

Abstract
In recent years, Fanconi anemia (FA) has been the subject of intense investigations, primarily in the DNA repair research field. Many discoveries have led to the notion of a canonical pathway, termed the FA pathway, where all FA proteins function sequentially in different protein complexes to repair DNA cross-link damages. Although a detailed architecture of this DNA cross-link repair pathway is emerging, the question of how a defective DNA cross-link repair process translates into the disease phenotype is unresolved. Other areas of research including oxidative metabolism, cell cycle progression, apoptosis, and transcriptional regulation have been studied in the context of FA, and some of these areas were investigated before the fervent enthusiasm in the DNA repair field. These other molecular mechanisms may also play an important role in the pathogenesis of this disease. In addition, several FA-interacting proteins have been identified with roles in these “other” nonrepair molecular functions. Thus, the goal of this paper is to revisit old ideas and to discuss protein-protein interactions related to other FA-related molecular functions to try to give the reader a wider perspective of the FA molecular puzzle.
1. The FA Clinical Phenotype
Fanconi anemia (FA) is a complex disease that is considered a congenital form of aplastic anemia. The genetic mode of transmission is both autosomal and X-linked, and a growing number of identified genes are distributed among the various chromosomes. The common clinical manifestation in most patients with FA, which may occur in all FA patients eventually, is life-threatening bone marrow failure (BMF) [1, 2]. FA is also associated with diverse birth defects and a predisposition to malignancies. FA-associated congenital malformations can affect many organ systems including the central nervous system, the gastrointestinal system, and the skeletal system [3–8]. Other findings in patients with FA include short stature, skin pigmentation abnormalities, and small facial features. In addition, more than 70% of patients with FA show endocrine dysfunctions including deficiencies in growth hormone and thyroid hormone as well as diabetes [9, 10]. All of these disease manifestations suggest a role for FA genes in mechanisms that bear on hematopoiesis, development, and neoplasia.
2. The FA Molecular Pathway
Patients with FA are classified into complementation groups (to date 14 groups from A to P have been identified), and all of these groups correspond to one of the following cloned genes: FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ/BRIP1/BACH1, FANCL/PHF9, FANCM/HEF, FANCN/PALB2, and FANCP/SLX4 [11–27]. Approximately 85% of FA patients have a defective FANCA, FANCC or FANCG gene, while the other genes account for less than 5% of the mutations found in FA patients. To date, some patients still remain unassigned indicating the possibility of novel FA genes [28]. Mutations in the RAD51C gene (provisionally termed FANCO) have been associated with a FA-like disorder, suggesting that this gene may represent yet another FA gene [29, 30]. Patients with mutations in one of the 15 FA and FA-like genes present clinical FA aspects to various degrees but show a common cellular phenotype: hypersensitivity to DNA cross-linking agents such as mitomycin C (MMC), diepoxybutane, and cisplatin [28, 31]. When exposed to those agents, cells from FA patients show an abnormally prolonged cell cycle arrest in the G2/M phase, increased chromosomal aberrations, and reduced survival. These cellular features define FA, and presumably, all FA proteins cooperate in a pathway, termed the FA pathway, to maintain chromosome integrity.
In the canonical FA pathway, FA proteins are subdivided into three complexes based on protein-protein interaction studies. The first complex, known as the core complex or complex I, is composed of seven FA proteins, including FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, and FANCL [22, 27, 32–40]. The FANCM protein was also considered part of this core complex; however, further analysis revealed confounding results in FANCM-mutated cells [41]. Other proteins found in association with the core complex include Fanconi anemia-associated protein 24 (FAAP24), FAAP100, FANCM-associated histone fold protein 1 (MHF1), MHF2, and hairy enhancer of split 1 (HES1) [42–44]. All of these associated proteins are required for efficient FA pathway activation, but disease-causing mutations have yet to be found. Following DNA cross-link damage, the core complex associates with the FANCM-FAAP24 heterodimer on the chromatin [27, 39, 45, 46] and monoubiquitinates, through the activity of the FANCL E3 ubiquitin ligase, the complex II (or ID complex) components FANCD2 and FANCI [22, 47–50]. This ubiquitin tag alters the cellular distribution of complex II and promotes its association with the FA pathway complex III components FANCD1, FANCJ, FANCN and FANCP, and the FA-like disorder protein RAD51C [29, 30]. Complex III, similar to FANCM, is dispensable for the monoubiquitination of ID complex components, supporting the role of FA proteins in a linear (or canonical) response pathway. The deubiquitination of FANCD2 and FANCI by the UAF1/USP1 deubiquitinating enzyme complex appears to be required for the completion of the repair process [50–52].
3. FA Protein Cellular Localization
One intriguing aspect of the FA molecular pathway is the cellular distribution of FA proteins. Although the well-characterized function of this pathway in DNA crosslink damage occurs in the nucleus, FA core complex proteins can be found in different cellular compartments in addition to the nucleus.
The first identified FA protein, FANCC, is principally found in the cytoplasm [53–56]. It was first reported that FANCC function in DNA cross-links resistance required its cytoplasmic location and that enforced nuclear FANCC expression abolished its ability to correct FA-C cells [55, 57]. It was later found that FANCC is partially localized to the nucleus, and this FANCC nuclear localization is critical for the function of the FA core complex in DNA crosslink responses [56, 58]. These conflicting results may suggest that FANCC is required in both cellular compartments, and that regulation of its protein level in each cellular compartment is important for its function. The molecular chaperone, glucose-related protein 94 (GRP94) involved in protein quality control, protein folding, and ER stress responses [59], directly binds FANCC and regulates its intracellular levels [60]. Reducing the levels of GRP94 through the use of a specific ribozyme affects FANCC stability and renders cells hypersensitive to MMC suggesting a possible mechanism of FANCC quality control and subsequent cellular localization.
FANCA cellular localization has been extensively studied [32, 36, 37, 61–63]. FANCA possesses a bipartite nuclear localization signal (NLS) domain that is required for its nuclear shuttling [64] and contains five nuclear export signal NES domains involved in chromosome region maintenance 1 (CRM1/exportin 1)-dependent nuclear export [65]. It has been proposed that the interaction of FANCA with the sorting nexin 5 protein (SNX5) may be involved in its subcellular trafficking [66]. Other studies have reported that FA core complex proteins could be found in the cytoplasm, in an unbound form, but also in a 600 kDa complex composed of at least four FA core complex proteins (i.e., FANCA, FANCC, FANCF, and FANCG) [57, 61, 67, 68]. Other studies have suggested the formation of different cytoplasmic subcomplexes, including FANCA/FANCG [36, 38], FANCB/FANCL [23], and FANCC/FANCE [33, 69–71]. It has been suggested that these subcomplexes are probably translocated to the nucleus independently before association into a core complex through FANCF, which acts as a linker protein [33, 72].
Because the nuclear presence of both FANCC and FANCE depends on each other, an interdependence between FA proteins has been proposed. Indeed, studies on FA mutant cell lines have shown that the core complex fails to form if one protein is absent or mutated [33]. Studies of synchronized cells have shown that FA proteins shuttle between the nucleus and the cytoplasm during the cell cycle. Early in the G1 phase, FA core complex proteins are localized to the cytoplasm, at the G1-S border, they are loaded onto chromatin, and throughout mitosis they migrate to the nuclear periphery to become completely excluded from the condensed chromosomes [67, 73, 74]. The exclusion of FA proteins from condensed chromosomes occurs in the absence of DNA damage, whereas treatment with MMC results in an increased binding of core complex proteins to chromatin [67, 73, 75]. Considering these results, it is not inconceivable to surmise that the cytoplasmic forms of FA proteins or FA subcomplexes are likely to be critical for cell signaling events in normative conditions.
4. Posttranslational Modification of FA Proteins
FA proteins undergo multiple posttranslational modifications, including monoubiquitination, phosphorylation and proteolytic processing. The most widely studied modification is the monoubiquitination of FANCD2 and FANCI. Although only two of the fifteen FA proteins are monoubiquitinated, several FA proteins, including FANCA, FANCE, FANCG, FANCD2, FANCI, and FANCM, are phosphorylated, and two FA proteins are regulated through a caspase-mediated proteolytic process. These data suggest that posttranslational modifications play an important role in FA proteins activity. In this section, we will only discuss FA protein modifications other than the ubiquitination of FANCD2 and FANCI because it has been extensively discussed [47, 76, 77].
