysplasia and whether any feature of dysplastic change predicts future progression to SCC,20,23,24 so the need to identify prognostic biomarkers is urgent.
In an earlier study,25 we detected coordinate induction of p16INK4A and overexpression of laminin γ2 (Lamγ2) in some oral and epidermal dysplasias, occurring at greater frequency in dysplasias of higher degrees of atypia and at margins of invasive SCC. Lamγ2 and the two partner subunits laminin α3 and laminin β3 form Lam332. After intracellular assembly of the three subunits, normal basal epithelial cells secrete the Lam332 trimer, proteolytically process it, and deposit it into the underlying basal lamina, where it serves as an essential component of hemidesmosomes, conferring stable, polarized epithelial cell adhesion.2 After tissue development is complete, basal keratinocytes stop synthesizing immunohistochemically detectable amounts of Lamγ2.25,26 Invading carcinoma cells, however, frequently reinitiate high rates of Lamγ2 synthesis.27–29 Because abnormal Lamγ2 expression begins in premalignant lesions,25 its immunohistologic detection has potential as a prognostic marker for lesions at high risk for progression to SCC. Determining the mechanism of neoplastic Lamγ2 dysregulation could identify ancillary prognostic markers and potential targets for early-intervention drug therapy.
We have identified a human cell culture model system that recapitulates the in vivo situation of Lamγ2 repression by normal keratinocytes and overexpression by premalignant and malignant keratinocytes. Using this system, we have identified EGFR/MAPK(ERK)-pathway hyperactivation as the driver of Lamγ2 overexpression by neoplastic cells, correlated with increased phosphorylation and activation of the translation factors S6 and eIF4B. Our data reveal that neoplastic Lamγ2 expression is the result of increased translation, not transcription. We find that in oral dysplasias in vivo, Lamγ2 overexpression occurs strictly within fields of basal cells that have abnormally increased levels of p-S6. These results serve to define a new, immunohistologically detectable stage of neoplastic progression in preinvasive dysplasias.
Materials and Methods
Cells and Cell Culture
The cell lines used are characterized in Table 1 and in the articles cited therein.1,30–39 Derivation and use of these cell lines were IRB-approved. The premalignant oral keratinocyte line D17 was provided by Dr. E. Kenneth Parkinson (Queen Mary University of London, UK). Cells were cultured in keratinocyte serum-free medium (Ksfm) (Invitrogen; Life Technologies, Carlsbad, CA) with 25 μg/mL bovine pituitary extract, 0.2 ng/mL epidermal growth factor (EGF), and CaCl2 to a final Ca2+ concentration of 0.4 mmol/L, as described previously.30 To maintain healthy confluent cultures, after cultures reached ∼40% confluence in Ksfm they were refed daily with 1:1 medium (1:1 vol/vol Ca2+-free Dulbecco's modified Eagle's medium from Invitrogen with Ksfm and supplemented as described above for Ksfm alone), as described previously.1 To avoid variation among cell lines with respect to stratification and differentiation, cultures approaching confluence were fed with 1:1 medium with 0.1 mmol/L CaCl2. To assess EGF-dependent growth, 2000 cells per 9-cm2 well were plated with or without EGF, refed every 2 to 3 days, and counted 6 to 8 days later. Average growth rate in terms of population doublings (PD) per day was calculated as log2(number of cells obtained at subculture/number of cells plated)/number of days cultured.
Table 1.
Human Cell Lines Used in This Study
| Cell line | Donor sex (age)⁎ | Special characteristics | References† |
|---|---|---|---|
| Epidermal keratinocyte | |||
| Strain N | M (0) | Normal | 1, 30–33 |
| N/HPV16E6E7 | M (0) | Immortal; p53- and pRb-deficient | 1, 31 |
| N/HPV16E6 | M (0) | All E6 functions, including p53-deficiency | |
| N/HPV16E6 (JH26 mutant) | M (0) | p53 normal but has other E6 functions | |
| N/HPV16E7 | M (0) | All E7 functions, including Rb-deficiency | 30 |
| N/p53DD/cdk4R24C | M (0) | p53-deficient and p16INK4A-resistant | 34 |
| N/TERT1 | M (0) | Immortal; loss of ability to express p16INK4A and p14ARF | 1, 30, 31 |
| N/TERT1/EGFRΔ (E746-A750) | M (0) | Immortal; EGF-independent for growth | |
| N/TERT1/HRAS (G12V) | M (0) | Immortal; EGF-independent for growth; Lamγ2 overexpressing | |
| N/TERT1/cRAF1 (22W) | M (0) | Immortal; EGF-independent for growth; Lamγ2 overexpressing | |
| N/TERT1/MEK1 (DD) | M (0) | Immortal; EGF-independent for growth; Lamγ2 overexpressing | |
| G5Ep | M (0) | Normal | 35 |
| O5Ep | M (0) | Normal | |
| Epidermal SCC | |||
| SCC-13 | F (56) | p16- and p53-deficient; immortal; tumorigenic | 31–33, 36, 37 |
| Oral keratinocyte | |||
| OKF6 | M (57) | Normal | 30 |
| Dysplastic oral keratinocyte | |||
| POE9n | M (65) | Homozygous deletion of p16INK4A and p14ARF; p53-deficient, nontumorigenic | 30, 31 |
| D17 | ND (ND) | p16INK4A-deficient; normal p53; nontumorigenic | 38, 39 |
| Oral SCC | |||
| SCC-15 | M (55) | p16- and p53-deficient; immortal; tumorigenic | 32, 36, 37 |
| SCC-25 | M (74) | p16- and p53-deficient; immortal; tumorigenic | 32, 36, 37 |
| SCC-68 | M (ND) | p16- and p53-deficient; immortal; tumorigenic | |
| SCC-71 | M (80) | p16- and p53-deficient; immortal; tumorigenic | 31, 36 |
F, female; M, male; ND, no data.
Ages are given in years; 0 indicates newborn.
References for derivation and characterization of previously published cell lines.
Protein Extracts and Conditioned Medium Samples
To analyze laminin protein expression in cells, cultures were suspended with trypsin/EDTA and lysed in protein extraction buffer consisting of 20 mmol/L Tris-HCl (pH 7.3), 2% SDS, EDTA-free protease inhibitor cocktail (Roche, Indianapolis, IN), and phosphatase inhibitor cocktail (Thermo Scientific, Rockford, IL). Alternatively, cultures were rinsed twice with PBS, drained, and lysed in protein extraction buffer to obtain both intracellular proteins and proteins that had been deposited onto the culture dish (matrix proteins). Extracts were sonically disrupted and stored at −80°C. The BCA protein assay (Thermo Scientific) was used to measure protein concentrations. For phosphoprotein analysis, cultures were lysed in reducing Laemmli SDS sample buffer.40 To analyze secreted laminin, conditioned medium of cultures was collected 24 hours after refeeding, passed through 0.2-μm filters, and stored at −80°C. After conditioned medium harvest, cultures were trypsinized and their proteins were extracted and measured to normalize loading volumes of conditioned medium samples for comparative Western blot analysis.
Western Blotting
Protein (20 μg per lane) in Laemmli sample buffer was separated by SDS-PAGE under reducing or nonreducing conditions on 4% to 20% gradient gels (Bio-Rad Laboratories, Hercules, CA), blotted to polyvinyl difluoride membranes (Bio-Rad Laboratories), blocked for 1 hour with 5% nonfat milk in Tris-buffered saline with 0.05% polysorbate 20 (Tween 20), incubated overnight with primary antibody, rinsed, and incubated for 1 hour with peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:2000) (Dako, Glostrup, Denmark) at room temperature. Blots were developed using ECL (Roche) or SuperSignal West Femto chemiluminescent substrate (Thermo Scientific) and exposed to HyBlot CL autoradiography film (Denville Scientific, Metuchen, NJ).
Some Lamγ2 Western blots were analyzed densitometrically using ImageJ software version 1.45 (NIH, Bethesda, MD; http://rsbweb.nih.gov/ij). Two different film exposure times were analyzed. The total intensity of each Lamγ2 band was normalized to the actin band intensity of the same extract and film exposure time.
Antibodies
The murine monoclonal antibodies used were laminin γ2 (clone D4B526; Chemicon, Billerica MA), laminin α3 (clone 12C4,41 provided by Jonathan Jones, Northwestern University, Chicago, IL), laminin β3 (clone 17) and c-myc (clone 9E10; BD Transduction Laboratories, Franklin Lakes, NJ), p53 (clone DO-1; Santa Cruz Biotechnology, Santa Cruz, CA), EGFR (clone 1F4; Cell Signaling Technology, Danvers, MA), and FLAG (clone M2; Sigma-Aldrich, St. Louis, MO). The rabbit monoclonal antibodies used were p-eIF4E(Ser209) (clone EP2151Y) from EMD Millipore (Billerica, MA) and p-AKT(Ser473) (clone D9E), p-S6(ser235/236) (clone 91B2), and p-EGFR(Tyr1068) (clone D7A5) from Cell Signaling Technology. The rabbit polyclonal antibodies used were β-actin (A-2066; Sigma-Aldrich), p-eIF4B(Ser422) (Abcam, Cambridge, MA), and ERK1/2, p-ERK1/2(Thr202/Tyr204), and p-4E-BP1(Ser65) (Cell Signaling Technology).
Signaling Pathway Inhibitors
The TGFβ receptor I kinase inhibitor TRI (discovered independently as Biogen HTS466284 and Lilly LY364947) was used at 1 μmol/L (EMD Millipore Chemicals, Gibbstown, NJ).31,42 The EGFR inhibitor gefitinib43,44 (provided by Pasi Janne, Dana-Farber Cancer Institute) was used at 1 μmol/L. The MEK inhibitors AZD624445 (Selleck Chemicals, Houston, TX) and U012646 (Cell Signaling Technology) were used at 100 nmol/L and 1 μmol/L, respectively. The mTORC1/2 inhibitor Ku-006379447,48 (Chemdea, Ridgewood, NJ) was used at 500 nmol/L. The ERK inhibitor CAY1056149 (Cayman Chemical, Ann Arbor, MI) and the RSK inhibitor BI-D187050 (Symansis, Auckland, NZ) were each used at 1 μmol/L. The translation inhibitor cycloheximide (Sigma-Aldrich) was used at 10 μg/mL and the protein transport inhibitor brefeldin A (BD Biosciences, San Jose, CA) at 1 μg/mL. Inhibitors were added to the medium at plating and each refeeding from 1000× concentrated solutions in dimethyl sulfoxide, stored frozen between uses.
Immunofluorescence Staining
Cultures were rinsed, fixed, stored in 100% methanol at −20°C, and air-dried before use. After 15 minutes of blocking in PBS with 10% goat serum (Invitrogen), primary antibodies diluted in PBS with 0.1% BSA were applied for 1 hour at room temperature. Cultures then were rinsed with PBS, incubated with fluorescent-labeled secondary antibodies (Invitrogen) for 1 hour, rinsed, and coverslip-mounted with ProLong Gold antifade mounting medium with DAPI (Invitrogen). Cells were examined under an inverted Nikon Eclipse Ti fluorescence microscope fitted with an RT-slider SPOT camera (SPOT Imaging Solutions, Sterling Heights, MI).
Tissue Samples, Immunohistochemical Staining, and Quantification
Selection and diagnosis of a set of 26 formalin-fixed, paraffin-embedded, lateral tongue tissue specimens from the University of Connecticut Oral Pathology archival collection was performed with IRB approval by an expert oral pathologist (E.N.). The cohort included normal epithelium, benign warts, dysplasias, and microinvasive to advanced SCCs.
