

Abstract
Human familial hypertrophic cardiomyopathy is the most common Mendelian cardiovascular disease worldwide. Among the most severe presentations of the disease are those in families heterozygous for the mutation R403Q in β-cardiac myosin. Mice heterozygous for this mutation in the α-cardiac myosin isoform display typical familial hypertrophic cardiomyopathy pathology. Here, we study cardiomyocytes from heterozygous 403/+ mice. The effects of the R403Q mutation on force-generating capabilities and dynamics of cardiomyocytes were investigated using a dual carbon nanofiber technique to measure single-cell parameters. We demonstrate the Frank-Starling effect at the single cardiomyocyte level by showing that cell stretch causes an increase in amplitude of contraction. Mutant 403/+ cardiomyocytes exhibit higher end-diastolic and end-systolic stiffness than +/+ cardiomyocytes, whereas active force generation capabilities remain unchanged. Additionally, 403/+ cardiomyocytes show slowed relaxation dynamics. These phenotypes are consistent with increased end-diastolic and end-systolic chamber elastance, as well as diastolic dysfunction seen at the level of the whole heart. Our results show that these functional effects of the R403Q mutation are cell-intrinsic, a property that may be a general phenomenon in familial hypertrophic cardiomyopathy.
Introduction
Familial hypertrophic cardiomyopathy (HCM) is the most common Mendelian cardiovascular disease, affecting 1 in 500 people worldwide (1,2). HCM is defined as hypertrophy of the heart that is unexplained by mechanical factors such as hypertension or valve disease. This abnormal hypertrophy is often accompanied by diminished cavity volume and impaired relaxation (3). Clinical outcomes of HCM are diverse, ranging from mild adjustments in lifestyle to severe heart failure and early-onset sudden cardiac death (2,4,5). To date, hundreds of mutations in multiple cardiomyocyte sarcomeric genes have been identified (6). These mutations cause alterations in proteins including those essential for force production, namely β-cardiac myosin and actin, regulatory elements like tropomyosin and troponin, and those primarily responsible for maintaining stability of the sarcomere, such as myosin-binding protein C.
The HCM mutation with one of the highest penetrance and most severe clinical phenotypes is a missense mutation in the β-cardiac myosin heavy chain gene that results in an amino acid change from arginine to glutamine at position 403 (R403Q). This mutation occurs at a highly conserved residue (7) in close proximity to the actin-binding interface of the myosin motor domain (8). Patients with the R403Q mutation have a high incidence of morbidity and early mortality (4).
Although some information has been obtained on the functional effects of the R403Q mutation on contractile function in purified myosin (9–19), at the tissue level (20–22), as well as at the whole heart level (23–26), only one study so far has looked at contractility in R403Q heart muscle cells (27) (cardiomyocytes). Investigating the effects of the mutation at the single cardiomyocyte level has the advantage of allowing us to determine which effects are cell-intrinsic (i.e., a property of the cell independent of its extracellular environment or interactions between the cells as part of the native tissue) and to tease apart the contributions of intracellular sarcomeric proteins and extracellular fibrosis or neurohormonal influence to the disease. Additionally, the use of intact cardiomyocytes allows us to study the interaction of the sarcomeric proteins in their native environment where they are highly ordered, and these studies can be compared to in vitro studies with purified proteins (28).
Although one ideally wants to study human cardiomyocytes, several limitations exist in their availability. Human biopsy samples, particularly those from HCM patients, are difficult to obtain and additionally provide very low yields (no more than 20%) of viable cardiomyocytes (29,30). Although technologies for generating differentiated cardiomyocytes from either human embryonic stem cells (31) or induced-pluripotent stem cells (32,33) have been established, they are still in their infancy. More importantly, these cardiomyocyte differentiation technologies, at least for now, are only able to generate cardiomyocytes that resemble the immature state and fail to mature them to an adult cardiomyocyte phenotype (34). Fortunately, a murine model is available for the R403Q mutation, and the model does recapitulate the HCM clinical phenotype (26). Hence, in this study we turned to this murine model of HCM from which fully differentiated adult cardiomyocytes are readily available.