The FANCA protein was the first FA protein shown to be phosphorylated [58, 63, 78]. The FANCA phosphorylation site was first thought to be located at S1149 [79], which harbors an AKT kinase consensus sequence; however, the FANCA S1149A mutant was shown to be more efficiently phosphorylated than wildtype FANCA. The FANCA phosphorylation site was later identified by mass spectrometry as serine 1449 [80]. The phosphorylation of FANCA at S1449 is functionally important because it was found to be defective in lymphoblasts from several patients with FA, and FANCA S1449A failed to fully rescue FA-A mutant cells. A novel wortmannin-sensitive protein kinase termed FANCA-PK was first suggested as the kinase responsible for FANCA phosphorylation [81]; however, the true kinase that phosphorylates FANCA in response to DNA damage was later identified as ATR [80]. FANCA was also shown to associate with the IKK signalosome through direct interaction with IKK2, the IκB Kinase-2 [82]. Although this kinase affected the phosphorylation state of several FANCA-associated proteins, no clear, direct phosphorylation of FANCA by IKK2 has been reported. Different groups have reported that FANCG is also phosphorylated. FANCG phosphorylation occurs at serines 7, 383, and 387, with the two latter sites being cell cycle-dependent and actively phosphorylated during mitosis [83–85]. The exclusion of FA proteins from chromosomes during mitosis appears to coincide with phosphorylated FANCG. All the FANCG phosphorylation sites are functionally important because mutant forms of these FANCG serine residues compromised its ability to rescue FA-G mutant cells. The FANCC-interacting protein kinase cdc2 [86] was shown to be required for the phosphorylation of at least the S387 FANCG residue. The kinases responsible for the phosphorylation of the other FANCG target sites are unknown.
The FANCD2 protein undergoes two different posttranslational modifications. In addition to monoubiquitination on lysine 561, FANCD2 is phosphorylated on several residues by different kinases depending on the cellular signal. In response to various DNA damaging agents, including ultraviolet light, MMC, hydroxyurea, and ionizing radiation, FANCD2 is phosphorylated on T691 and S717 followed by S222 in an ATM/ATR-dependent mechanism [87, 88]. S222 phosphorylation triggers the activation of an intra-S-phase checkpoint response. In addition, the FANCD2-T691A/S717A double mutant does not complement the MMC sensitivity in FA-D2 cells, suggesting that this posttranslational modification is required for FANCD2 function in the DNA damage response. Other ATM-dependent FANCD2 phosphorylation sites have been described and have been shown to be functional in in vitro assays, including S1401, S1404, and S1418, and only S1401 has been confirmed in vivo. S331 of FANCD2 has been shown to be phosphorylated by the checkpoint kinase 1 (CHK1) and is essential for MMC resistance [89]. Although FANCD2 phosphorylation is independent of other posttranslational modifications, it promotes or enhances the monoubiquitination process. For instance, ATR-mediated phosphorylation of FANCD2 is essential for its monoubiquitination in response to DNA damage as shown by absence of FANCD2 monoubiquitination in ATR-deficient cells and cells from patients with Seckel syndrome, a disease resembling FA [90]. FANCI was identified in the search for ATR-inducible phosphorylated proteins in response to ionizing radiation [50]. Three phosphorylation sites were detected in the human FANCI protein (S730, T952, and S1121), and two other sites were detected in the mouse protein (S555 and T558). FANCI phosphorylation is essential for the FA pathway activation following DNA damage as measured by FANCD2 monoubiquitination [91].
FANCE and FANCM phosphorylation has also been studied in the context of induced DNA damage. In response to DNA damage, CHK1 was shown to phosphorylate FANCE on two residues, notably T346 and S374 [92]. FANCE phosphorylation is required for MMC resistance but is dispensable for FA pathway activation as measured by FANCD2 monoubiquitination. The CHK1-induced phosphorylation of FANCE promotes its assembly with FANCD2 into nuclear foci and promotes its degradation, serving as a potential negative regulatory mechanism of the FA pathway. FANCM is a phosphoprotein that contains multiple predicted ATR phosphorylation sites and becomes hyperphosphorylated in response to DNA damage and during mitosis [27, 93]. FANCM phosphorylation occurs independently of the FA core complex activation leading to the monoubiquitination of the ID complex.
Finally, another posttranslational modification found in FANCC is caspase-mediated proteolytic processing [94]. Similar to many proteins involved in signaling mechanisms, FANCC is cleaved by a caspase during apoptosis. This proteolytic modification of FANCC is not required for its function in DNA repair or DNA damage signaling, but the cleavage of FANCC inhibits its suppressor of apoptosis function. Recently, a second FA protein, FANCD2 was shown to be regulated by a caspase-mediated proteolytic processing in response to DNA crosslink damage and the non-DNA damaging agent TNF-α [95]. Both the TNF-α and DNA crosslink-mediated disappearance of FANCD2 was blocked by caspase inhibitors but not by proteasome inhibitors, suggesting that FANCD2 is regulated through a caspase-dependent mechanism in response to cellular stress.
5. FA Proteins Partners with Roles in Oxidative Metabolism
The abnormal sensitivity of FA cells to reactive oxygen species (ROS) was first suggested in 1977 by Nordenson [96] who showed reduced chromosomal breaks in FA lymphocytes cultured in the presence of superoxide dismutase, catalase, or both enzymes. The role of oxygen on chromosomal instability in FA mutant cells was confirmed by Joenje et al. in 1981; they showed attenuated chromosomal aberrations at low oxygen tension (5%) but aggravated chromosomal aberrations at high concentrations of oxygen [97]. Subsequently, several reports indicated that FA cells were hypersensitive to oxygen radicals showing reduced growth and blockage in the G2 phase of the cell cycle [98–101]. Increased ROS in FA leucocytes has also been reported [99]. In addition, the overexpression of detoxifying enzymes, the inhibition of enzymes involved in oxidation or the use of antioxidants in FA cells reduced the rate of spontaneous chromosomal breakage and abolished the DNA damaging effects of MMC [98, 102–106]. Other studies have established a link between an altered redox state and reduced proliferation, reduced growth, and altered cytokine responses in FA cells including hematopoietic progenitors [107–110]. Studies from FancC/Sod1 double mutant mice exhibiting defects in hematopoiesis including bone marrow hypocellularity and cytopenia, which is reminiscent of phenotypes observed in patients with FA, suggest that an abnormal redox state contribute to BMF in FA [111].
Together, these data indicate that FA proteins may be involved in responses to endogenous oxidative stress or in the regulation of the cell redox state. This hypothesis is further supported by studies showing interactions between FA proteins and proteins involved in oxygen metabolism [112]. For instance, FANCC has protein partners with roles in redox metabolisms, including glutathione S-transferase π I (GSTπI) and NADPH cytochrome-P450 reductase (RED) [113, 114], whereas FANCG interacts with cytochrome P450 2E1 (CYP2E1) and the mitochondrial peroxiredoxin-3 (PRDX3) [68, 112, 115]. Through these interactions, FA proteins were shown to attenuate the redox activation of xenobiotics and to prevent apoptosis. Consequently, in FA mutant cells, a loss of interaction between FA proteins and these molecular antioxidants leads to an aberrant redox metabolism that translates into ROS-mediated DNA damage and cell death (see Table 1).
Table 1.