Paraffin sections (5 μm thick) on slides were warmed to 37°C for 30 minutes, then deparaffinized and hydrated through xylene, ethanol, and PBS; to better preserve antigens, slides were not baked at 50°C after sectioning, as is a common practice of histopathology labs.25 Antigen retrieval was performed in 10 mmol/L sodium citrate buffer (pH 6.0) autoclaved for 15 minutes. Endogenous peroxidase was inactivated by incubating slides in 0.3% H2O2 in methanol for 10 minutes. After a PBS rinse, slides were blocked with PBS with 10% horse serum (Invitrogen) for 20 minutes, incubated with primary antibody for 1 hour at room temperature, biotinylated secondary antibody (1:100) for 1 hour, and avidin/biotin/peroxidase complex (ABC) reagent (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA) for 45 minutes, with three PBS rinses between each step. Slides were incubated with Vector Red (Vector Laboratories) for 1 to 5 minutes, the reaction was stopped by rinsing in water, and slides were coverslip-mounted with Fluoromount-G mounting medium (SouthernBiotech, Birmingham, AL). Slides were examined under a Nikon Eclipse TE2000-S microscope and were photographed as described above.
Twenty-one dysplasia specimens for which sequential sections were stained for Lamγ2 and p-S6 were quantified for percentage basal p-S6 and Lamγ2 expression. The total length of dysplastic areas in each specimen was determined by examining an adjacent H&E-stained slide, and the proportion of the dysplastic region containing p-S6+ and Lamγ2+ basal cells was determined.
Retroviral Vectors and Transduction
Retroviral vector plasmids pBABE.puro/EGFRΔ(E746-A750)-FLAG, [EGFRΔ (746–750) retroviral vector was provided by Dr. Pasi Janne (Dana Farber Cancer Institute, Boston)],43 pBABE.puro/cRAF-1(22w),51,52 pLHCX/MEK1(S218D,S222D) [MEK1(DD)],53,54 pBABE.puro/HRAS (G12V),55,56 pL(HPV16E6)SN [HPV16E6 retroviral vector was provided by Dr. Denise Galloway (Fred Hutchinson Cancer Center, Seattle)],57 and pL(E6(JH26))SN58–60 [HPV16E6(JH26) retroviral vector was provided by Drs. Karl Munger and Peter Howley (Harvard Medical School, Boston)] were transfected into Phoenix packaging cells61 using TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI). Retroviral supernatants were collected in 1:1 medium (as above) at 8- to 16-hour intervals from 1 to 3 days after transfection, passed through 0.45-μm pore filters, and stored at −80°C. Cells plated 1 day previously at 105 cells/9 cm2 well in Ksfm were transduced for 6 to 7 hours with retroviral supernatants containing 2 μg/mL polybrene (Sigma-Aldrich), as described previously.30 Transduced cells were subcultured the next day into p100 tissue culture dishes with Ksfm containing 15 μg/mL hygromycin, 1 μg/mL puromycin, or 0.2 mg/mL G418 and drug-selected for 5 to 7 days to obtain pure populations of stable transductants.
Quantitative PCR
Total RNA was isolated from cells using an RNeasy Plus mini kit (Qiagen, Valencia, CA) and cDNA synthesized from 500 ng total RNA using the iScript cDNA synthesis kit (Bio-Rad Laboratories). mRNA levels were quantified by quantitative real-time PCR (qPCR) using Applied Biosystems (ABI) FAST SYBR Green PCR master mix (Life Technologies, Foster City, CA) on an ABI StepONE Plus instrument (40 cycles of 95°C for 15 seconds and 58°C for 30 seconds). Amplicons were as follows: laminin γ2 (LAMC2 gene), forward 5′-CTCTGCTTCTCGCTCCTCC-3′ and reverse 5′-TCTGTGAAGTTCCCGATCAA-3′; laminin α3 (LAMA3), forward 5′-TCAGCACATATTATCTGGGAGG-3′ and reverse 5′-AAATTTTTCATGCAGCCTCG-3′; laminin β3 (LAMB3), forward 5′-CCAAGCCTGAGACCTACTGC-3′ and reverse 5′-GCCACATTCTCTACTCGGTGA-3′); and GAPDH, forward 5′-GAGCCTCAAGATCATCAGCA-3′ and reverse 5′-ACAGTCTTCTGGGTGGCAGT-3′). Relative expression was calculated using the ΔΔCT method, normalizing values to GAPDH within each sample; standard error of the mean was calculated from the results of triplicate aliquots.
Results
A Human Cell Culture Model of Neoplastic Laminin γ2 Overexpression
Our earlier study that found abnormal Lamγ2 expression in oral and epidermal dysplasias25 extended previous reports that Lamγ2 is frequently expressed by cells at the invading fronts of SCC and certain other carcinomas.27–29 Examples of such Lamγ2 immunostaining patterns are shown in Figure 1A. To investigate the mechanism of neoplastic Lamγ2 overexpression, we sought to identify culture conditions under which normal primary keratinocytes would cease, but SCC cells would continue, expressing Lamγ2. Cell lines used are characterized in Table 1. At low cell density, both normal keratinocytes (G5Ep) and SCC cells (SCC-68) synthesized Lamγ2 (intracellular staining) and secreted this protein onto the culture dish (extracellular staining) (Figure 1B). Substratum-deposited Lamγ2 remained stably adherent to the culture dish for many days after secretion, as evidenced by the equal staining intensity of Lamγ2 adjacent to and at a distance from the current position of cells (Figure 1B). (Note that during proliferation in culture keratinocytes migrate randomly over short distances, with the extent of this movement over the course of many days detectable by immunostaining for the Lam332 that the cells deposit on the substratum.31)
Figure 1.

Laminin γ2 overexpression by neoplastic epithelial cells in vivo and at high cell density in culture. A: H&E-stained and Lamγ2-immunostained sections of the margin between normal oral epithelium and a premalignant dysplasia (top panels) and an invasive oral SCC (bottom panels). Note absence of Lamγ2 expression in normal epithelium, in contrast to focal expression of Lamγ2 by basal cells in the dysplastic lesion and in the SCC. Scale bar = 250 μm. B: Preconfluent and confluent cultures of normal primary keratinocytes (G5Ep) and SCC cells (SCC-68) showing phase-contrast images and the same (preconfluent) or similar (confluent) fields immunostained for Lamγ2 (green fluorescence). Note substantial intracellular (endoplasmic reticulum/Golgi) Lamγ2 content and deposition onto the culture dish by preconfluent normal keratinocytes and SCC cells, whereas only the SCC cells continued to express immunodetectable Lamγ2 at confluence. Scale bar = 50 μm. C: Western blot analysis of Lamγ2 expression by G5Ep and SCC-68 cells as a function of cell density. Protein extracts of replicate cultures were harvested at cell densities indicated by the phase contrast microscopic images shown beneath (Scale bar = 500 μm), either as trypsin/EDTA-detached cell extracts (C) or scraped extracts (ie, total cell plus extracellular matrix material) (C+M). The upper band in blots is the 155-kDa unprocessed form and the lower band is the 105-kDa postsecretion processed form of Lamγ2. Note reduction of Lamγ2 expression by normal keratinocytes with increasing cell density, in contrast to density-independent expression by SCC cells. D: Harvests of conditioned medium at 24 hours from confluent cultures of normal keratinocytes (G5Ep), premalignant keratinocytes (N/E6E7 and POE9n), and SCC cells (SCC-13, SCC-68) separated on a reducing gel, Western blotted, and probed for Lamγ2. Note increased production rates of Lamγ2 by premalignant and SCC cells, and also Lamγ2 processing deficiencies in POE9n, SCC-13, and SCC-68.
As normal keratinocytes grew to higher density, they began to reduce Lamγ2 synthesis and at confluence no longer expressed this protein at detectable levels (Figure 1, B and C). In contrast, SCC cells expressed Lamγ2 at the same levels independent of density (Figure 1, B and C). Lamγ2 previously deposited by cells onto the culture dish remained adherent after trypsin/EDTA-detachment of cells but was solubilized together with intracellular protein by direct addition of SDS lysis buffer to cultures (compare detached cell extracts with scraped extracts in Figure 1C). The scraped extract contained both intracellular, unprocessed (155 kDa) and extracellular, proteolytically processed (105 kDa) forms of Lamγ2. Western blot analysis of the conditioned medium of confluent N/E6E7 cultures showed an increased rate of Lamγ2 synthesis and secretion, similar to that of SCC cells (Figure 1D).
To determine whether different rates of intracellular Lamγ2 protein turnover contribute to the apparent differences In Lamγ2 expression between normal keratinocytes and SCC cells, we examined intracellular Lamγ2 stability using cycloheximide. Because Lamγ2 is normally secreted, we treated cells with brefeldin A, an inhibitor of endoplasmic reticulum-to-Golgi transport, to retain the protein inside cells. Treatment with brefeldin A alone increased intracellular Lamγ2, as expected (see Supplemental Figure S1 at http://ajp.amjpathol.org). Normal and SCC cells treated with brefeldin A and cycloheximide showed little or no decrease in Lamγ2 over 6 hours, with no indication of a shorter half-life of the protein in normal cells (see Supplemental Figure S1 at http://ajp.amjpathol.org). In contrast, levels of MYC, a protein known to have a short half-life, decreased progressively in normal keratinocytes and SCC cells with time after cycloheximide treatment (see Supplemental Figure S1 at http://ajp.amjpathol.org). We concluded that Lamγ2 overexpression is the result of increased rates of synthesis of this protein by SCC cells.
Unbalanced Expression of the Three Laminin-332 Chains Results in Secretion of Laminin γ2 Monomers by SCC Cells
We next examined expression of the other two Lam332 subunits, Lamα3 and Lamβ3, to determine whether their synthesis is differentially regulated in normal keratinocytes and SCC cells. Normal keratinocytes (G5Ep, O5Ep, and strain N) at high cell density greatly reduced Lamα3 expression, as well as Lamγ2 expression, and slightly reduced Lamβ3 expression, compared with low density cells (Figure 2A). SCC cells showed density-related changes in Lamα3 and Lamβ3 expression similar to that of normal keratinocytes, but continued to express Lamγ2 at high levels at confluence (Figure 2A). The consequence of excess Lamγ2 expression in SCC cells was revealed by nonreducing Western blots, which resolve laminin monomers from the disulfide-bonded heterodimer assembly intermediates and the ultimate Lam332 trimer.62 Cell extracts of confluent normal keratinocytes contained Lamγ2 and Lamβ3 monomers and the ∼240 kDa Lamβ3γ2 assembly intermediate, in addition to fully formed ∼440 kDa Lam332 trimer (Figure 2B). In contrast, SCC cells contained mostly Lamγ2 and Lamβ3 monomers and heterodimers, with little trimer (Figure 2B), presumably the result of unbalanced subunit expression (Figure 2A). Analysis of conditioned medium (Figure 2C) revealed that normal keratinocytes secreted Lamγ2 exclusively assembled into Lam332 trimers, whereas a fraction of the total Lamγ2 in SCC conditioned medium was in the form of monomers, both unprocessed (155 kDa) and processed (105 kDa).63
Figure 2.

Unbalanced Lam332 subunit expression by SCC cells and secretion of abnormal assembly intermediates. A: Western blot analysis of the three Lam332 subunits in low-density (L) versus high-density (H) cultures of normal keratinocytes (G5Ep, O5Ep, strain N) and SCC cells (SCC-25, SCC-15, SCC-68). Note strong density repression of Lamγ2 by normal keratinocytes and lack of it by SCC cells. B: Nonreducing gels of cell extracts of confluent normal keratinocyte and SCC cultures Western blotted for Lamγ2 (upper gel) and Lamβ3 (lower gel). Note that SCCs have little or no intracellular Lam332 trimer, compared with normal keratinocytes, and that the Lamβ3 antibody does not bind well to Lamβ3 after assembly into the Lam332 trimer in normal cells. C: Nonreducing gels of conditioned medium from confluent normal keratinocyte and SCC cultures Western blotted for Lamγ2, disclosing relative proportions of normal Lam332 trimer, β3γ2 dimer, and Lamγ2 monomer. Note secretion by SCC cells of Lamγ2 monomer in processed (105 kDa; γ2) and unprocessed (155 kDa; γ2′) forms.