A mouse model of the HCM-causing R403Q mutation was established by Geisterfer-Lowrance et al. (26). It is important to emphasize that although the mutation is in the predominant mouse α-cardiac myosin heavy chain isoform, these mice do display typical HCM pathology. This is perhaps not surprising, given that the mouse α-cardiac myosin heavy chain is highly homologous to human β-cardiac myosin heavy chain (92% identical overall, 100% identical for 30 amino acids flanking residue 403). Humans with cardiomyopathies are generally heterozygous for the mutation, and homozygous 403/403 mice do not survive past 1 week. Adult heterozygous mice (403/+, >15 weeks old) exhibit cardiomyocyte hypertrophy, cellular disarray and fibrosis (26), myofibrillar disarray (27), as well as diastolic dysfunction (23–25). These phenotypes are not usually seen in wild-type (+/+) mice. Thus, this murine model of HCM can provide useful insights into the effects of the R403Q mutation on sarcomere function.
In a previously published study on R403Q cardiomyocytes (27), the authors examined the unloaded contractile properties of single cardiomyocytes from adult heterozygous 403/+ mice that were placed on a regular glass coverslip surface. We note that the close proximity of the cardiomyocytes to the coverslip could potentially interfere with contractility due to friction from the surface. Cardiomyocytes can also adhere to glass surfaces with time, and hence the contact of the cardiomyocytes with the coverslip at the start of the experiment is variable, and can also vary during the course of the experiment. Additionally, cardiomyocytes contract against a variety of loads in vivo and hence the absence of any loading on the cardiomyocytes in the experiment is clearly unphysiological. To provide further insight into the behavior of cardiomyocytes under more physiological conditions, we used a dual carbon nanofiber setup (35) to suspend single cardiomyocytes in solution, away from the coverslip surface to minimize any interfering friction during cell contraction. We compared the forces generated by cardiomyocytes from +/+ and 403/+ mice, as well as the dynamics of contraction and relaxation. Most importantly, our setup allowed us to apply preload (stretch) to the cardiomyocytes by moving the CFs apart, which enabled us to determine the stiffness or elasticity of the cardiomyocyte both while relaxed (diastole) and at maximal contraction (systole). Application of preload also allows us to probe the Frank-Starling effect at a single-cell level. The Frank-Starling effect (36,37) is the ability of the heart to increase its force of contraction and hence stroke volume in response to an increase in the volume of blood filling the heart; at the single-cell level this corresponds to an increase in active contraction force generation with increased preload (stretch).
We find that 403/+ cardiomyocytes have increased end-diastolic as well as end-systolic stiffness compared to +/+ cardiomyocytes. However, the ability to generate increased active force with increasing preload remains unchanged. 403/+ cardiomyocytes also showed slowed relaxation dynamics compared to +/+ cardiomyocytes. These findings at the single-cell level are consistent with increased myocardial elastance, both in diastole and systole, as well as diastolic dysfunction (23–25) at the level of the whole heart in 403/+ mice. This suggests that these functional effects of the R403Q mutation are cell-intrinsic, a property that may be a general phenomenon in HCM.
Methods and Materials
Animals
The mouse model of familial HCM used in this study contained an Arg to Gln missense mutation at residue 403 of the mouse α-cardiac myosin heavy chain (MHC) (26). Mouse genotypes were determined by restriction enzyme digestion of polymerase chain reaction-amplified tail DNA as previously described (26). Heterozygous male α-MHC403/+ and wild-type male α-MHC+/+ littermates were used for the study. All mice were maintained according to protocols approved by the Administrative Panel on Laboratory Animal Care of Stanford University.