List of FA protein partners.
| Functional class | Specific function | Protein name | Interacts with | References |
|---|---|---|---|---|
| Transcription | Transcriptional repressor | FAZF | FANCC | [112, 147] |
| HES1 | FANCA, F, G, L | [44] | ||
| Stress-induced chaperone | Hsp70 | FANCC | [112, 140] | |
| GRP94 | FANCC | [60, 112] | ||
| Chromatin modifier | BRG1 | FANCA | [112, 154] | |
|
| ||||
| Cell cycle | Serine/threonine kinase | cdc2 | FANCC | [86, 112] |
|
| ||||
| Cell signaling | Cytokine response | STAT1 | FANCC | [133] |
| IKK2 | FANCA | [79, 82, 112] | ||
| Secondary modification | Akt kinase | FANCA | [79, 112] | |
|
| ||||
| Oxidative metabolism | Electron transfer | RED | FANCC | [112, 114] |
| Cytosolic Detoxifying enzyme | GSTP1 | FANCC | [112, 113] | |
| Metabolism of xenobiotics | CYP2E1 | FANCG | [112, 115] | |
| Antioxidant enzyme | PRDX3 | FANCG | [68] | |
|
| ||||
| Transporter | Intracellular trafficking | SNX5 | FANCA | [66, 112] |
6. FA Proteins Partners with Roles in Cytokine Signaling and Apoptosis
It is well established that FA mutant cells are prone to apoptosis. The FA literature is rich in reports pertaining to FA mutant cells (human bone-marrow-derived cells, lymphocytes, fibroblasts, and mouse embryonic fibroblasts) that show increased apoptosis or reduced cell growth in response to various agents including ROS inducers, DNA damaging agents, growth factor withdrawal, and cytokines. It is clear from many studies of patient-derived cells and cells from FA mouse models that FA proteins are involved in pathways that regulate cell survival or cell death [116–121]. For instance, two FA proteins, FANCC and FANCD2, are caspase targets [94, 95], and FANCC overexpression or the inhibition of its caspase-mediated cleavage prevents or delays apoptosis, even in wildtype cells supporting the idea of a cell survival function of the FA proteins [94, 119, 122]. The role of FANCC in cell survival has been linked to oxidative metabolism as described above but it may also be linked to cytokine-mediated cellular responses because many cytokine-mediated signaling events lead to apoptosis. It has been suggested that abnormal cytokine regulation may account for the progressive BMF observed in patients with FA because TNF-α overproduction and underproduction of Il-6 have been detected in the sera of patients with FA [123–125]. FA-C mutant cells and FancC−/− progenitor and stem cells are hypersensitive to the inhibitory cytokines including TNF-α and IFN-γ, and show suppressed growth and increased apoptosis at doses that do not affect normal cells [116, 122, 126, 127]. In addition, the continuous injection of low IFN-γ doses in vivo leads to BMF in FA mice [128, 129], whereas TNF-α leads to clonal evolution and leukemia in this FA mouse model [130]. In support of these altered cytokine responses in FA cells, the cytokine-response genes myxovirus A (MxA), IFN response factor 1 (IRF1), p21CIP/WAF, and IFN-stimulated gene factor 3 (ISGF3γ) were highly expressed in FA mutant cells without exogenous cytokine stimulation, while corrected cells suppressed this overproduction and restored their MMC resistance [116, 131, 132]. These data suggest that FA proteins, or at least FANCC, function to modulate a cytokine-mediated signal. Indeed, FANCC was shown to directly interact with signal transducer and activator of transcription 1 (STAT1), which is an IFN signal transducer [133]. FANCC functions as a control factor for STAT1 docking at the IFN-γR complex and subsequent activation of the IFN type II signaling cascade [133]. Thus, the STAT1 activation defect observed in FA-C cells results in an altered nuclear STAT1-DNA complex, which diminishes the expression of IRF1. The STAT1-FANCC interaction is also induced by other cytokines, including IFN-α, granulocyte-macrophage colony-stimulating factor (GM-CSF), and stem cell factor, whereas mutant FANCC does not associate with STAT1 in cells stimulated with these factors. FANCC seems to regulate IFNγ-inducible genes (e.g., IRF1, p21WAF, and ISGF3γ) independently of STAT1 binding. An altered response to type I IFN was also observed in FANCC mutant and FancC−/− cells, as shown by the reduced phosphorylation of the Janus kinases, Jak1 and Tyk2, and the subsequently diminished phosphorylation of STAT1, STAT3, and STAT5 [134]. This altered Tyk2 response translates into reduced numbers of CD4-positive cells in FancC−/− mice. Because Tyk2 plays a role in the differentiation and maintenance of T helper cells, failure of FANCC to normally activate Jak/STAT signaling may result in impaired immune cell differentiation and immune defects, as reported in patients with FA [135–139].
FANCC has been shown to physically interact with Hsp70 [140]. This interaction appears to be required for protection against TNF-α and IFN-γ-induced apoptosis because reduced Hsp70 expression sensitizes normal cells to these cytokines but does not further augment the hypersensitivity to apoptosis in FA-C cells. Because Hsp70 is known to suppress the IFN-inducible double-stranded RNA (dsRNA)-dependent protein kinase (PKR) activation [141] and FA-C cells have constitutively activated PKR [142], FANCC was shown to inhibit the kinase activity of PKR through physical interaction with Hsp70 [143]. Although this activity is independent of a functional FA complex, the FA core complex protein FANCA was found to interact with IKK2 (or IKKβ) a kinase and a component of the IKK signalosome [82]. The IKK signalosome is a critical mediator of the cellular response to stressors such as dsRNA and cytokines [144, 145]. Deletion of IKK2 has been shown to affect the development of CD4-positive cells [146]. Because FancC−/− mice have reduced numbers of CD4+ cells and two FA proteins have partners that participate in cytokine-activated signaling cascades affecting the development of these lymphocytes, we can speculate that FA proteins may act as converging key molecules.
7. FA Protein Partners with Roles in Transcription
Another FA protein role less considered is the regulation of transcription. Several FA proteins have interacting partners directly involved in transcriptional regulation. The first FA protein partner identified that acts in transcription is FAZF (FA Zinc Finger) [147]. FAZF, also known as RoG (for repressor of GATA) [148], PLZP (for PLZF-like zinc finger protein) [149] and TZFP (for testis zinc finger) [150], is a transcriptional repressor that belongs to the BTB/POZ family of proteins and is similar to the PLZF protein [147]. This family of transcriptional repressors was shown to be important for several developmental processes including tissue proliferation and differentiation and tumor formation. FAZF was identified in a yeast 2-hybrid screen with FANCC. FAZF was shown to be highly expressed in CD34-positive progenitor cells; it further increased during proliferation of these cells and decreased during their terminal differentiation [151]. FAZF acts as a negative regulator of transcription. Because a disease-causing mutation in FANCC interferes with FAZF binding [147], and FancC−/− hematopoietic stem/progenitor cells show increased cycling and aberrant cell cycle control [152], a plausible hypothesis is that the FANCC-FAZF interaction in hematopoietic stem/progenitor cells leads to the repression of critical target genes required for growth suppression.
A second transcriptional repressor identified as a FA-binding protein is the hairy enhancer of split 1 (HES1) [44]. HES1 is a member of a highly conserved family of hairy-related basic helix-loop-helix (bHLH)-type transcriptional repressors. HES1 was shown to interact directly with several components of the FA core complex. The FA core complex was shown to contribute to the transcriptional regulation of HES1-responsive genes, both positively (HES1) and negatively (cyclin-dependent kinase inhibitor p21cip1/waf1). Two mechanisms of regulation by FA proteins have been proposed. The first proposed mechanisms is that by interacting with HES1, FA core complex proteins antagonize HES1-mediated transcriptional repression by interfering with the assembly of the HES1/transducing-like-enhancer of split (TLE) corepressor complex at the HES1 promoter [153]. The second proposed mechanism involves an indirect mechanism where the binding of FA proteins with HES1 influences HES1 affinity or its specificity for promoters, such as that of p21cip1/waf1.
The brahma-related gene 1 protein (BRG1) has also been identified as a FA-binding partner through a yeast 2-hybrid screen [154]. BRG1 is the central catalytic subunit of the SWI/SNF family of ATP-dependent chromatin remodeling complexes [155]. BRG1 is a major coregulator of transcription, both in activation and repression, through chromatin modulation. Although the FANCA-BRG1 interaction has been shown in cells, the functional impact of this interaction remains unclear.
In view of these FA protein partners with roles in transcriptional regulation and the fact that the FA core complex possesses E3 ubiquitin ligase activity, it is possible that FA proteins act as transcriptional coregulators through the posttranslational modification of these transcriptional regulators.
8. Conclusion
Since the discovery of FANCC, the first identified FA gene in 1992 [15], there have been significant advances in the FA molecular biology field. These advances mostly include characterization of the canonical FA pathway, which is activated in response to DNA crosslink damage. It is clear that FA proteins are required for DNA crosslink repair; however, the question of how a defective FA protein leads to BMF, and developmental abnormalities remains elusive. It is obvious that absence of a functional FA protein affects many cellular and molecular functions and leads to an array of cellular phenotypes (see Figure 1). A perplexing question is whether FA proteins interactions with their nonrepair partners act only as modifiers of the clinical manifestation of FA. Once we reconcile all the notions related to FA proteins role in these various cellular and molecular activities, we may then obtain a clearer picture of the complexities of this molecular puzzle.