Abnormal Laminin γ2 Regulation Begins Early in Neoplastic Progression as a Characteristic of Premalignant Keratinocytes
In vivo, Lamγ2 overexpression begins in regions of preinvasive dysplasias25 (Figure 1A). We therefore asked whether cells cultured from dysplasias display abnormal Lamγ2 expression as a stable characteristic, as is the case for SCC cells. The POE9n and D17 cell lines cultured from human oral dysplasias30,38,39 proved to maintain Lamγ2 expression at confluence, in contrast to the normal keratinocytes OKF6 and strain N (Figure 3, A and B).
Figure 3.

Abnormal laminin γ2 expression by premalignant keratinocytes. A: Lamγ2 immunofluorescence-stained confluent cultures of normal oral keratinocyte line OKF6, premalignant dysplastic oral keratinocyte lines POE9n and D17, normal epidermal keratinocyte line strain N, and strain N transduced to express the HPV16 E6 and E7 oncoproteins (N/E6E7). Note continued Lamγ2 expression at confluence by POE9n, D17, and N/E6E7 in contrast to repression of Lamγ2 under this condition by normal keratinocytes. Scale bar = 200 μm. B: Left: Lamγ2 Western blots of low-density (L) and high-density (H) cultures of the same cell lines, confirming lack of γ2 repression at confluence by POE9n, D17, and N/E6E7. Right: Lamγ2 Western blots of N/E7, N/E6, N/E6JH26, and of strain N transduced to express a dominant-negative mutant p53 and a p16INK4A-resistant mutant cdk4 (N/p/c), showing that E6 alone can dysregulate Lamγ2, and that functional loss of p53 is not involved in this dysregulation. Densitometric quantification of each band is shown below the blots. The Lamγ2 level (normalized to its actin band) of the preconfluent culture of each cell line is set as 1 (gray bars), with the relative Lamγ2 level of the confluent culture shown for each cell line (black bars). Error bars indicate the results of quantifying scans of two exposures of each gel. The error bars show the results of the scans of two different exposure times of the same Western blot. The gray and black bars show the average of these two scans.
In certain stratified epithelia (ie, cervix, tonsil, and vulva), early neoplastic development is sometimes linked to infection with human papillomavirus (HPV) type 16 or 18, leading to integration of viral genomic DNA and expression of their E6 and E7 oncogenes.5–8 Normal primary keratinocytes (strain N) transduced to express HPV16 E6 and E7 (the N/E6E7 cell line) maintained Lamγ2 expression at confluence, similar to SCC cells (Figures 1 and 2) and POE9n and D17 cells (Figure 3, A and B).
Important activities of the HPV16 oncoproteins are to target p53 for degradation (accomplished by the E6 oncoprotein60) and to inactivate pRB and thereby confer resistance to the CDK4 inhibitor p16INK4A (accomplished by E764). We wished to determine whether loss of p53-dependent and pRB/p16INK4A-dependent growth control achieved by genetic alterations not involving viral oncogenes, as is the case for most oral and epidermal SCCs, would result in loss of normal Lamγ2 regulation. We therefore assessed the Lamγ2 expression characteristics of strain N keratinocytes engineered to express a dominant-negative mutant p53 and a p16-insensitive mutant cdk4 (N/p/c cells34) and found that N/p/c cells reduced Lamγ2 expression at confluence similar to normal primary keratinocytes. This finding demonstrated that activities of E6 and/or E7 other than disabling p53 or pRB/p16INK4A functions are responsible for Lamγ2 dysregulation. Comparing the properties of strain N cells engineered to express either E7 or E6 alone, we found that E6 alone caused loss of Lamγ2 regulation (Figure 3B). Finally, examining the effects of expressing E6(JH26), a mutant of E6 that lacks p53-targeting activity but retains E6 transcriptional transactivation activity,58,60 we found that an activity of E6 other than p53 degradation is responsible for Lamγ2 dysregulation (Figure 3B; see also Supplemental Figure S2 available at http://ajp.amjpathol.org). Notably, E6 activates transcription of a variety of genes, including hTERT65,66 and EGFR,67,68 and has been reported recently to activate mTOR.69
Hyperactivity of Signaling Kinases Downstream of EGFR Correlates with Lamin γ2 Overexpression
Hyperactivity of the EGFR/(MAPK)ERK and PI3K/AKT/mTOR pathways are frequent abnormalities of advanced oral and oropharyngeal SCC.9–11 We sought to identify signaling alterations in these two pathways (Figure 4A) in confluent cultures of premalignant keratinocytes and SCCs that correlate with Lamγ2 dysregulation. EGFR overexpression, accompanied by increased levels of the activated p-(Y1068) form of EGFR, was common to all cell lines that overexpressed Lamγ2 (Figure 4B). Levels of p-ERK, a downstream target of EGFR activation mediated by RAS, RAF, and MEK, also were elevated, compared with normal cells, in all of the Lamγ2-overexpressing neoplastic cell lines, although not proportional to p-EGFR levels (Figure 4B).
Figure 4.

Neoplastic cells that lack density-dependent Lamγ2 repression display EGFR/ERK hyperactivation. A: The EGFR/ERK and PI3K/AKT/mTOR kinase cascade signaling pathways and their downstream targets analyzed in this study. Arrows indicate activation; T-bars indicate inhibition. Dashed arrows indicate a less important role for S6K1 in activating these targets. Note that S6K1 must first phosphorylate S6 at ser(240/244) before either RSK or S6K1 can phosphorylate S6 at ser(235/236). Underlining indicates proteins that were analyzed for phosphorylation/activation by Western blotting. For some experiments, some proteins (shown in blue) were expressed or hyperactivated by engineering normal keratinocytes. Inhibitors that specifically block the associated kinase are shown in red. B: Confluent cultures of normal (G5Ep, OKF6, strain N) and premalignant (N/E6E7, POE9n, D17) keratinocytes and SCC cells (SCC-13, SCC-71, SCC-15) analyzed by Western blotting for Lamγ2 and selected phosphoproteins. Note consistent p-EGFR, p-ERK, and p-S6 increases in all of the Lamγ2-overexpressing cell lines. C: Effects of kinase inhibitors on Lamγ2 and Lamβ3 expression and on signaling molecules. Confluent cultures of SCC-68, SCC-13, POE9n, and N/E6E7 and preconfluent cultures of G5Ep were treated for 2 days with kinase inhibitors and then were analyzed by Western blotting. TRI is the TGFβ receptor I inhibitor, gefitinib is an EGFR inhibitor, U0126 and AZD6244 are MEK inhibitors, and Ku-0063794 is a TORC1/2 inhibitor. Note reduction of Lamγ2 expression, but not of Lamβ3 expression, by all kinase inhibitors tested except Ku-0063794. Lamγ2 reduction correlated consistently with decreased p-ERK.
We also analyzed the phosphorylation states of AKT, a signaling mediator in the PI3K/mTOR pathway, and of S6, a downstream target of both EGFR/ERK and PI3K/mTOR pathways. p-AKT was elevated in only three of the six Lamγ2 dysregulated cell lines tested, but p-S6 was substantially elevated in all of them (Figure 4B). The p-S6 antibody we used recognizes p-S6(ser235/236), which can be phosphorylated either by the ERK-activated RSK kinase or by the TORC1-activated S6 kinase (S6K1).70 We interpreted the lack of correlation between p-AKT and p-S6 levels as indicating that S6 phosphorylation in Lamγ2-overexpressing cells results from EGFR/ERK pathway hyperactivity.
We next assessed the effects of small-molecule inhibitors of pathway kinases on Lamγ2 overexpression by POE9n, NE6E7, and two SCC cell lines. The EGFR inhibitor gefitinib and the MEK inhibitors U0126 and AZD6244 substantially reduced p-ERK levels and Lamγ2 expression (Figure 4C). Gefitinib and U0126 greatly reduced p-S6 levels in all four lines, although AZD6244 did not affect S6 activation in POE9n and N/E6E7 (Figure 4C). These results indicate that ERK-dependent RSK is the major activator of S6. The TORC1/2 inhibitor Ku-0063794 greatly reduced p-S6 levels, but did not change Lamγ2 expression, revealing that S6 activation is not essential for Lamγ2 overexpression (Figure 4C). S6K1 is thought to phosphorylate S6 on ser235/236 and on ser240/244, whereas the ERK-activated RSK phosphorylates S6 only on ser235/236.71,72 Prior phosphorylation of S6 on ser240/244 by S6K1 increases the ability of RSK to phosphorylate S6 on ser235/236.67 The reduction of p-S6(ser235/236) levels by Ku-0063794 we observed is consistent with those studies.
We also tested the effects of a TGFβ receptor I kinase inhibitor, TRI, because the LAMC2 gene, which encodes Lamγ2, is transcriptionally activated by TGFβ.73 As expected, TRI reduced Lamγ2 expression in all cell lines (Figure 4C). None of the kinase inhibitors we tested reduced Lamβ3 expression (Figure 4C), consistent with the lack of correlation between Lamγ2 and Lamβ3 expression in preconfluent and confluent normal keratinocytes and SCC cells (Figure 2A).
Normal keratinocytes transiently overexpress Lamγ2 during wound re-epithelialization in vivo (see Supplemental Figure S3B at http://ajp.amjpathol.org).31,74,75 The effects of inhibitors on Lamγ2 expression and p-ERK and p-S6 levels in preconfluent normal keratinocytes were the same as for confluent premalignant and SCC cells (Figure 4C), demonstrating that Lamγ2 expression is ERK-dependent in both normal and neoplastic settings.
Laminin γ2 Expression in Neoplastic Cells Is Regulated at the Level of Translation and Correlates with the Activation States of Translation Initiation Factors
ERK activation initially was found to have effects in the nucleus, activating several transcriptional regulators, including Elk-1.76 We used qPCR to investigate whether the reduction of Lamγ2 protein expression caused by EGFR and MEK inhibition (Figure 4C) is the result of reduced LAMC2 gene transcription. Confluent SCC cultures treated with EGFR or MEK inhibitors showed no substantial change in Lamγ2 mRNA levels, compared with untreated controls (Figure 5A). TRI reduced Lamγ2 mRNA levels to 40% of control levels (Figure 5A), consistent with TRI effects on Lamγ2 protein expression (Figure 4C) and confirming the role of TGFβ signaling in Lamγ2 transcription.73
Figure 5.

Lamγ2 overexpression by neoplastic cells is unrelated to Lamγ2 mRNA levels, but correlates with ERK-dependent activation of translation initiation factors. A: Effects of kinase inhibitors on Lamγ2 mRNA levels in SCC cells. Confluent cultures were treated for 2 days with the inhibitors TRI, gefitinib, U0126, AZD6244, and Ku-0063794 and then were analyzed by qPCR for Lamγ2 mRNA, with results for each cell line internally normalized to GAPDH mRNA levels. Note the decrease in Lamγ2 mRNA levels resulting from treatment with TRI, but not with the EGFR inhibitor gefitinib or the MEK inhibitors U0126 and AZD6244, which block Lamγ2 protein synthesis in confluent SCC cultures. B: qPCR comparison of Lamγ2 mRNA levels in preconfluent and confluent cultures of normal and premalignant keratinocytes and SCC cells. Results shown are relative to Lamγ2 mRNA levels in preconfluent cultures of strain N keratinocytes, set at an arbitrary unit of 1. Note lower levels of Lamγ2 mRNA in neoplastic cells than in normal keratinocytes and absence of cell density-dependent mRNA decreases in neoplastic cells, compared with normal keratinocytes. C: Western blot analysis of activated forms of translation initiation factors in normal and premalignant keratinocytes and SCC cells. Note increased levels of p-eIF4B, p-eIF4E, and p-4E-BP1 in premalignant keratinocytes and SCC cells, compared with normal keratinocytes. D: Western blot analysis of effects of 2-day treatment of confluent SCC cultures with kinase inhibitors on levels of Lamγ2, c-myc, and phosphorylated states of translation initiation factors. Note strong reduction of Lamγ2, c-myc, and p-eIF4B levels in response to the EGFR inhibitor gefitinib and the MEK inhibitors U0126 and AZD6244, in contrast to small or no effect of the TORC1/2 inhibitor Ku-0063794. E: Western blot analysis of the effects on levels of Lamγ2 and p-eIF4B of 2-day treatments of confluent SCC cultures with ERK and RSK inhibitors, compared with U0126 treatment. Note that ERK and RSK inhibitors substantially reduced Lamγ2 and p-eIF4B levels.