Figure 1.
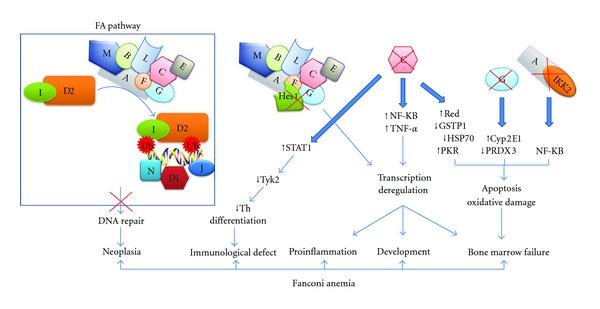
Putative roles of FA proteins through their interacting partners. The involvement of FA proteins with their protein partners in the different molecular mechanisms that lead to regulation of transcription, cell cycle regulation, ROS detoxification, DNA repair, and cell survival. Loss of protein interactions between FA proteins and their partners through disease causing mutations in a FA gene could lead to a defective molecular function resulting in an array of phenotypes including BMF and congenital malformations.
Acknowledgments
The authors acknowledge laboratories whose fine work could not be cited owing to space constraints. T. Kadder was supported by a fellowship from the CIHR and the Canadian Fanconi anemia Research Fund. M. Carreau was funded by the Canadian Blood Services/CIHR Blood Utilisation and Conservation Initiative grant.
References
- 1.Bagby GC, Lipton JM, Sloand EM, Schiffer CA. Marrow failure. Hematology. 2004:318–336. doi: 10.1182/asheducation-2004.1.318. [DOI] [PubMed] [Google Scholar]
- 2.Buchwald M, Carreau M. Genetic basis of Fanconi anemia. In: Schrezenmeier H, Bacigalupo A, editors. Aplastic Anemia. Cambridge, UK: Cambridge University Press; 1999. p. 403. [Google Scholar]
- 3.Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi anemia Registry Study. American Journal of Medical Genetics. 1997;68(1):58–61. [PubMed] [Google Scholar]
- 4.Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, Auerbach AD. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the international Fanconi anemia registry. Pediatrics. 1993;91(6):1116–1120. [PubMed] [Google Scholar]
- 5.Tsilou ET, Giri N, Weinstein S, Mueller C, Savage SA, Alter BP. Ocular and orbital manifestations of the inherited bone marrow failure syndromes: Fanconi anemia and dyskeratosis congenita. Ophthalmology. 2010;117(3):615–622. doi: 10.1016/j.ophtha.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vale MJ, Dinis MJ, Bini-Antunes M, Porto B, Barbot J, Coutinho MB. Audiologic abnormalities of Fanconi anaemia. Acta Oto-laryngologica. 2008;128(9):992–996. doi: 10.1080/00016480701793750. [DOI] [PubMed] [Google Scholar]
- 7.Tischkowitz M, Dokal I. Fanconi anaemia and leukaemia—clinical and molecular aspects. British Journal of Haematology. 2004;126(2):176–191. doi: 10.1111/j.1365-2141.2004.05023.x. [DOI] [PubMed] [Google Scholar]
- 8.Sari N, Akyuz C, Aktas D, et al. Wilms tumor, AML and medulloblastoma in a child with cancer prone syndrome of total premature chromatid separation and Fanconi anemia. Pediatric Blood and Cancer. 2009;53(2):208–210. doi: 10.1002/pbc.21966. [DOI] [PubMed] [Google Scholar]
- 9.Wajnrajch MP, Gertner JM, Huma Z, et al. Evaluation of growth and hormonal status in patients referred to the international Fanconi anemia registry. Pediatrics. 2001;107(4 I):744–754. doi: 10.1542/peds.107.4.744. [DOI] [PubMed] [Google Scholar]
- 10.Giri N, Batista DL, Alter BP, Stratakis CA. Endocrine abnormalities in patients with Fanconi anemia. Journal of Clinical Endocrinology and Metabolism. 2007;92(7):2624–2631. doi: 10.1210/jc.2007-0135. [DOI] [PubMed] [Google Scholar]
- 11.Kang M. Cancel all hollidays for SLX4 mutations: identification of a new Fanconi anemia subtype, FANCP. Clinical Genetics. 2011;80(1):28–30. doi: 10.1111/j.1399-0004.2011.01679.x. [DOI] [PubMed] [Google Scholar]
- 12.Crossan GP, Van Der Weyden L, Rosado IV, et al. Disruption of mouse SLX4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nature Genetics. 2011;43(2):147–152. doi: 10.1038/ng.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallmeier E, Hucl T, Brody JR, et al. High-throughput screening identifies novel agents eliciting hypersensitivity in Fanconi pathway-deficient cancer cells. Cancer Research. 2007;67(5):2169–2177. doi: 10.1158/0008-5472.CAN-06-2711. [DOI] [PubMed] [Google Scholar]
- 14.De Winter JP, Rooimans MA, Van Der Weel L, et al. The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nature Genetics. 2000;24(1):15–16. doi: 10.1038/71626. [DOI] [PubMed] [Google Scholar]
- 15.Strathdee CA, Gavish H, Shannon WR, Buchwald M. Cloning of cDNAs for Fanconi’s anaemia by functional complementation. Nature. 1992;356(6372):763–767. doi: 10.1038/356763a0. [DOI] [PubMed] [Google Scholar]
- 16.De Winter JP, Waisfisz Q, Rooimans MA, et al. The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nature Genetics. 1998;20(3):281–283. doi: 10.1038/3093. [DOI] [PubMed] [Google Scholar]
- 17.Foe JR, Rooimans MA, Bosnoyan-Collins L, et al. Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nature genetics. 1996;14(4):p. 488. doi: 10.1038/ng1296-488. [DOI] [PubMed] [Google Scholar]
- 18.De Winter JP, Leveille F, Van Berkel CGM, et al. Isolation of a cDNA representing the Fanconi anemia complementation group E gene. American Journal of Human Genetics. 2000;67(5):1306–1308. doi: 10.1016/s0002-9297(07)62959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fanconi anaemia/Breast Cancer Consortium. Positional cloning of the Fanconi anaemia group A gene. Nature Genetics. 1996;14(3):324–328. doi: 10.1038/ng1196-324. [DOI] [PubMed] [Google Scholar]
- 20.Timmers C, Taniguchi T, Hejna J, et al. Positional cloning of a novel Fanconi anemia gene, FANCD2. Molecular Cell. 2001;7(2):241–248. doi: 10.1016/s1097-2765(01)00172-1. [DOI] [PubMed] [Google Scholar]
- 21.Howlett NG, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297(5581):606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 22.Meetei AR, De Winter JP, Medhurst AL, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nature Genetics. 2003;35(2):165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 23.Meetei AR, Levitus M, Xue Y, et al. X-linked inheritance of Fanconi anemia complementation group B. Nature Genetics. 2004;36(11):1219–1224. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- 24.Levran O, Attwooll C, Henry RT, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nature Genetics. 2005;37(9):931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 25.Levitus M, Waisfisz Q, Godthelp BC, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nature Genetics. 2005;37(9):934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 26.Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nature Genetics. 2005;37(9):953–957. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 27.Meetei AR, Medhurst AL, Ling C, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nature Genetics. 2005;37(9):958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinto FO, Leblanc T, Chamousset D, et al. Diagnosis of Fanconi anemia in patients with bone marrow failure. Haematologica. 2009;94(4):487–495. doi: 10.3324/haematol.13592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nature Genetics. 2010;42(5):410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 30.Vaz F, Hanenberg H, Schuster B, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nature Genetics. 2010;42(5):406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- 31.Auerbach AD. Fanconi anemia and its diagnosis. Mutation Researchs. 2009;668(1-2):4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kupfer GM, Näf D, Suliman A, Pulsipher M, D’Andrea AD. The Fanconi anaemia proteins, FAA and FAC, interact to form a nuclear complex. Nature Genetics. 1997;17(4):487–490. doi: 10.1038/ng1297-487. [DOI] [PubMed] [Google Scholar]