We next examined the density dependence of Lamγ2 mRNA levels. Lamγ2 mRNA levels in normal keratinocytes at confluence were 25% to 40% of that of preconfluent cells (Figure 5B), consistent with transcription or mRNA stability playing a significant role in normal density-related Lamγ2 repression. However, Lamγ2 mRNA levels in premalignant and SCC cell lines did not change between preconfluence and confluence and were typically lower than those of normal keratinocytes at confluence (Figure 5B). We concluded that Lamγ2 mRNA is translated much more efficiently in neoplastic cells than in normal cells at high cell density. In contrast, Lamα3 mRNA decreased at confluence in both normal keratinocytes and SCC cells, and Lamβ3 mRNA slightly decreased in normal keratinocytes and increased in SCC cells at confluence (see Supplemental Figure S4A at http://ajp.amjpathol.org). This confirmed that the mechanism regulating expression of Lamγ2 is different from the mechanism or mechanisms regulating expression of the other Lam332 subunits.
Translation initiation is the rate-limiting step for protein synthesis. Efficient initiation requires translation initiation factors of the eIF4 family, the activities of which are regulated by the EGFR/ERK and PI3K/mTOR signaling pathways71,77,78 (Figure 4A). We asked whether Lamγ2-overexpressing neoplastic cells contain higher levels of the phosphorylated active forms of any of these factors, compared with normal cells. Western blot analysis of confluent cultures revealed higher levels of p-eIF4B, p-eIF4E, and p-4E-BP1 in neoplastic cells than in normal keratinocytes (Figure 5C). The EGFR kinase inhibitor gefitinib and the MEK inhibitors U0126 and AZD6244 greatly reduced p-eIF4B and p-4E-BP1 levels and had a lesser effect on p-eIF4E levels in SCC cells (Figure 5D). The TORC1/2 inhibitor Ku-0063794 greatly reduced p-4E-BP1 levels, slightly reduced p-eIF4B levels, and had no effect on p-eIF4E levels (Figure 5D). As expected, the TGFBRI kinase inhibitor TRI had no effect on levels of any of these phosphoproteins (Figure 5D).
The effects of these inhibitors on c-myc expression proved to be the same as on Lamγ2 expression (Figure 5D). Expression of c-myc is regulated at the level of translation by eIF4B/eIF4A, which aids translation initiation of mRNAs having very long 5′-UTR sequences that form stem-loop secondary structures.78,79 The Lamγ2 5′-UTR proved to have such a long and complex structure, similar to that of c-myc and much longer than those of Lamα3 and Lamβ3 mRNAs (see Supplemental Figure S4B at http://ajp.amjpathol.org).
We extended our analysis by testing the effects of the ERK1/2 inhibitor CAY1056149 and of the RSK inhibitor BI-D1870.50 These inhibitors reduced Lamγ2 expression associated with substantially reduced levels of p-S6 and p-eIF4B (Figure 5E). These results are consistent with ERK/RSK activation of p-eIF4B-dependent Lamγ2 mRNA translation as the mechanism of Lamγ2 protein overexpression in neoplastic keratinocytes.
Hyperactive RAS, RAF, or MEK Signaling Is Sufficient to Cause Abnormal Laminin γ2 Expression
The above experiments clearly demonstrated that EGFR/ERK/RSK activity is essential for Lamγ2 overexpression in neoplastic cells. We wished to determine whether activation of the EGFR/ERK signaling pathway in normal keratinocytes is sufficient to cause loss of density-dependent Lamγ2 regulation. Our earlier study had revealed that primary keratinocytes expressing activated HRAS grow well in the fibroblast feeder layer culture system, but undergo growth arrest if switched to a semidefined keratinocyte medium,80 and strain N and G5Ep primary keratinocyte lines transduced to express HRAS(G12V) proved unable to proliferate in Ksfm medium. Constitutively active mutant HRAS has been reported to induce p16INK4A-enforced senescence in normal cells under some conditions.56,81 For the following experiments, therefore, we used our TERT-immortalized normal keratinocyte line, N/TERT1,30 which is unable to express p16INK4A or p14ARF but displays normal Lamγ2 repression at confluence (Figure 6A).
Figure 6.
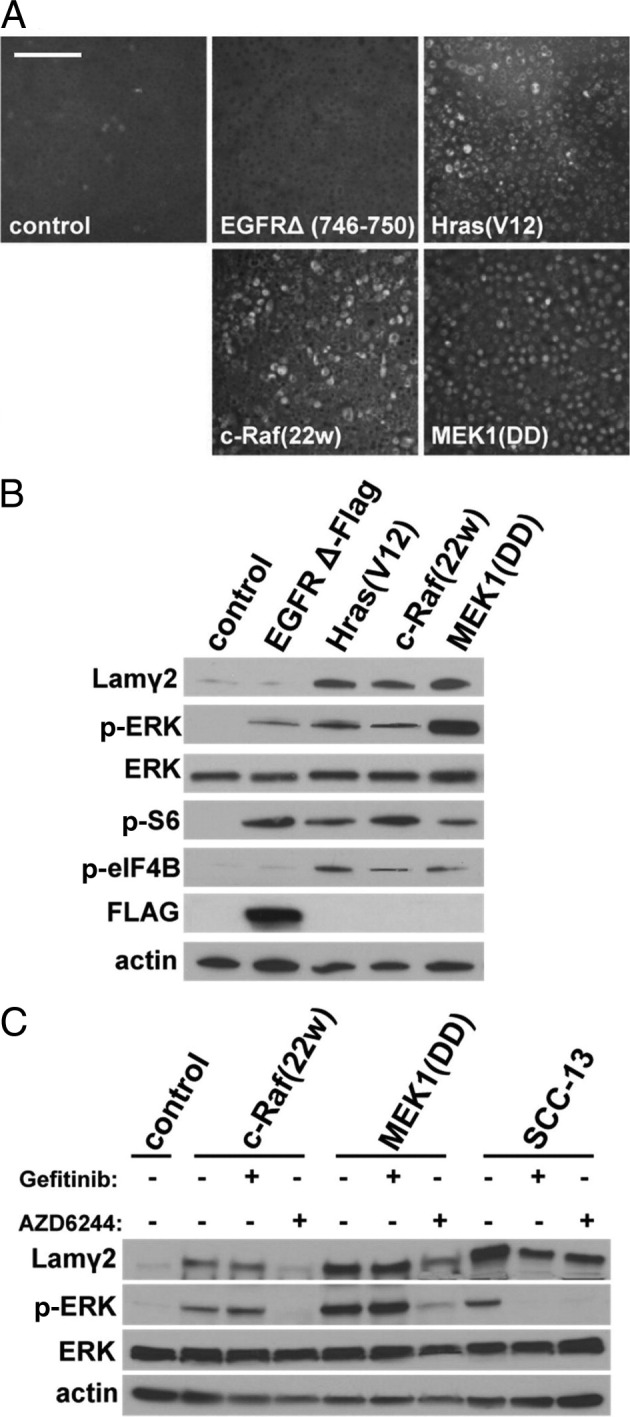
Hyperactivation of signaling pathway kinases upstream of ERK results in Lamγ2 dysregulation in normal keratinocytes. A: Lamγ2 immunostaining of confluent cultures of N/TERT-1 keratinocytes engineered to express constitutively active HRAS(G12V), cRAF1(22w), or MEK1(DD), and EGFRΔ(E746-A750)-FLAG. Scale bar = 200 μm. B: Western blot analysis of Lamγ2, signaling pathway phosphoproteins, and activation state of translation initiation factors of the same cell lines. Control indicates N/TERT-1 cells. Note that expression of constitutively active HRAS, cRAF1, or MEK1 increased p-ERK, p-S6, and p-eIF4B levels, whereas constitutively active EGFR slightly increased levels of p-ERK, robustly increased p-S6, but did not increase p-eIF4B and did not cause Lamγ2 overexpression. C: Western blot analysis of effects of EGFR and MEK inhibitors on Lamγ2 and p-ERK levels in control N/TERT-1 cells, cRAF1(22w) and MEK1(DD) transductants, and SCC-13 cells. Note that Lamγ2 overexpression and increased p-ERK levels in keratinocytes expressing activated mutant cRAF1 or MEK are normalized by treatment with the MEK inhibitor AZD6244, but not with the EGFR kinase inhibitor gefitinib.
N/TERT-1 cells transduced to express constitutively active mutant forms of either EGFR, HRAS, cRAF1, or MEK1 grew rapidly and through serial passages in Ksfm and proved to be independent of EGF for optimal growth (see Supplemental Figure S5 at http://ajp.amjpathol.org). This EGF independence was expected from the mutant ras result of our previous study80 and confirmed the biological activity of all four EGFR signaling pathway mutants in keratinocytes. Immunostaining and Western blot analysis of confluent cultures revealed that the constitutively active mutant forms of HRAS, cRAF1, and MEK1, but not of EGFR, conferred loss of density-dependent Lamγ2 repression (Figure 6, A and C). We analyzed the mutant cRAF1 and MEK1 transductants further by assessing the effects of kinase inhibitors. As expected, p-ERK levels and Lamγ2 expression in the cRAF1(22w) and MEK1(DD) transductants were unaffected by gefitinib inhibition of the upstream EGFR kinase, but were reduced by AZD6244 inhibition of MEK (Figure 6C). Both inhibitors reduced Lamγ2 and p-ERK levels in SCC-13 (Figures 6C and 4C), consistent with an essential role of EGFR hyperactivity for ERK activation and consequent Lamγ2 overexpression in this SCC cell line.
Having established that expression of either HRAS(G12V) or cRAF1(22w) or MEK1(DD) is sufficient to cause Lamγ2 overexpression, we asked whether such oncogene-driven ERK hyperactivation results in activation of the downstream translation factors that we had found (as described above) to correlate closely with ERK hyperactivity. Levels of p-eIF4B and p-S6 proved to be elevated in the mutant HRAS, cRAF1, and MEK1 transductants (Figure 6B). The mutant EGFR transductant, which showed normal density repression of Lamγ2, displayed a rather low level of p-ERK activation and no increase in p-eIF4B. Its p-S6 levels, however, were increased to the same degree as in the mutant HRAS, cRAF1, and MEK1 transductants (Figure 6B), suggesting that lower levels of ERK activation are sufficient to produce maximum p-S6. These results demonstrate that ERK hyperactivation is sufficient to instigate neoplastic Lamγ2 overexpression and provide further correlative evidence that translation initiation factor activation produces this response.
Basal Cells in Oral Dysplasias That Overexpress Laminin γ2 Have Elevated Levels of p-S6
Finally, we asked whether the abnormal activation of ERK, translation factors, and S6 that we had identified in premalignant dysplastic and SCC cells in culture is detectable in vivo and correlates with Lamγ2 overexpression. We used immunostaining to examine a set of oral dysplastic lesions, some of which included regions of normal epithelium as well as SCC. Lamγ2 was detectable in basal cells of some regions in 23/26 dysplasias and 8/8 invasive SCCs, but was undetectable in normal oral epithelium (Figure 7) or in two benign hyperplastic warts (see Supplemental Figure S3A at http://ajp.amjpathol.org). Intensity of Lamγ2 immunostaining varied among and within specimens of dysplasia, without apparent relation to any morphological features of the dysplasias (Figure 7). The p-ERK, p-EIF4B, p-eIF4E, and p-4E-BP1 antibodies yielded weak uniform staining or scattered suprabasal cell nuclear staining and did not identify any differences among normal, dysplastic, and SCC specimens (data not shown).
Figure 7.

Immunohistologic detection of Lamγ2 overexpression in vivo and its association with increased p-S6 and p-eIF4B levels in basal cells of oral premalignant dysplasias. In normal human oral mucosal epithelium (A), oral mucosal dysplasias (B–D), and a microinvasive oral SCC arising from a dysplasia (E), sections were stained with H&E and immunostained for Lamγ2 and p-S6. Enlargements of the areas demarcated by small rectangles are shown as corresponding insets, with the basal lamina traced by dotted lines. Scale bar = 200 μm. A: Normal oral epithelium showing no Lamγ2 and only suprabasal p-S6 staining. B: Margin between normal epithelium (left half) and dysplasia (right half), showing no Lamγ2 and p-S6 confined to suprabasal cells in the normal region, contrasting with Lamγ2 and basal cell p-S6 in the dysplastic region. C: Dysplasia in which not all basal cells are p-S6+, not all p-S6+ cells overexpress Lamγ2, and the intensity of Lamγ2 immunostaining varies greatly. D: Region of a dysplasia in which many basal cells are p-S6+, but none express Lamγ2. E: Dysplasia overlying microinvasive SCC (indicated by arrowheads in the H&E-stained panel), showing variable Lamγ2 and basal p-S6 staining in the dysplasia and strong Lamγ2 and basal p-S6 staining in areas of microinvasive SCC.
In contrast, the p-S6 antibody identified clear differences between normal and neoplastic epithelium, associated with Lamγ2 overexpression. In normal epithelium and benign warts, p-S6 immunostaining was always confined to suprabasal cell layers (Figure 7; see also Supplemental Figure S3A at http://ajp.amjpathol.org). In contrast, discontinuous basal cell p-S6 staining appeared in 20% to 100% of the dysplastic regions of 20/21 specimens evaluated quantitatively (Figures 7 and 8). Lamγ2 immunostaining was always associated with basal p-S6 immunostaining, in a specific way. Regions containing Lamγ2+ cells always were within regions that contained p-S6+ basal cells. However, only a subfraction of the basal p-S6+ regions also contained Lamγ2+ basal cells, ranging from zero to 100% (Figures 7 and 8).
Figure 8.
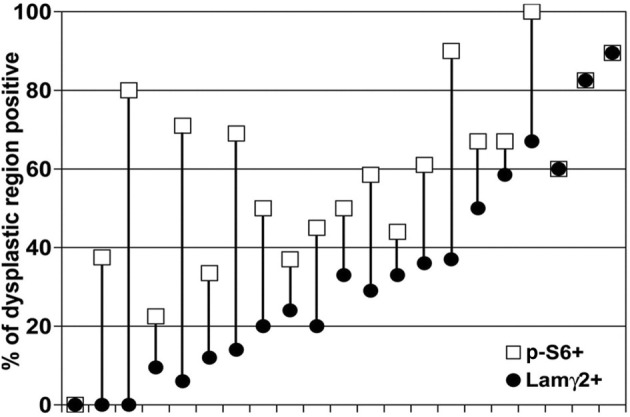
Association of Lamγ2 overexpression with increased basal cell p-S6 levels in dysplastic lesions. Twenty-one oral lesions with dysplasia were evaluated microscopically for the fraction of the dysplastic region of each that contained p-S6+ and Lamγ2+ basal cells. Specimens were charted in order of increasing percentage of Lamγ2+ and p-S6+, to better show the range of immunostaining patterns.
As reported previously,31,74,75 normal keratinocytes at the edge of wounds or ulcers expressed Lamγ2 (see Supplemental Figure S3B at http://ajp.amjpathol.org). These regions also showed basal p-S6 immunostaining (see Supplemental Figure S3B at http://ajp.amjpathol.org), consistent with our finding that normal keratinocytes in preconfluent cultures, which express Lamγ2, have ERK-dependent increased p-S6 (Figure 4C). The dysplasias we examined sometimes contained regions of ulceration or microerosion, in which the epithelium did not completely cover the underlying connective tissue. The Lamγ2/PS-6 immunostaining patterns of ulcerated regions of dysplasia (see Supplemental Figure S3C at http://ajp.amjpathol.org) resembled that of a wound edge in normal epithelium, with Lamγ2 and basal cell p-S6 immunostaining. To avoid possible wound responses in our quantitative immunostaining analysis (Figure 8), we did not score regions of dysplasias within ∼20 cells of an ulcer margin. We concluded from our analysis that immunohistologically detectable increased p-S6 levels in basal cells of dysplasias precede and accompany Lamγ2 overexpression in vivo, consistent with a causal relationship between ERK-dependent activation of translation factors and Lamγ2 overexpression.
Discussion
The present study extends our earlier study identifying Lamγ2 overexpression in many premalignant oral and epidermal dysplasias,25 which had followed studies by others finding Lamγ2 overexpression in advanced SCCs.28,29 Here, we have investigated the mechanism of neoplastic Lamγ2 overexpression using a set of normal primary and premalignant human keratinocyte and SCC lines that recapitulate in culture this difference in Lamγ2 regulation between normal and neoplastic epithelial cells. ERK hyperactivity proved to be necessary and sufficient to cause Lamγ2 overexpression, strongly correlating with ERK-dependent phosphorylation and activation of the translation factors S6 and eIF4B. Consistent with this finding, neoplastic Lamγ2 overexpression in culture proved to be at the level of translation, not transcription. Our cell culture findings were confirmed by immunostaining analysis of clinical material, which revealed increased p-S6 preceding and accompanying Lamγ2 overexpression in the basal cell layer of oral dysplasias. Most importantly, the present study identifies EGFR/ERK hyperactivity and consequent Lamγ2 overexpression as a rather early step during progression to SCC, occurring in some regions of preinvasive dysplasias.
Laminin γ2 Overexpression as an Indicator of ERK Hyperactivity
Hyperactivity of the EGFR/ERK82–84 and PI3K/AKT/mTOR11,85,86 signaling pathways is a frequent feature of oral and oropharyngeal SCC. We found increased levels of EGFR and of its activated, phosphorylated tyr1068 state in all of the SCC and premalignant cell lines we tested. However, we found increased p-AKT levels in only some SCC lines, consistent with a previous study,85 and no increase of p-AKT in the premalignant lines. Small-molecule inhibitors of the EGFR and MEK kinases blocked Lamγ2 overexpression, but an mTORC1/2 inhibitor did not, supporting the conclusion that hyperactive EGFR/ERK signaling is the cause of dysregulated Lamγ2 expression. Reinforcing this conclusion was our finding that ERK hyperactivity achieved by engineering normal keratinocytes to express constitutively active HRAS, cRAF1, or MEK1 was sufficient to cause Lamγ2 overexpression. Activating HRAS gene mutations are found in ∼5% of human oral and oropharyngeal SCCs,86 and amplifications of KRAS and ERK1(MAPK3) are occasionally detected also,84 but EGFR overexpression is the most frequent instigator of ERK hyperactivity in oral and oropharyngeal SCCs.82–84
Engineered expression of a constitutively active EGFR deletion mutant [EGFRΔ(E746-A750)], cloned from a tracheobronchial SCC,43 did not yield Lamγ2 overexpression, but resulted in a modest increase of p-ERK levels and EGF independence for growth. This finding suggests that ERK stimulation above a certain threshold is required for the Lamγ2 phenotype, a hypothesis testable in future experiments examining the effects of other activating EGFR mutations. The E6 and E7 viral oncoproteins of HPV16 and HPV18 initiate neoplastic progression in a high percentage of tonsil/oropharynx SCCs, but are rarely involved in oral cavity and epidermal SCC.6 Consistent with reports that HPV16 E6, potentiated by E7, increases EGFR levels and activation in keratinocytes,67,68 we found that primary keratinocytes engineered to express E6 and E7 displayed hyperactive EGFR and ERK and overexpressed Lamγ2.
The same system responsible for Lamγ2 overexpression in dysplasias and SCCs proved to be essential for Lamγ2 expression in normal keratinocytes in preconfluent cultures, when they are expressing high levels of Lamγ2. Under this condition, the EGFR/ERK pathway was active and S6 was phosphorylated in normal cells (as expected, because the cells are grown with EGF as a mitogen), and EGFR and MEK inhibitors repressed Lamγ2 expression. This cell culture finding is a manifestation of the in vivo wound re-epithelialization response, because we found that basal keratinocytes at normal wound edges have increased Lamγ2 and p-S6 levels.
Neoplastic Overexpression of Laminin γ2 and eIF4B-Dependent Regulation of mRNA Translation
Consistent with a previous report of selective overexpression of the Lamγ2 subunit of Lam332 in carcinoma cells in vivo,27 confluent cultures of premalignant keratinocytes and SCC cells displayed ERK hyperactivity-dependent Lamγ2 overexpression, whereas regulation of their Lamα3 and Lamβ3 expression was normal. In the process of normal Lam332 assembly, β3γ2 dimer formation necessarily precedes the addition of Lamα3 and ultimate secretion of Lam332 trimer.62 Lamγ2 overexpression resulted in abnormal secretion of Lamγ2 monomers, with most of the Lamγ2 molecules remaining in the unprocessed 155-kDa form. Basal epithelial cells adhere to normally processed mature Lam332 by interaction of α6β4 integrin to the C-terminus of the Lamα3 chain.2,41 We reported previously that Lam332 trimers having unprocessed Lamγ2 subunits induce sustained directional motility in keratinocytes.31 Whether autocrine or paracrine action of secreted Lamγ2 monomer or Lam332 with unprocessed Lamγ2 produces any neoplastic phenotype in vivo remains to be determined.
We were surprised to find from qPCR analysis that neoplastic Lamγ2 overexpression is not the result of increased mRNA levels. Transcription of the genes encoding the three subunits of Lam332 is known to require TGFβ pathway signaling,73 such that a TGFβRI kinase inhibitor greatly inhibits Lamγ2 expression at both the protein level 31 and also the mRNA level (present study). ERK activates transcriptional regulators in the nucleus,76 so we had expected that Lamγ2 gene expression might be ERK-dependent. However, ERK activates the signaling kinases RSK and MNK1 which, in turn, activate the ribosomal protein S6 and several subunits of the cytoplasmic eIF4 complex required for translation initiation of some mRNAs.77,78 eIF4E permits translation of mRNAs that have the common 5′ cap (also known as the m7G or 7-methylguanosine cap). eIF4E activity requires inactivation of the eIF4E inhibitor 4E-BP1 by TORC1-catalyzed phosphorylation.87 In our experiments, the TORC1/2 inhibitor Ku-0063794 blocked 4E-BP1 phosphorylation, but had no effect on Lamγ2 synthesis, ruling out an essential role for eIF4E. Another protein of the eIF4 complex is eIF4B, an essential activator of the eIF4A helicase, which unwinds and opens mRNAs that have long 5′-UTRs with a densely hydrogen-bonded stem-loop structure. eIF4B must be phosphorylated to be active, and our kinase inhibitor experiments revealed that this is accomplished primarily by the ERK-dependent RSK kinase. In our experiments, p-eIF4B levels proved to correlate most closely with the level of Lamγ2 expression. MYC mRNA has a long and highly structured 5′-UTR and requires eIF4B activation for translation.79 The 5′-UTR of Lamγ2 mRNA has a similar length and structure, and our results clearly implicate eIF4B-dependent translation initiation as the determining factor for Lamγ2 expression. This hypothesis can be tested by determining whether the Lamγ2 5′-UTR imposes ERK/RSK-dependent translation control on reporter constructs.
S6 Phosphorylation as a Biomarker of ERK Hyperactivity in Neoplastic Keratinocytes
S6 is a component of the 40S ribosome subunit, and its phosphorylation at ser235/236 appears to be important under conditions that require very high rates of protein synthesis and to ensure normal cell size.88 The high level of p-S6(ser235/236) present in all of the SCC and premalignant lines at confluence indicates that S6 kinases are active in these cells. The S6(ser235/236) phosphorylation, essential for S6 activity, can be accomplished either by TORC1-dependent S6K1 or by ERK-dependent RSK.70 The relative importance of ERK-dependent and TORC1-dependent kinases on S6 phosphorylation and activation has been incompletely studied and may vary among different cell types and circumstances. In a study of HeLa cells, most S6(ser235/236) phosphorylation was ERK-dependent.72 Little or no S6(ser235/236) phosphorylation occurred in keratinocytes and SCC cells when ERK activity was blocked by a MEK inhibitor, indicating that in these cells the critical ser235/236 phosphorylation is ERK-dependent. The ability of RSK to accomplish the ser235/236 phosphorylation is greatly enhanced by prior S6K1-catalyzed ser240/244 phosphorylation of S6.71,72 Treatment with the TORC1/2 inhibitor Ku-0063794 resulted in complete absence of p-S6(ser234/235), consistent with the precondition of TORC1-dependent S6K1 phosphorylation at ser(240/244). The absence of an effect of Ku-0063794 on Lamγ2 overexpression shows that p-S6(ser235/236) levels are not limiting for Lamγ2 synthesis. Despite being nonessential for Lamγ2 overexpression, high levels of p-S6(235/236) always accompanied it in our cell culture systems in the absence of kinase inhibitors. We found that p-S6(235/236) was the most useful biomarker of signaling pathway hyperactivity associated with Lamγ2 overexpression in vivo.