- 33.De Winter JP, Van Der Weel L, De Groot J, et al. The Fanconi anemia protein FANCF forms a nuclear complex with FANCA, FANCC and FANCG. Human Molecular Genetics. 2000;9(18):2665–2674. doi: 10.1093/hmg/9.18.2665. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Higuera I, Kuang Y, Näf D, Wasik J, D’Andrea AD. Fanconi anemia proteins FANCA, FANCC, and FANCG/XRCC9 interact in a functional nuclear complex. Molecular and Cellular Biology. 1999;19(7):4866–4873. doi: 10.1128/mcb.19.7.4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medhurst AL, Huber PAJ, Waisfisz Q, De Winter JP, Mathew CG. Direct interactions of the five known Fanconi anaemia proteins suggest a common functional pathway. Human Molecular Genetics. 2001;10(4):423–429. doi: 10.1093/hmg/10.4.423. [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Higuera I, Kuang Y, Denham J, D’Andrea AD. The Fanconi anemia proteins FANCA and FANCG stabilize each other and promote the nuclear accumulation of the Fanconi anemia complex. Blood. 2000;96(9):3224–3230. [PubMed] [Google Scholar]
- 37.Kruyt FAE, Abou-Zahr F, Mok H, Youssoufian H. Resistance to mitomycin C requires direct interaction between the Fanconi anemia proteins FANCA and FANCG in the nucleus through an arginine-rich domain. Journal of Biological Chemistry. 1999;274(48):34212–34218. doi: 10.1074/jbc.274.48.34212. [DOI] [PubMed] [Google Scholar]
- 38.Waisfisz Q, De Winter JP, Kruyt FAE, et al. A physical complex of the Fanconi anemia proteins FANCG/XRCC9 and FANCA. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(18):10320–10325. doi: 10.1073/pnas.96.18.10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciccia A, Ling C, Coulthard R, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Molecular Cell. 2007;25(3):331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Ling C, Ishiai M, Ali AM, et al. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO Journal. 2007;26(8):2104–2114. doi: 10.1038/sj.emboj.7601666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh TR, Bakker ST, Agarwal S, et al. Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood. 2009;114(1):174–180. doi: 10.1182/blood-2009-02-207811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh TR, Saro D, Ali AM, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Molecular Cell. 2010;37(6):879–886. doi: 10.1016/j.molcel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan Z, Delannoy M, Ling C, et al. A histone-fold complex and FANCM forma conserved DNA-remodeling complex to maintain genome stability. Molecular Cell. 2010;37(6):865–878. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tremblay CS, Huang FF, Habi O, et al. HES1 is a novel interactor of the Fanconi anemia core complex. Blood. 2008;112(5):2062–2070. doi: 10.1182/blood-2008-04-152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson LH, Hinz JM. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights. Mutation Research. 2009;668(1-2):54–72. doi: 10.1016/j.mrfmmm.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xue Y, Li Y, Guo R, Ling C, Wang W. FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Human Molecular Genetics. 2008;17(11):1641–1652. doi: 10.1093/hmg/ddn054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Higuera I, Taniguchi T, Ganesan S, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular Cell. 2001;7(2):249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 48.Meetei AR, Yan Z, Wang W. FANCL replaces BRCA1 as the likely ubiquitin ligase responsible for FANCD2 monoubiquitination. Cell Cycle. 2004;3(2):179–181. [PubMed] [Google Scholar]
- 49.Sims AE, Spiteri E, Sims RJ, et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nature Structural and Molecular Biology. 2007;14(6):564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- 50.Smogorzewska A, Matsuoka S, Vinciguerra P, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129(2):289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nijman SMB, Huang TT, Dirac AMG, et al. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Molecular Cell. 2005;17(3):331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 52.Cohn MA, Kowal P, Yang K, et al. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Molecular Cell. 2007;28(5):786–797. doi: 10.1016/j.molcel.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 53.Yamashita T, Barber DL, Zhu Y, Wu N, D’Andrea AD. The Fanconi anemia polypeptide FACC is localized to the cytoplasm. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(14):6712–6716. doi: 10.1073/pnas.91.14.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Youssoufian H. Localization of Fanconi anemia c protein to the cytoplasm of mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(17):7975–7979. doi: 10.1073/pnas.91.17.7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Youssoufian H. Cytoplasmic localization of FAC is essential for the correction of a prerepair defect in Fanconi anemia group C cells. Journal of Clinical Investigation. 1996;97(9):2003–2010. doi: 10.1172/JCI118635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoatlin ME, Christianson TA, Keeble WW, et al. The Fanconi anemia group C gene product is located in both the nucleus and cytoplasm of human cells. Blood. 1998;91(4):1418–1425. [PubMed] [Google Scholar]
- 57.Kruyt FAE, Youssoufian H. The Fanconi anemia proteins FAA and FAC function in different cellular compartments to protect against cross-linking agent cytotoxicity. Blood. 1998;92(7):2229–2236. [PubMed] [Google Scholar]
- 58.Yamashita T, Kupfer GM, Naf D, et al. The Fanconi anemia pathway requires FAA phosphorylation and FAA/FAC nuclear accumulation. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(22):13085–13090. doi: 10.1073/pnas.95.22.13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eletto D, Dersh D, Argon Y. GRP94 in ER quality control and stress responses. Seminars in Cell and Developmental Biology. 2010;21(5):479–485. doi: 10.1016/j.semcdb.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoshino T, Wang J, Devetten MP, et al. Molecular chaperone GRP94 binds to the Fanconi anemia group C protein and regulates its intracellular expression. Blood. 1998;91(11):4379–4386. [PubMed] [Google Scholar]
- 61.Kruyt FAE, Waisfisz Q, Dijkmans LM, et al. Cytoplasmic localization of a functionally active Fanconi anemia group A-green fluorescent protein chimera in human 293 cells. Blood. 1997;90(9):3288–3295. [PubMed] [Google Scholar]
- 62.Näf D, Kupfer GM, Suliman A, Lambert K, D’Andrea AD. Functional activity of the Fanconi anemia protein FAA requires FAC binding and nuclear localization. Molecular and Cellular Biology. 1998;18(10):5952–5960. doi: 10.1128/mcb.18.10.5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kupfer G, Naf D, Garcia-Higuera I, et al. A patient-derived mutant form of the Fanconi anemia protein, FANCA, is defective in nuclear accumulation. Experimental Hematology. 1999;27(4):587–593. doi: 10.1016/s0301-472x(99)00022-3. [DOI] [PubMed] [Google Scholar]
- 64.Lightfoot J, Alon N, Bosnoyan-Collins L, Buchwald M. Characterization of regions functional in the nuclear localization of the Fanconi anemia group A protein. Human Molecular Genetics. 1999;8(6):1007–1015. doi: 10.1093/hmg/8.6.1007. [DOI] [PubMed] [Google Scholar]
- 65.Ferrer M, Rodríguez JA, Spierings EA, de Winter JP, Giaccone G, Kruyt FAE. Identification of multiple nuclear export sequences in Fanconi anemia group A protein that contribute to CRM1-dependent nuclear export. Human Molecular Genetics. 2005;14(10):1271–1281. doi: 10.1093/hmg/ddi138. [DOI] [PubMed] [Google Scholar]