Immunodetection of Abnormal p-S6 and Laminin γ2 in Clinical Specimens of Oral Dysplasia
This initial analysis of signaling protein phosphorylation status during oral epithelial neoplastic progression in vivo has revealed the potential power of immunohistochemical staining for detecting signaling abnormalities that may have diagnostic or prognostic value, as well as the limitations. Cytoplasmic content of the secreted protein Lamγ2 and the phosphorylated versions of signaling mediators are excellent candidates for biomarkers of neoplastic progression, because they indicate the current or very recent physiological state of individual cells at the time of fixation. Clinical and experimental utility depends, of course, on reliable detectability. Phosphorylations of signaling pathway proteins are subject to continual turnover, owing to their susceptibility to intracellular phosphatases.89,90 Current procedures for handling of biopsy and surgical specimens and for formalin fixation may not consistently preserve the phosphorylated states of some proteins. Several immunohistochemical analyses of signaling pathway activation in tissue microarrays of oropharyngeal SCCs have produced inconclusive results, owing to inconsistent, weak, and/or predominantly differentiated cell staining for phosphoproteins and the challenge of finding suitable criteria for quantifying the results.91,92 As an integral ribosomal protein, S6 is much more abundant, and its phosphorylated form may also be more stable, than that of eIF4B and mediators of the EGFR/ERK/RSK pathway.
p-S6 immunostaining proved to be intense, preserved in all archival pathology specimens we tested, and showing clear differences between normal or hyperplastic versus dysplastic or SCC tissue. p-S6 was readily detectable in suprabasal cell layers of normal stratified epithelia, but not in the basal cell layer. In contrast, periodic or continuous basal cell p-S6 was detectable in portions of all but one of the dysplasias examined, ranging from ∼20% to 100% of the length of dysplastic regions, consistent with a previous study.93 We found that Lamγ2 overexpression was consistently confined to a subfraction, ranging from zero to 100%, of the basal p-S6+ regions. There are at least two explanations for this finding. First, levels of p-S6 (as a surrogate for ERK hyperactivity and consequent eIF4B activity) may fluctuate substantially in vivo and may need to remain elevated continuously for a sufficiently long time for enough Lamγ2 translation to occur to be detectable. Second, increased p-S6(ser235/236) levels may in some cases result from increased AKT/mTOR activity instead of increased ERK/RSK activity, but only the latter will result in increased Lamγ2 expression. We did not detect basal cell p-AKT by immunostaining, which could have identified a role for AKT/mTOR hyperactivity in p-S6+/Lamγ2− regions.
The present study demonstrates that ERK hyperactivity and consequent Lamγ2 overexpression begins relatively early, in the preinvasive stages of oral epithelial neoplastic progression. These results provide the rationale for larger studies to seek correlations between p-S6/Lamγ2 immunostaining patterns in dysplasias and future progression (and its time frame) to invasive SCC. Such studies would aim to identify a specific pattern or extent of p-S6 and Lamγ2 staining that correlates with future SCC, thus providing a definitive prognosis of dysplastic lesions. Our results also support the idea of early EGFR or MEK kinase inhibitor therapy for patients whose oral lesions have a high probability of progressing to invasive SCC but cannot be completely resected surgically without compromising function of the tongue. Clinical studies to date using targeted kinase inhibitors systemically for treatment of advanced SCC have yielded modest or temporary responses and serious side effects.12,94 Preinvasive lesions at high risk of malignant progression, and of relatively small total mass, may be more amenable to complete cure by local and/or short-term kinase inhibitor treatment.
Footnotes
Supported by NIH grant R01-DE13178 and a Brigham and Women's Hospital BRI grant (J.G.R.), by NIH grant P30-AR42689 (Thomas S. Kupper) on which J.G.R. had one of the projects, and by postdoctoral fellowships from the Swiss National Science Foundation-SNSF (Schweizerische Nationalfonds), Novartis Foundation, and the Swiss Foundation for Grants in Biology and Medicine (Schweizerische Stiftung für Biologisch-Medizinische Stipendien) (M.D.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.02.028.
Supplementary data
Similar stability of Lamγ2 protein in normal keratinocytes and SCC cells. Replicate preconfluent and confluent cultures of normal keratinocytes (N/TERT-1) and SCC-13 cells were pretreated for 1 hour with brefeldin A and then cycloheximide was added for 0.5, 1, 3, or 6 hours. At each time point, proteins were extracted and Western blotted for Lamγ2 and c-myc. In contrast to c-myc, Lamγ2 is very stable and shows a modest decrease in levels only after 6 hours of cycloheximide treatment, to a similar extent in preconfluent and confluent N/TERT-1 cells and in confluent SCC-13 cells. BfA, brefeldin A; CHX, cycloheximide.
An activity of the HPV16 E6 oncoprotein not involving p53 degradation causes Lamγ2 dysregulation in keratinocytes. Western blot analysis of Lamγ2 and p53 levels in the cell lines strain N, N/E6E7 [early (90) and later (250) passage after transduction], N/E6, N/E7, N/E6JH26, POE9n, SCC-15, and SCC-68. Note that p53 levels in the E6E7 and E6 transductant are reduced, but are normal in the E6JH26 transductant.
Increased basal p-S6 and Lamγ2 expression is not a feature of benign hyperplasia, but rather is induced coordinately in normal epithelial cells participating in wound repair. Areas demarcated by rectangles in the p-S6 panels are enlarged as insets in the same panels, with the basal lamina indicated by a dotted line. A: Benign oral wart. Note absence of Lamγ2 and p-S6 staining in basal cells. B: Slightly atypical, hyperplastic oral mucosal epithelium at the left edge of an ulcer. Note basal p-S6 and Lamγ2 immunostaining in the migrating tongue of cells (arrow) next to the clot (red superficial material marked by asterisk in the H&E panel), in contrast to suprabasal p-S6 and absence of Lamγ2 immunostaining in the epithelium at a distance from the wound edge. C: Note a microerosion (arrow) at the margin between normal epithelium and a dysplasia, showing a pattern of p-S6 and Lamγ2 staining reminiscent both of dysplasia and of the wound edge response. Scale bar = 200 μm.
Levels of Lamα3 and Lamβ3 mRNA in preconfluent versus confluent cultures of normal keratinocytes and SCC cells and predicted secondary structures of the 5′-UTRs of laminin mRNAs. A: qPCR comparative analysis of Lamα3 and Lamβ3 mRNA levels in preconfluent and confluent cultures of normal keratinocytes and SCC cells. Laminin mRNA levels were normalized to GAPDH mRNA levels in the same sample. All cell lines were compared with the Lamα3 and Lamβ3 mRNA levels in preconfluent cultures of the normal primary keratinocyte line strain N, set at an arbitrary unit of 1. Note that levels of Lamα3 mRNA, but not of Lamβ3, decrease in confluent cultures of both normal keratinocytes and SCC cells, correlating with the reduction in Lamα3 protein and maintenance of Lamβ3 protein at confluence by normal and SCC cells (Figure 2). B: Predicted secondary structures of the 5′UTR regions of the mRNAs encoding Lamγ2, Lamα3, Lamβ3, and c-myc. Note the long, stem-loop hydrogen bonded structure of the 5′UTR of Lamγ2, similar to that of c-myc.
Independence from exogenous EGF for optimal growth resulting from constitutively active EGFR, HRAS, cRAF1, or MEK1 expression. Cells were plated in medium with and without EGF, as described under Materials and Methods. Note that all of the engineered EGFR/ERK hyperactive cell lines grew at their maximum rates in the absence of EGF; in contrast, the N/TERT-1 parent cell line required EGF to grow optimally.
References
- 1.Dabelsteen S., Hercule P., Barron P., Rice M., Dorsainville G., Rheinwald J.G. Epithelial cells derived from human embryonic stem cells display p16INK4A senescence, hypermotility, and differentiation properties shared by many p63+ somatic cell types. Stem Cells. 2009;27:1388–1399. doi: 10.1002/stem.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tzu J., Marinkovich M.P. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol. 2008;40:199–214. doi: 10.1016/j.biocel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.American Cancer Society; Atlanta, GA: 2011. American Cancer Society: Cancer Facts and Figures 2011. [Google Scholar]
- 4.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Murray T., Thun M.J. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 5.Syrjanen S. Human papillomavirus (HPV) in head and neck cancer. J Clin Virol. 2005;32(Suppl 1):S59–S66. doi: 10.1016/j.jcv.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 6.Hobbs C.G., Sterne J.A., Bailey M., Heyderman R.S., Birchall M.A., Thomas S.J. Human papillomavirus and head and neck cancer: a systematic review and meta-analysis. Clin Otolaryngol. 2006;31:259–266. doi: 10.1111/j.1749-4486.2006.01246.x. [DOI] [PubMed] [Google Scholar]
- 7.McLaughlin-Drubin M.E., Munger K. Oncogenic activities of human papillomaviruses. Virus Res. 2009;143:195–208. doi: 10.1016/j.virusres.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghittoni R., Accardi R., Hasan U., Gheit T., Sylla B., Tommasino M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes. 2010;40:1–13. doi: 10.1007/s11262-009-0412-8. [DOI] [PubMed] [Google Scholar]
- 9.Klein J.D., Grandis J.R. The molecular pathogenesis of head and neck cancer. Cancer Biol Ther. 2010;9:1–7. doi: 10.4161/cbt.9.1.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leemans C.R., Braakhuis B.J., Brakenhoff R.H. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 11.Molinolo A.A., Amornphimoltham P., Squarize C.H., Castilho R.M., Patel V., Gutkind J.S. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–334. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L.F., Cohen E.E., Grandis J.R. New strategies in head and neck cancer: understanding resistance to epidermal growth factor receptor inhibitors. Clin Cancer Res. 2010;16:2489–2495. doi: 10.1158/1078-0432.CCR-09-2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng H., Xu X., Costa D.B., Powell C.A., Halmos B. Molecular testing in lung cancer: the time is now. Curr Oncol Rep. 2010;12:335–348. doi: 10.1007/s11912-010-0118-z. [DOI] [PubMed] [Google Scholar]
- 14.Cooper J.B., Cohen E.E. Mechanisms of resistance to EGFR inhibitors in head and neck cancer. Head Neck. 2009;31:1086–1094. doi: 10.1002/hed.21109. [DOI] [PubMed] [Google Scholar]
- 15.Mao L., Hong W.K., Papadimitrakopoulou V.A. Focus on head and neck cancer. Cancer Cell. 2004;5:311–316. doi: 10.1016/s1535-6108(04)00090-x. [DOI] [PubMed] [Google Scholar]
- 16.Argiris A., Karamouzis M.V., Raben D., Ferris R.L. Head and neck cancer. Lancet. 2008;371:1695–1709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Napier S.S., Speight P.M. Natural history of potentially malignant oral lesions and conditions: an overview of the literature. J Oral Pathol Med. 2008;37:1–10. doi: 10.1111/j.1600-0714.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 18.Bremmer J.F., Brakenhoff R.H., Broeckaert M.A., Belien J.A., Leemans C.R., Bloemena E., van der Waal I., Braakhuis B.J. Prognostic value of DNA ploidy status in patients with oral leukoplakia. Oral Oncol. 2011;47:950–960. doi: 10.1016/j.oraloncology.2011.07.025. [DOI] [PubMed] [Google Scholar]
- 19.Holmstrup P., Vedtofte P., Reibel J., Stoltze K. Long-term treatment outcome of oral premalignant lesions. Oral Oncol. 2006;42:461–474. doi: 10.1016/j.oraloncology.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 20.van der Waal I. Potentially malignant disorders of the oral and oropharyngeal mucosa; terminology, classification and present concepts of management. Oral Oncol. 2009;45:317–323. doi: 10.1016/j.oraloncology.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 21.Poh C.F., Ng S., Berean K.W., Williams P.M., Rosin M.P., Zhang L. Biopsy and histopathologic diagnosis of oral premalignant and malignant lesions. J Can Dent Assoc. 2008;74:283–288. [PubMed] [Google Scholar]
- 22.Natarajan E., Eisenberg E. Contemporary concepts in the diagnosis of oral cancer and precancer. Dent Clin North Am. 2011;55:63–68. doi: 10.1016/j.cden.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Holmstrup P., Vedtofte P., Reibel J., Stoltze K. Oral premalignant lesions: is a biopsy reliable. J Oral Pathol Med. 2007;36:262–266. doi: 10.1111/j.1600-0714.2007.00513.x. [DOI] [PubMed] [Google Scholar]
- 24.Karabulut A., Reibel J., Therkildsen M.H., Praetorius F., Nielsen H.W., Dabelsteen E. Observer variability in the histologic assessment of oral premalignant lesions. J Oral Pathol Med. 1995;24:198–200. doi: 10.1111/j.1600-0714.1995.tb01166.x. [DOI] [PubMed] [Google Scholar]
- 25.Natarajan E., Saeb M., Crum C.P., Woo S.B., McKee P.H., Rheinwald J.G. Co-expression of p16INK4A and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am J Pathol. 2003;163:477–491. doi: 10.1016/s0002-9440(10)63677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizushima H., Koshikawa N., Moriyama K., Takamura H., Nagashima Y., Hirahara F., Miyazaki K. Wide distribution of laminin-5 gamma 2 chain in basement membranes of various human tissues. Horm Res. 1998;50(Suppl 2):7–14. doi: 10.1159/000053118. [DOI] [PubMed] [Google Scholar]
- 27.Koshikawa N., Moriyama K., Takamura H., Mizushima H., Nagashima Y., Yanoma S., Miyazaki K. Overexpression of laminin gamma2 chain monomer in invading gastric carcinoma cells. Cancer Res. 1999;59:5596–5601. [PubMed] [Google Scholar]
- 28.Ono Y., Nakanishi Y., Ino Y., Niki T., Yamada T., Yoshimura K., Saikawa M., Nakajima T., Hirohashi S. Clinocopathologic significance of laminin-5 gamma2 chain expression in squamous cell carcinoma of the tongue: immunohistochemical analysis of 67 lesions. Cancer. 1999;85:2315–2321. [PubMed] [Google Scholar]
- 29.Pyke C., Rømer J., Kallunki P., Lund L.R., Ralfkiaer E., Dano K., Tryggvason K. The gamma 2 chain of kalinin/laminin 5 is preferentially expressed in invading malignant cells in human cancers. Am J Pathol. 1994;145:782–791. [PMC free article] [PubMed] [Google Scholar]
- 30.Dickson M.A., Hahn W.C., Ino Y., Ronfard V., Wu J.Y., Weinberg R.A., Louis D.N., Li F.P., Rheinwald J.G. Human keratinocytes that express hTERT and also bypass a p16INK4a-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Natarajan E., Omobono J.D., 2nd, Guo Z., Hopkinson S., Lazar A.J., Brenn T., Jones J.C., Rheinwald J.G. A keratinocyte hypermotility/growth-arrest response involving Laminin 5 and p16INK4A activated in wound healing and senescence. Am J Pathol. 2006;168:1821–1837. doi: 10.2353/ajpath.2006.051027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rheinwald J.G., Beckett M.A. Defective terminal differentiation in culture as a consistent and selectable character of malignant human keratinocytes. Cell. 1980;22:629–632. doi: 10.1016/0092-8674(80)90373-6. [DOI] [PubMed] [Google Scholar]
- 33.Rollins B.J., O'Connell T.M., Bennett G., Burton L.E., Stiles C.D., Rheinwald J.G. Environment-dependent growth inhibition of human epidermal keratinocytes by recombinant human transforming growth factor-beta. J Cell Physiol. 1989;139:455–462. doi: 10.1002/jcp.1041390302. [DOI] [PubMed] [Google Scholar]
- 34.Rheinwald J.G., Hahn W.C., Ramsey M.R., Wu J.Y., Guo Z., Tsao H., De Luca M., Catricala C., O'Toole K.M. A two-stage, p16INK4A- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Mol Cell Biol. 2002;22:5157–5172. doi: 10.1128/MCB.22.14.5157-5172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei W., Barron P.D., Rheinwald J.G. Modulation of TGF-beta-inducible hypermotility by EGF and other factors in human prostate epithelial cells and keratinocytes. In Vitro Cell Dev Biol Anim. 2010;46:841–855. doi: 10.1007/s11626-010-9353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu L., Crowe D.L., Rheinwald J.G., Chambon P., Gudas L.J. Abnormal expression of retinoic acid receptors and keratin 19 by human oral and epidermal squamous cell carcinoma cell lines. Cancer Res. 1991;51:3972–3981. [PubMed] [Google Scholar]
- 37.Rheinwald J.G., Beckett M.A. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultures from human squamous cell carcinomas. Cancer Res. 1981;41:1657–1663. [PubMed] [Google Scholar]
- 38.McGregor F., Muntoni A., Fleming J., Brown J., Felix D.H., MacDonald D.G., Parkinson E.K., Harrison P.R. Molecular changes associated with oral dysplasia progression and acquisition of immortality: potential for its reversal by 5-azacytidine. Cancer Res. 2002;62:4757–4766. [PubMed] [Google Scholar]
- 39.Muntoni A., Fleming J., Gordon K.E., Hunter K., McGregor F., Parkinson E.K., Harrison P.R. Senescing oral dysplasias are not immortalized by ectopic expression of hTERT alone without other molecular changes, such as loss of INK4A and/or retinoic acid receptor-beta: but p53 mutations are not necessarily required. Oncogene. 2003;22:7804–7808. doi: 10.1038/sj.onc.1207085. [DOI] [PubMed] [Google Scholar]
- 40.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 41.Goldfinger L.E., Hopkinson S.B., deHart G.W., Collawn S., Couchman J.R., Jones J.C. The alpha3 laminin subunit, alpha6beta4 and alpha3beta1 integrin coordinately regulate wound healing in cultured epithelial cells and in the skin. J Cell Sci. 1999;112:2615–2629. doi: 10.1242/jcs.112.16.2615. [DOI] [PubMed] [Google Scholar]
- 42.Singh J., Ling L.E., Sawyer J.S., Lee W.C., Zhang F., Yingling J.M. Transforming the TGFbeta pathway: convergence of distinct lead generation strategies on a novel kinase pharmacophore for TbetaRI (ALK5) Curr Opin Drug Discov Devel. 2004;7:437–445. [PubMed] [Google Scholar]
- 43.Greulich H., Chen T.H., Feng W., Janne P.A., Alvarez J.V., Zappaterra M., Bulmer S.E., Frank D.A., Hahn W.C., Sellers W.R., Meyerson M. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wakeling A.E., Guy S.P., Woodburn J.R., Ashton S.E., Curry B.J., Barker A.J., Gibson K.H. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62:5749–5754. [PubMed] [Google Scholar]
- 45.Yeh T.C., Marsh V., Bernat B.A., Ballard J., Colwell H., Evans R.J., Parry J., Smith D., Brandhuber B.J., Gross S., Marlow A., Hurley B., Lyssikatos J., Lee P.A., Winkler J.D., Koch K., Wallace E. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 46.Favata M.F., Horiuchi K.Y., Manos E.J., Daulerio A.J., Stradley D.A., Feeser W.S., Van Dyk D.E., Pitts W.J., Earl R.A., Hobbs F., Copeland R.A., Magolda R.L., Scherle P.A., Trzaskos J.M. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 47.Dowling R.J., Topisirovic I., Fonseca B.D., Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta. 2010;1804:433–439. doi: 10.1016/j.bbapap.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Garcia-Martinez J.M., Moran J., Clarke R.G., Gray A., Cosulich S.C., Chresta C.M., Alessi D.R. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009;421:29–42. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aronov A.M., Baker C., Bemis G.W., Cao J., Chen G., Ford P.J., Germann U.A., Green J., Hale M.R., Jacobs M., Janetka J.W., Maltais F., Martinez-Botella G., Namchuk M.N., Straub J., Tang Q., Xie X. Flipped out: structure-guided design of selective pyrazolylpyrrole ERK inhibitors. J Med Chem. 2007;50:1280–1287. doi: 10.1021/jm061381f. [DOI] [PubMed] [Google Scholar]
- 50.Sapkota G.P., Cummings L., Newell F.S., Armstrong C., Bain J., Frodin M., Grauert M., Hoffmann M., Schnapp G., Steegmaier M., Cohen P., Alessi D.R. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J. 2007;401:29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McFall A., Ulku A., Lambert Q.T., Kusa A., Rogers-Graham K., Der C.J. Oncogenic Ras blocks anoikis by activation of a novel effector pathway independent of phosphatidylinositol 3-kinase. Mol Cell Biol. 2001;21:5488–5499. doi: 10.1128/MCB.21.16.5488-5499.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanton V.P., Jr, Nichols D.W., Laudano A.P., Cooper G.M. Definition of the human raf amino-terminal regulatory region by deletion mutagenesis. Mol Cell Biol. 1989;9:639–647. doi: 10.1128/mcb.9.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boehm J.S., Zhao J.J., Yao J., Kim S.Y., Firestein R., Dunn I.F., Sjostrom S.K., Garraway L.A., Weremowicz S., Richardson A.L., Greulich H., Stewart C.J., Mulvey L.A., Shen R.R., Ambrogio L., Hirozane-Kishikawa T., Hill D.E., Vidal M., Meyerson M., Grenier J.K., Hinkle G., Root D.E., Roberts T.M., Lander E.S., Polyak K., Hahn W.C. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 54.Brunet A., Pages G., Pouyssegur J. Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene. 1994;9:3379–3387. [PubMed] [Google Scholar]
- 55.Feig L.A., Corbley M., Pan B.T., Roberts T.M., Cooper G.M. Structure/function analysis of ras using random mutagenesis coupled with functional screening assays. Mol Endocrinol. 1987;1:127–136. doi: 10.1210/mend-1-2-127. [DOI] [PubMed] [Google Scholar]
- 56.Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4A. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 57.Halbert C.L., Demers G.W., Galloway D.A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Foster S.A., Demers G.W., Etscheid B.G., Galloway D.A. The ability of human papillomavirus E6 proteins to target p53 for degradation in vivo correlates with their ability to abrogate actinomycin D-induced growth arrest. J Virol. 1994;68:5698–5705. doi: 10.1128/jvi.68.9.5698-5705.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huibregtse J.M., Beaudenon S.L. Mechanism of HPV E6 proteins in cellular transformation. Semin Cancer Biol. 1996;7:317–326. doi: 10.1006/scbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- 60.Mietz J.A., Unger T., Huibregtse J.M., Howley P.M. The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. EMBO J. 1992;11:5013–5020. doi: 10.1002/j.1460-2075.1992.tb05608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swift S., Lorens J., Achacoso P., Nolan G.P. Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.im1017cs31. May; Chapter 10:Unit 10.17C. [DOI] [PubMed] [Google Scholar]
- 62.Matsui C., Wang C.K., Nelson C.F., Bauer E.A., Hoeffler W.K. The assembly of laminin-5 subunits. J Biol Chem. 1995;270:23496–23503. doi: 10.1074/jbc.270.40.23496. [DOI] [PubMed] [Google Scholar]