- 66.Otsuki T, Kajigaya S, Ozawa K, Liu JM. SNX5, a new member of the sorting nexin family, binds to the Fanconi anemia complementation group A protein. Biochemical and Biophysical Research Communications. 1999;265(3):630–635. doi: 10.1006/bbrc.1999.1731. [DOI] [PubMed] [Google Scholar]
- 67.Thomashevski A, High AA, Drozd M, et al. The Fanconi anemia core complex forms four complexes of different sizes in different subcellular compartments. Journal of Biological Chemistry. 2004;279(25):26201–26209. doi: 10.1074/jbc.M400091200. [DOI] [PubMed] [Google Scholar]
- 68.Mukhopadhyay SS, Leung KS, Hicks MJ, Hastings PJ, Youssoufian H, Plon SE. Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. Journal of Cell Biology. 2006;175(2):225–235. doi: 10.1083/jcb.200607061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pace P, Johnson M, Tan WM, et al. FANCE: the link between Fanconi anaemia complex assembly and activity. EMBO Journal. 2002;21(13):3414–3423. doi: 10.1093/emboj/cdf355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taniguchi T, D’Andrea AD. The Fanconi anemia protein, FANCE, promotes the nuclear accumulation of FANCC. Blood. 2002;100(7):2457–2462. doi: 10.1182/blood-2002-03-0860. [DOI] [PubMed] [Google Scholar]
- 71.Leveille F, Ferrer M, Medhurst AL, et al. The nuclear accumulation of the Fanconi anemia protein FANCE depends on FANCC. DNA Repair. 2006;5(5):556–565. doi: 10.1016/j.dnarep.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 72.Leveille F, Blom E, Medhurst AL, et al. The Fanconi anemia gene product FANCF is a flexible adaptor protein. Journal of Biological Chemistry. 2004;279(38):39421–39430. doi: 10.1074/jbc.M407034200. [DOI] [PubMed] [Google Scholar]
- 73.Mi J, Kupfer GM. The Fanconi anemia core complex associates with chromatin during S phase. Blood. 2005;105(2):759–766. doi: 10.1182/blood-2004-01-0001. [DOI] [PubMed] [Google Scholar]
- 74.Qiao F, Moss A, Kupfer GM. Fanconi anemia proteins localize to chromatin and the nuclear matrix in a DNA damage- and cell cycle-regulated manner. Journal of Biological Chemistry. 2001;276(26):23391–23396. doi: 10.1074/jbc.M101855200. [DOI] [PubMed] [Google Scholar]
- 75.Akkari YMN, Bateman RL, Reifsteck CA, Olson SB, Grompe M. DNA replication is required to elicit cellular responses to psoralen-induced DNA interstrand cross-links. Molecular and Cellular Biology. 2000;20(21):8283–8289. doi: 10.1128/mcb.20.21.8283-8289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garner E, Smogorzewska A. Ubiquitylation and the Fanconi anemia pathway. Febs Letters. 2011 doi: 10.1016/j.febslet.2011.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alpi AF, Patel KJ. Monoubiquitylation in the Fanconi anemia DNA damage response pathway. DNA Repair. 2009;8(4):430–435. doi: 10.1016/j.dnarep.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 78.Adachi D, Oda T, Yagasaki H, et al. Heterogenous activation of the Fanconi anemia pathway by patient-derived FANCA mutants. Human Molecular Genetics. 2002;11(25):3125–3134. doi: 10.1093/hmg/11.25.3125. [DOI] [PubMed] [Google Scholar]
- 79.Otsuki T, Nagashima T, Komatsu N, et al. Phosphorylation of Fanconi anemia protein, FANCA, is regulated by akt kinase. Biochemical and Biophysical Research Communications. 2002;291(3):628–634. doi: 10.1006/bbrc.2002.6504. [DOI] [PubMed] [Google Scholar]
- 80.Collins NB, Wilson JB, Bush T, et al. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood. 2009;113(10):2181–2190. doi: 10.1182/blood-2008-05-154294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yagasaki H, Adachi D, Oda T, et al. A cytoplasmic serine protein kinase binds and may regulate the Fanconi anemia protein FANCA. Blood. 2001;98(13):3650–3657. doi: 10.1182/blood.v98.13.3650. [DOI] [PubMed] [Google Scholar]
- 82.Otsuki T, Young DB, Sasaki DT, et al. Fanconi anemia protein complex is a novel target of the IKK signalsome. Journal of Cellular Biochemistry. 2002;86(4):613–623. doi: 10.1002/jcb.10270. [DOI] [PubMed] [Google Scholar]
- 83.Mi J, Qiao F, Wilson JB, et al. FANCG is phosphorylated at serines 383 and 387 during mitosis. Molecular and Cellular Biology. 2004;24(19):8576–8585. doi: 10.1128/MCB.24.19.8576-8585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Qiao F, Mi J, Wilson JB, et al. Phosphorylation of Fanconi anemia (FA) complementation group G protein, FANCG, at serine 7 is important for function of the FA pathway. Journal of Biological Chemistry. 2004;279(44):46035–46045. doi: 10.1074/jbc.M408323200. [DOI] [PubMed] [Google Scholar]
- 85.Futaki M, Watanabe S, Kajigaya S, Liu JM. Fanconi anemia protein, FANCG, is a phosphoprotein and is upregulated with FANCA after TNF-α treatment. Biochemical and Biophysical Research Communications. 2001;281(2):347–351. doi: 10.1006/bbrc.2001.4359. [DOI] [PubMed] [Google Scholar]
- 86.Kupfer GM, Yamashita T, Naf D, Suliman A, Asano S, D’Andrea AD. The Fanconi anemia polypeptide, FAC, binds to the cyclin-dependent kinase, cdc2. Blood. 1997;90(3):1047–1054. [PubMed] [Google Scholar]
- 87.Taniguchi T, Garcia-Higuera I, Xu B, et al. Convergence of the Fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109(4):459–472. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
- 88.Ho GPH, Margossian S, Taniguchi T, D’Andrea AD. Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Molecular and Cellular Biology. 2006;26(18):7005–7015. doi: 10.1128/MCB.02018-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhi G, Wilson JB, Chen X, et al. Fanconi anemia complementation group FANCD2 protein serine 331 phosphorylation is important for Fanconi anemia pathway function and BRCA2 interaction. Cancer Research. 2009;69(22):8775–8783. doi: 10.1158/0008-5472.CAN-09-2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage responsey. Genes and Development. 2004;18(16):1958–1963. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ishiai M, Kitao H, Smogorzewska A, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nature Structural and Molecular Biology. 2008;15(11):1138–1146. doi: 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D’Andrea AD. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Molecular and Cellular Biology. 2007;27(8):3098–3108. doi: 10.1128/MCB.02357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim JM, Kee Y, Gurtan A, D’Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111(10):5215–5222. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brodeur I, Goulet I, Tremblay CS, et al. Regulation of the Fanconi anemia group C protein through proteolytic modification. Journal of Biological Chemistry. 2004;279(6):4713–4720. doi: 10.1074/jbc.M301291200. [DOI] [PubMed] [Google Scholar]
- 95.Park SJ, Beck BD, Saadatzadeh MR, Haneline LS, Clapp DW, Lee SH. Fanconi anemia D2 protein is an apoptotic target mediated by caspases. Journal of Cellular Biochemistry. 2011;112(9):2383–2391. doi: 10.1002/jcb.23161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nordenson I. Effect of oxide dismutase and catalase on spontaneously occurring chromosome breaks in patients with fanconi’s anemia. Hereditas. 1977;86(2):147–150. doi: 10.1111/j.1601-5223.1977.tb01223.x. [DOI] [PubMed] [Google Scholar]
- 97.Joenje H, Arwert F, Eriksson AW. Oxygen-dependence of chromosomal aberrations in fanconi’s anaemia. Nature. 1981;290(5802):142–143. doi: 10.1038/290142a0. [DOI] [PubMed] [Google Scholar]
- 98.Gille JJP, Wortelboer HM, Joenje H. Antioxidant status of Fanconi anemia fibroblasts. Human Genetics. 1987;77(1):28–31. doi: 10.1007/BF00284708. [DOI] [PubMed] [Google Scholar]