- 63.Marinkovich M.P., Lunstrum G.P., Burgeson R.E. The anchoring filament protein kalinin is synthesized and secreted as a high molecular weight precursor. J Biol Chem. 1992;267:17900–17906. [PubMed] [Google Scholar]
- 64.Farthing A.J., Vousden K.H. Functions of human papillomavirus E6 and E7 oncoproteins. Trends Microbiol. 1994;2:170–174. doi: 10.1016/0966-842x(94)90667-x. [DOI] [PubMed] [Google Scholar]
- 65.Klingelhutz A.J., Foster S.A., McDougall J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380:79–82. doi: 10.1038/380079a0. [DOI] [PubMed] [Google Scholar]
- 66.Howie H.L., Katzenellenbogen R.A., Galloway D.A. Papillomavirus E6 proteins. Virology. 2009;384:324–334. doi: 10.1016/j.virol.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sizemore N., Choo C.K., Eckert R.L., Rorke E.A. Transcriptional regulation of the EGF receptor promoter by HPV16 and retinoic acid in human ectocervical epithelial cells. Exp Cell Res. 1998;244:349–356. doi: 10.1006/excr.1998.4179. [DOI] [PubMed] [Google Scholar]
- 68.Akerman G.S., Tolleson W.H., Brown K.L., Zyzak L.L., Mourateva E., Engin T.S., Basaraba A., Coker A.L., Creek K.E., Pirisi L. Human papillomavirus type 16 E6 and E7 cooperate to increase epidermal growth factor receptor (EGFR) mRNA levels, overcoming mechanisms by which excessive EGFR signaling shortens the life span of normal human keratinocytes. Cancer Res. 2001;61:3837–3843. [PubMed] [Google Scholar]
- 69.Spangle J.M., Munger K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J Virol. 2010;84:9398–9407. doi: 10.1128/JVI.00974-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pende M., Um S.H., Mieulet V., Sticker M., Goss V.L., Mestan J., Mueller M., Fumagalli S., Kozma S.C., Thomas G. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–3124. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anjum R., Blenis J. The RSK family of kinases: emerging roles in cellular signaling. Nat Rev Mol Cell Biol. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- 72.Roux P.P., Shahbazian D., Vu H., Holz M.K., Cohen M.S., Taunton J., Sonenberg N., Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korang K., Christiano A.M., Uitto J., Mauviel A. Differential cytokine modulation of the genes LAMA3, LAMB3, and LAMC2, encoding the constitutive polypeptides, alpha 3, beta 3, and gamma 2, of human laminin 5 in epidermal keratinocytes. FEBS Lett. 1995;368:556–558. doi: 10.1016/0014-5793(95)00740-z. [DOI] [PubMed] [Google Scholar]
- 74.Dabelsteen E., Gron B., Mandel U., Mackenzie I. Altered expression of epithelial cell surface glycoconjugates and intermediate filaments at the margins of mucosal wounds. J Invest Dermatol. 1998;111:592–597. doi: 10.1046/j.1523-1747.1998.00346.x. [DOI] [PubMed] [Google Scholar]
- 75.Kainulainen T., Hakkinen L., Hamidi S., Larjava K., Kallioinen M., Peltonen J., Salo T., Larjava H., Oikarinen A. Laminin-5 expression is independent of the injury and the microenvironment during reepithelialization of wounds. J Histochem Cytochem. 1998;46:353–360. doi: 10.1177/002215549804600309. [DOI] [PubMed] [Google Scholar]
- 76.Gille H., Sharrocks A.D., Shaw P.E. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature. 1992;358:414–417. doi: 10.1038/358414a0. [DOI] [PubMed] [Google Scholar]
- 77.Ma X.M., Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 78.Shahbazian D., Parsyan A., Petroulakis E., Hershey J., Sonenberg N. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle. 2010;9:4106–4109. doi: 10.4161/cc.9.20.13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shahbazian D., Parsyan A., Petroulakis E., Topisirovic I., Martineau Y., Gibbs B.F., Svitkin Y., Sonenberg N. Control of cell survival and proliferation by mammalian eukaryotic initiation factor 4B. Mol Cell Biol. 2010;30:1478–1485. doi: 10.1128/MCB.01218-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Henrard D.R., Thornley A.T., Brown M.L., Rheinwald J.G. Specific effects of ras oncogene expression on the growth and histogenesis of human epidermal keratinocytes. Oncogene. 1990;5:475–481. [PubMed] [Google Scholar]
- 81.Lin A.W., Barradas M., Stone J.C., van Aelst L., Serrano M., Lowe S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ishitoya J., Toriyama M., Oguchi N., Kitamura K., Ohshima M., Asano K., Yamamoto T. Gene amplification and overexpression of EGF receptor in squamous cell carcinomas of the head and neck. Br J Cancer. 1989;59:559–562. doi: 10.1038/bjc.1989.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Temam S., Kawaguchi H., El-Naggar A.K., Jelinek J., Tang H., Liu D.D., Lang W., Issa J.P., Lee J.J., Mao L. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol. 2007;25:2164–2170. doi: 10.1200/JCO.2006.06.6605. [DOI] [PubMed] [Google Scholar]
- 84.Sheu J.J., Hua C.H., Wan L., Lin Y.J., Lai M.T., Tseng H.C., Jinawath N., Tsai M.H., Chang N.W., Lin C.F., Lin C.C., Hsieh L.J., Wang T.L., Shih Ie M., Tsai F.J. Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer Res. 2009;69:2568–2576. doi: 10.1158/0008-5472.CAN-08-3199. [DOI] [PubMed] [Google Scholar]
- 85.Amornphimoltham P., Sriuranpong V., Patel V., Benavides F., Conti C.J., Sauk J., Sausville E.A., Molinolo A.A., Gutkind J.S. Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res. 2004;10:4029–4037. doi: 10.1158/1078-0432.CCR-03-0249. [DOI] [PubMed] [Google Scholar]
- 86.Stransky N., Egloff A.M., Tward A.D., Kostic A.D., Cibulskis K., Sivachenko A., Kryukov G.V., Lawrence M.S., Sougnez C., McKenna A., Shefler E., Ramos A.H., Stojanov P., Carter S.L., Voet D., Cortes M.L., Auclair D., Berger M.F., Saksena G., Guiducci C., Onofrio R.C., Parkin M., Romkes M., Weissfeld J.L., Seethala R.R., Wang L., Rangel-Escareno C., Fernandez-Lopez J.C., Hidalgo-Miranda A., Melendez-Zajgla J., Winckler W., Ardlie K., Gabriel S.B., Meyerson M., Lander E.S., Getz G., Golub T.R., Garraway L.A., Grandis J.R. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beretta L., Gingras A.C., Svitkin Y.V., Hall M.N., Sonenberg N. Rapamycin blocks phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 1996;15:658–664. [PMC free article] [PubMed] [Google Scholar]
- 88.Ruvinsky I., Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–348. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 89.Honkanen R.E., Golden T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era. Curr Med Chem. 2002;9:2055–2075. doi: 10.2174/0929867023368836. [DOI] [PubMed] [Google Scholar]
- 90.Westermarck J., Hahn W.C. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med. 2008;14:152–160. doi: 10.1016/j.molmed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 91.Fury M.G., Drobnjak M., Sima C.S., Asher M., Shah J., Lee N., Carlson D., Wendel H.G., Pfister D.G. Tissue microarray evidence of association between p16 and phosphorylated eIF4E in tonsillar squamous cell carcinoma. Head Neck. 2010;33:1340–1345. doi: 10.1002/hed.21621. [DOI] [PubMed] [Google Scholar]
- 92.Molinolo A.A., Hewitt S.M., Amornphimoltham P., Keelawat S., Rangdaeng S., Meneses Garcia A., Raimondi A.R., Jufe R., Itoiz M., Gao Y., Saranath D., Kaleebi G.S., Yoo G.H., Leak L., Myers E.M., Shintani S., Wong D., Massey H.D., Yeudall W.A., Lonardo F., Ensley J., Gutkind J.S. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res. 2007;13:4964–4973. doi: 10.1158/1078-0432.CCR-07-1041. [DOI] [PubMed] [Google Scholar]
- 93.Amornphimoltham P., Patel V., Sodhi A., Nikitakis N.G., Sauk J.J., Sausville E.A., Molinolo A.A., Gutkind J.S. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res. 2005;65:9953–9961. doi: 10.1158/0008-5472.CAN-05-0921. [DOI] [PubMed] [Google Scholar]
- 94.Hartmann J.T., Haap M., Kopp H.G., Lipp H.P. Tyrosine kinase inhibitors—a review on pharmacology, metabolism and side effects. Curr Drug Metab. 2009;10:470–481. doi: 10.2174/138920009788897975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Similar stability of Lamγ2 protein in normal keratinocytes and SCC cells. Replicate preconfluent and confluent cultures of normal keratinocytes (N/TERT-1) and SCC-13 cells were pretreated for 1 hour with brefeldin A and then cycloheximide was added for 0.5, 1, 3, or 6 hours. At each time point, proteins were extracted and Western blotted for Lamγ2 and c-myc. In contrast to c-myc, Lamγ2 is very stable and shows a modest decrease in levels only after 6 hours of cycloheximide treatment, to a similar extent in preconfluent and confluent N/TERT-1 cells and in confluent SCC-13 cells. BfA, brefeldin A; CHX, cycloheximide.
An activity of the HPV16 E6 oncoprotein not involving p53 degradation causes Lamγ2 dysregulation in keratinocytes. Western blot analysis of Lamγ2 and p53 levels in the cell lines strain N, N/E6E7 [early (90) and later (250) passage after transduction], N/E6, N/E7, N/E6JH26, POE9n, SCC-15, and SCC-68. Note that p53 levels in the E6E7 and E6 transductant are reduced, but are normal in the E6JH26 transductant.
Increased basal p-S6 and Lamγ2 expression is not a feature of benign hyperplasia, but rather is induced coordinately in normal epithelial cells participating in wound repair. Areas demarcated by rectangles in the p-S6 panels are enlarged as insets in the same panels, with the basal lamina indicated by a dotted line. A: Benign oral wart. Note absence of Lamγ2 and p-S6 staining in basal cells. B: Slightly atypical, hyperplastic oral mucosal epithelium at the left edge of an ulcer. Note basal p-S6 and Lamγ2 immunostaining in the migrating tongue of cells (arrow) next to the clot (red superficial material marked by asterisk in the H&E panel), in contrast to suprabasal p-S6 and absence of Lamγ2 immunostaining in the epithelium at a distance from the wound edge. C: Note a microerosion (arrow) at the margin between normal epithelium and a dysplasia, showing a pattern of p-S6 and Lamγ2 staining reminiscent both of dysplasia and of the wound edge response. Scale bar = 200 μm.
Levels of Lamα3 and Lamβ3 mRNA in preconfluent versus confluent cultures of normal keratinocytes and SCC cells and predicted secondary structures of the 5′-UTRs of laminin mRNAs. A: qPCR comparative analysis of Lamα3 and Lamβ3 mRNA levels in preconfluent and confluent cultures of normal keratinocytes and SCC cells. Laminin mRNA levels were normalized to GAPDH mRNA levels in the same sample. All cell lines were compared with the Lamα3 and Lamβ3 mRNA levels in preconfluent cultures of the normal primary keratinocyte line strain N, set at an arbitrary unit of 1. Note that levels of Lamα3 mRNA, but not of Lamβ3, decrease in confluent cultures of both normal keratinocytes and SCC cells, correlating with the reduction in Lamα3 protein and maintenance of Lamβ3 protein at confluence by normal and SCC cells (Figure 2). B: Predicted secondary structures of the 5′UTR regions of the mRNAs encoding Lamγ2, Lamα3, Lamβ3, and c-myc. Note the long, stem-loop hydrogen bonded structure of the 5′UTR of Lamγ2, similar to that of c-myc.
Independence from exogenous EGF for optimal growth resulting from constitutively active EGFR, HRAS, cRAF1, or MEK1 expression. Cells were plated in medium with and without EGF, as described under Materials and Methods. Note that all of the engineered EGFR/ERK hyperactive cell lines grew at their maximum rates in the absence of EGF; in contrast, the N/TERT-1 parent cell line required EGF to grow optimally.