- 99.Korkina LG, Samochatova EV, Maschan AA, Suslova TB, Cheremisina ZP, Afanas’ev IB. Release of active oxygen radicals by leukocytes of Fanconi anemia patients. Journal of Leukocyte Biology. 1992;52(3):357–362. doi: 10.1002/jlb.52.3.357. [DOI] [PubMed] [Google Scholar]
- 100.Scarpa M, Rigo A, Momo F, Isacchi G, Novelli G, Dallapiccola B. Increased rate of superoxide ion generation in Fanconi anemia erythrocytes. Biochemical and Biophysical Research Communications. 1985;130(1):127–132. doi: 10.1016/0006-291x(85)90391-2. [DOI] [PubMed] [Google Scholar]
- 101.Degan P, Bonassi S, De Caterina M, et al. In vivo accumulation of 8-hydroxy-2’-deoxyguanosine in DNA correlates with release of reactive oxygen species in Fanconi’s anaemia families. Carcinogenesis. 1995;16(4):735–741. doi: 10.1093/carcin/16.4.735. [DOI] [PubMed] [Google Scholar]
- 102.Ruppitsch W, Meißlitzer C, Hirsch-Kauffmann M, Schweiger M. Overexpression of thioredoxin in Fanconi anemia fibroblasts prevents the cytotoxic and DNA damaging effect of mitomycin C and diepoxybutane. Febs Letters. 1998;422(1):99–102. doi: 10.1016/s0014-5793(97)01608-6. [DOI] [PubMed] [Google Scholar]
- 103.Nagasawa H, Little JB. Suppression of cytotoxic effect of mitomycin-C by superoxide dismutase in Fanconi’s anemia and dyskeratosis congenita fibroblasts. Carcinogenesis. 1983;4(7):795–799. doi: 10.1093/carcin/4.7.795. [DOI] [PubMed] [Google Scholar]
- 104.Dallapiccola B, Porfirio B, Mokini V, Alimena G, Isacchi G, Gandini E. Effect of oxidants and antioxidants on chromosomal breakage in Fanconi anemia lymphocytes. Human Genetics. 1985;69(1):62–65. doi: 10.1007/BF00295530. [DOI] [PubMed] [Google Scholar]
- 105.Ruppitsch W, Meißlitzer C, Weirich-Schwaiger H, et al. The role of oxygen metabolism for the pathological phenotype of Fanconi anemia. Human Genetics. 1997;99(6):710–719. doi: 10.1007/s004390050437. [DOI] [PubMed] [Google Scholar]
- 106.Clarke AA, Philpott NJ, Gordon-Smith EC, Rutherford TR. The sensitivity of Fanconi anaemia group C cells to apoptosis induced by mitomycin C is due to oxygen radical generation, not DNA crosslinking. British Journal of Haematology. 1997;96(2):240–247. doi: 10.1046/j.1365-2141.1997.d01-2023.x. [DOI] [PubMed] [Google Scholar]
- 107.Pearl-Yafe M, Halperin D, Halevy A, Kalir H, Bielorai B, Fabian I. An oxidative mechanism of interferon induced priming of the FAS pathway in Fanconi anemia cells. Biochemical Pharmacology. 2003;65(5):833–842. doi: 10.1016/s0006-2952(02)01620-9. [DOI] [PubMed] [Google Scholar]
- 108.Hadjur S, Jirik FR. Increased sensitivity of FANCC-deficient hematopoietic cells to nitric oxide and evidence that this species mediates growth inhibition by cytokines. Blood. 2003;101(10):3877–3884. doi: 10.1182/blood-2002-10-3147. [DOI] [PubMed] [Google Scholar]
- 109.Saadatzadeh MR, Bijangi-Vishehsaraei K, Hong P, Bergmann H, Haneline LS. Oxidant hypersensitivity of Fanconi anemia type C-deficient cells is dependent on a redox-regulated apoptotic pathway. Journal of Biological Chemistry. 2004;279(16):16805–16812. doi: 10.1074/jbc.M313721200. [DOI] [PubMed] [Google Scholar]
- 110.Bijangi-Vishehsaraei K, Saadatzadeh MR, Werne A, et al. Enhanced TNF-α-induced apoptosis in Fanconi anemia type C-deficient cells is dependent on apoptosis signal-regulating kinase 1. Blood. 2005;106(13):4124–4130. doi: 10.1182/blood-2005-05-2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hadjur S, Ung K, Wadsworth L, et al. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding FANCC and Cu/Zn superoxide dismutase. Blood. 2001;98(4):1003–1011. doi: 10.1182/blood.v98.4.1003. [DOI] [PubMed] [Google Scholar]
- 112.Reuter TY, Medhurst AL, Waisfisz Q, et al. Yeast two-hybrid screens imply involvement of Fanconi anemia proteins in transcription regulation, cell signaling, oxidative metabolism, and cellular transport. Experimental Cell Research. 2003;289(2):211–221. doi: 10.1016/s0014-4827(03)00261-1. [DOI] [PubMed] [Google Scholar]
- 113.Cumming RC, Lightfoot J, Beard K, Youssoufian H, O’brien PJ, Buchwald M. Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nature Medicine. 2001;7(7):814–820. doi: 10.1038/89937. [DOI] [PubMed] [Google Scholar]
- 114.Kruyt FAE, Hoshino T, Liu JM, Joseph P, Jaiswal AK, Youssoufian H. Abnormal microsomal detoxification implicated in Fanconi anemia group C by interaction of the FAC protein with ANDPH cytochrome P450 reductase. Blood. 1998;92(9):3050–3056. [PubMed] [Google Scholar]
- 115.Futaki M, Igarashi T, Watanabe S, et al. The FANCG Fanconi anemia protein interacts with CYP2E1: possible role in protection against oxidative DNA damage. Carcinogenesis. 2002;23(1):67–72. doi: 10.1093/carcin/23.1.67. [DOI] [PubMed] [Google Scholar]
- 116.Rathbun RK, Faulkner GR, Ostroski MH, et al. Inactivation of the Fanconi anemia group C gene augments interferon-γ- induced apoptotic responses in hematopoietic cells. Blood. 1997;90(3):974–985. [PubMed] [Google Scholar]
- 117.Whitney MA, Royle G, Low MJ, et al. Germ cell defects and hematopoietic hypersensitivity to γ-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88(1):49–58. [PubMed] [Google Scholar]
- 118.Segal GM, Magenis RE, Brown M, et al. Repression of Fanconi anemia gene (FACC) expression inhibits growth of hematopoietic progenitor cells. Journal of Clinical Investigation. 1994;94(2):846–852. doi: 10.1172/JCI117405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cumming RC, Liu JM, Youssoufian H, Buchwald M. Suppression of apoptosis in hematopoietic factor-dependent progenitor cell lines by expression of the FAC gene. Blood. 1996;88(12):4558–4567. [PubMed] [Google Scholar]
- 120.Kruyt FAE, Dijkmans LM, Van Den Berg TK, Joenje H. Fanconi anemia genes act to suppress a cross-linker-inducible p53-independent apoptosis pathway in lymphoblastoid cell lines. Blood. 1996;87(3):938–948. [PubMed] [Google Scholar]
- 121.Marathi UK, Howell SR, Ashmun RA, Brent TP. The Fanconi anemia complementation group C protein corrects DNA interstrand cross-link-specific apoptosis in HSC536N cells. Blood. 1996;88(6):2298–2305. [PubMed] [Google Scholar]
- 122.Haneline LS, Broxmeyer HE, Cooper S, et al. Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from FAC(-/-) mice. Blood. 1998;91(11):4092–4098. [PubMed] [Google Scholar]
- 123.Rosselli F, Sanceau J, Wietzerbin J, Moustacchi E. Abnormal lymphokine production: a novel feature of the genetic disease Fanconi anemia. I. Involvement of interleukin-6. Human Genetics. 1992;89(1):42–48. doi: 10.1007/BF00207040. [DOI] [PubMed] [Google Scholar]
- 124.Schultz JC, Shahidi NT. Tumor necrosis factor-α overproduction in Fanconi’s anemia. American Journal of Hematology. 1993;42(2):196–201. doi: 10.1002/ajh.2830420211. [DOI] [PubMed] [Google Scholar]
- 125.Rosselli F, Sanceau J, Gluckman E, Wietzerbin J, Moustacchi E. Abnormal lymphokine production: a novel feature of the genetic disease Fanconi anemia. II. in vitro and in vivo spontaneous overproduction of tumor necrosis factor α . Blood. 1994;83(5):1216–1225. [PubMed] [Google Scholar]
- 126.Wang J, Otsuki T, Youssoufian H, et al. Overexpression of the Fanconi anemia group C gene (FAC) protects hematopoietic progenitors from death induced by Fas-mediated apoptosis. Cancer Research. 1998;58(16):3538–3541. [PubMed] [Google Scholar]
- 127.Koh PS, Hughes GC, Faulkner GR, Keeble WW, Bagby GC. The Fanconi anemia group C gene product modulates apoptotic responses to tumor necrosis factor-α and fas ligand but does not suppress expression of receptors of the tumor necrosis factor receptor superfamily. Experimental Hematology. 1999;27(1):1–8. doi: 10.1016/s0301-472x(98)00064-2. [DOI] [PubMed] [Google Scholar]
- 128.Kurre P, Anandakumar P, Grompe M, Kiem HP. In vivo administration of interferon gamma does not cause marrow aplasia in mice with a targeted disruption of FANCC. Experimental Hematology. 2002;30(11):1257–1262. doi: 10.1016/s0301-472x(02)00932-3. [DOI] [PubMed] [Google Scholar]
- 129.Li X, Yang Y, Yuan J, et al. Continuous in vivo infusion of interferon-gamma (IFN-γ) preferentially reduces myeloid progenitor numbers and enhances engraftment of syngeneic wild-type cells in FANCC-/- mice. Blood. 2004;104(4):1204–1209. doi: 10.1182/blood-2004-03-1094. [DOI] [PubMed] [Google Scholar]
- 130.Li J, Sejas DP, Zhang X, et al. Tnf-α induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. Journal of Clinical Investigation. 2007;117(11):3283–3295. doi: 10.1172/JCI31772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li Y, Youssoufian H. MxA overexpression reveals a common genetic link in four Fanconi anemia complementation groups. Journal of Clinical Investigation. 1997;100(11):2873–2880. doi: 10.1172/JCI119836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fagerlie SR, Diaz J, Christianson TA, et al. Functional correction of FA-C cells with FANCC suppresses the expression of interferon γ-inducible genes. Blood. 2001;97(10):3017–3024. doi: 10.1182/blood.v97.10.3017. [DOI] [PubMed] [Google Scholar]
- 133.Pang Q, Fagerlie S, Christianson TA, et al. The Fanconi anemia protein FANCC binds to and facilitates the activation of STAT1 by gamma interferon and hematopoietic growth factors. Molecular and Cellular Biology. 2000;20(13):4724–4735. doi: 10.1128/mcb.20.13.4724-4735.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fagerlie SR, Koretsky T, Torok-Storb B, Bagby GC. Impaired type I IFN-induced JAK/STAT signaling in FA-C cells and abnormal CD4+ th cell subsets in FANCC-/- mice. Journal of Immunology. 2004;173(6):3863–3870. doi: 10.4049/jimmunol.173.6.3863. [DOI] [PubMed] [Google Scholar]
- 135.Castello G, Gallo C, Napolitano M, Ascierto PA. Immunological phenotype analysis of patients with Fanconi’s anaemia and their family members. Acta Haematologica. 1998;100(1):39–43. doi: 10.1159/000040861. [DOI] [PubMed] [Google Scholar]
- 136.Kaplan MJ, Sabio H, Wanebo HJ, Cantrell RW. Squamous cell carcinoma in the immunosuppressed patient: fanconi’s anemia. Laryngoscope. 1985;95(7 I):771–775. [PubMed] [Google Scholar]
- 137.Froom P, Aghai E, Dobinsky JB, Quitt M, Lahat N. Reduced natural killer activity in patients with Fanconi's anemia and in family members. Leukemia Research. 1987;11(2):197–199. doi: 10.1016/0145-2126(87)90026-9. [DOI] [PubMed] [Google Scholar]
- 138.Standen GR, HugHES IA, Geddes AD, Jones BM, Wardrop CAJ. Myelodysplastic syndrome with trisomy 8 in an adolescent with Fanconi anaemia and selective IgA deficiency. American Journal of Hematology. 1989;31(4):280–283. doi: 10.1002/ajh.2830310413. [DOI] [PubMed] [Google Scholar]
- 139.Johansson E, Niemi KM, Siimes M, Pyrhonen S. Fanconi’s anemia. tumor-like warts, hyperpigmentation associated with deranged keratinocytes, and depressed cell-mediated immunity. Archives of Dermatology. 1982;118(4):249–252. doi: 10.1001/archderm.118.4.249. [DOI] [PubMed] [Google Scholar]
- 140.Pang Q, Keeble W, Christianson TA, Faulkner GR, Bagby GC. FANCC interacts with Hsp70 to protect hematopoietic cells from IFN-γ/TNF-α-mediated cytotoxicity. EMBO Journal. 2001;20(16):4478–4489. doi: 10.1093/emboj/20.16.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Melville MW, Tan SL, Wambach M, Song J, Morimoto RI, Katze MG. The cellular inhibitor of the PKR protein kinase, p58(IPK), is an influenza virus-activated co-chaperone that modulates heat shock protein 70 activity. Journal of Biological Chemistry. 1999;274(6):3797–3803. doi: 10.1074/jbc.274.6.3797. [DOI] [PubMed] [Google Scholar]
- 142.Pang Q, Keeble W, Diaz J, et al. Role of double-stranded RNA-dependent protein kinase in mediating hypersensitivity of Fanconi anemia complementation group C cells to interferon γ, tumor necrosis factor-α, and double-stranded RNA. Blood. 2001;97(6):1644–1652. doi: 10.1182/blood.v97.6.1644. [DOI] [PubMed] [Google Scholar]
- 143.Pang Q, Christianson TA, Keeble W, Koretsky T, Bagby GC. The anti-apoptotic function of Hsp70 in the interferon-inducible double-stranded RNA-dependent protein kinase-mediated death signaling pathway requires the Fanconi anemia protein, FANCC. Journal of Biological Chemistry. 2002;277(51):49638–49643. doi: 10.1074/jbc.M209386200. [DOI] [PubMed] [Google Scholar]
- 144.Shifera AS. Protein-protein interactions involving IKKγ (NEMO) that promote the activation of NF-κB. Journal of Cellular Physiology. 2010;223(3):558–561. doi: 10.1002/jcp.22105. [DOI] [PubMed] [Google Scholar]
- 145.Schmid JA, Birbach A. Iκb kinase β (IKKβ/IKK2/ikbkb)-a key molecule in signaling to the transcription factor NF-κB. Cytokine and Growth factor Reviews. 2008;19(2):157–165. doi: 10.1016/j.cytogfr.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 146.Schmidt-Supprian M, Tian J, Grant EP, et al. Differential dependence of CD4+CD25+ regulatory and natural killer-like T cells on signals leading to NF-κB activation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(13):4566–4571. doi: 10.1073/pnas.0400885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hoatlin ME, Yu Z, Ball H, et al. A novel BTB/POZ transcriptional repressor protein interacts with the Fanconi anemia group C protein and PLZF. Blood. 1999;94(11):3737–3747. [PubMed] [Google Scholar]
- 148.Miaw SC, Choi A, Yu E, Kishikawa H, Ho IC. ROG, repressor of GATA, regulates the expression of cytokine genes. Immunity. 2000;12(3):323–333. doi: 10.1016/s1074-7613(00)80185-5. [DOI] [PubMed] [Google Scholar]
- 149.Piazza F, Costoya JA, Merghoub T, Hobbs RM, Pandolfi PP. Disruption of PLZP in mice leads to increased T-lymphocyte proliferation, cytokine production, and altered hematopoietic stem cell homeostasis. Molecular and Cellular Biology. 2004;24(23):10456–10469. doi: 10.1128/MCB.24.23.10456-10469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lin WC, Lai CH, Tang CJC, Huang CJ, Tang TK. Identification and gene structure of a novel human PLZF-related transcription factor gene, TZFP. Biochemical and Biophysical Research Communications. 1999;264(3):789–795. doi: 10.1006/bbrc.1999.1594. [DOI] [PubMed] [Google Scholar]
- 151.Dai MS, Chevallier N, Stone S, et al. The effects of the Fanconi anemia zinc finger (FAZF) on cell cycle, apoptosis, and proliferation are differentiation stage-specific. Journal of Biological Chemistry. 2002;277(29):26327–26334. doi: 10.1074/jbc.M201834200. [DOI] [PubMed] [Google Scholar]
- 152.Li X, Plett PA, Yang Y, et al. Fanconi anemia type C-deficient hematopoietic stem/progenitor cells exhibit aberrant cell cycle control. Blood. 2003;102(6):2081–2084. doi: 10.1182/blood-2003-02-0536. [DOI] [PubMed] [Google Scholar]
- 153.Tremblay CS, Huard CC, Huang FF, et al. The Fanconi anemia core complex acts as a transcriptional co-regulator in hairy enhancer of split 1 signaling. Journal of Biological Chemistry. 2009;284(20):13384–13395. doi: 10.1074/jbc.M807921200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Otsuki T, Furukawa Y, Ikeda K, et al. Fanconi anemia protein, FANCA, associates with BRG1, a component of the human SWI/SNF complex. Human Molecular Genetics. 2001;10(23):2651–2660. doi: 10.1093/hmg/10.23.2651. [DOI] [PubMed] [Google Scholar]
- 155.Trotter KW, Archer TK. The BRG1 transcriptional coregulator. Nuclear Receptor Signaling. 2008;6:p. e004. doi: 10.1621/nrs.06004. [DOI] [PMC free article] [PubMed] [Google Scholar]