Cardiomyocyte isolation
Mice, 20–25 weeks old, were anesthetized with isoflurane (2%) and given an intraperitoneal injection of heparin (200 units/mouse). Lack of withdraw reflex with toe pinch was confirmed before proceeding. Hearts were excised and promptly cannulated (within 70–90 s) via the aorta and Langendorff perfused at a rate of 4 ml/min. Hearts were first perfused with main solution (120.4 mM NaCl, 14.7 mM KCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 1.2 mM MgSO4, 4.6 mM NaHCO3, 10 mM HEPES, 30 mM taurine, 10 mM 2,3-butanedione monoxime and 5.5 mM glucose, pH 7.0) for 4 min, followed by collagenase solution (main solution + 600 units/ml Collagenase Type 2, Worthington Biochemical, Lakewood, NJ) for 2 min, and finally Ca2+-collagenase solution (collagenase solution + 50 μM CaCl2) for 6 min. During the last 1–2 min of perfusion, the Ca2+-collagenase solution was collected as it dripped off the heart, which was pale and flaccid by the end of perfusion.
Ventricles were harvested and placed into the collected Ca2+-collagenase solution. The tissue was cut into small pieces with scissors and then further dissociated by gentle pipetting through transfer pipettes. Stop solution 1 (main solution + 10% fetal bovine serum (FBS) + 12.5 μM CaCl2) was added and the dissociated tissue was then poured through a mesh filter into a 15 ml conical tube. Cells were allowed to sit for 5 min to allow myocytes to settle by gravity before centrifuging at ∼25 × g (400 rpm in Beckman Coulter Allegra 6R centrifuge with GH-3.8 rotor) for 3 min. The supernatant was decanted and the cells were gently resuspended in stop solution 2 (main solution + 10% FBS + 100 μM CaCl2). Settling, centrifugation, and resuspension steps were repeated with stop solutions 3 and 4 (main solution + 10% FBS + 400 or 900 μM CaCl2, respectively), and finally with contractility buffer (137 mM NaCl, 20 mM HEPES, 15 mM glucose, 5.4 mM KCl, 1.3 mM MgCl2, 1.2 mM NaH2PO4, 1 mM CaCl2). Isolations consistently yielded >40% rod-shaped cardiomyocytes with clear sarcomeric banding patterns. Experiments were conducted within 6 h of excision of hearts from mice.
Cell attachment and contractility measurements
All measurements were performed on a modified Myocyte Contractility System (IonOptix, Milton, MA). An add-on to the system (similar to that described in Iribe et al. (35)) allowed bidirectional manipulation of two carbon fibers (CFs) mounted in glass capillaries using hydraulic manipulators (Siskiyou, Grants Pass, OR) and piezo translators (Physik Instruments, Karlsruhe, Germany). CFs (12–14 μm in diameter) were generous gifts from Christian Bollensdorf and Peter Kohl at Imperial College London.
All contractility measurements were performed at 37°C in contractility buffer. Cells were placed at extremely low concentrations in a voltage-stimulation chamber with poly-HEMA (2-hydroxyethyl methacrylate, Sigma-Aldrich, St. Loius, MO) coated coverslips to prevent adhesion to the coverslip surface. Criteria used for selection of cardiomyocytes for experiments were i), rod-shaped, ii), clear and organized sarcomeres, iii), sarcomere lengths in the range of 1.6–1.85 μm, iv), bleb- and ruffle-free membranes, v), consistent ∼4–5% sarcomere shortening upon voltage stimulation before attachment of CFs, and vi), absence of spontaneous contractions when unstimulated, throughout the course of the experiment. These cardiomyocytes would fall primarily under “type I” according to the classification defined by Kim et al. (27). Non-type I cardiomyocytes frequently exhibited spontaneous contractions in the absence of stimulation, resulting in data that were unreliable. The selected cardiomyocyte was subject to field voltage stimulation (paced) at 1 Hz while the two CF tips were lightly pressed down in succession, at a distance of 55–60 μm from each other. Initial adhesion was verified by lifting the CFs vertically; CFs with cardiomyocyte attached were subsequently returned to the surface, and pacing continued for at least 3 min to allow enhancement of adhesion. Finally, cardiomyocytes were lifted ∼5–10 μm off the coverslip surface.
While the cell was suspended between the two CFs away from the coverslip surface and before application of any preload, measurements of resting length parameters were taken. Preload was then applied by axial stretch of the cell during diastole, in 2–4 μm step increments. The positions of the CFs were controlled by piezo translators and custom MATLAB (The MathWorks, Natick, MA) software, courtesy of Peter Lee from the Kohl lab. The cell was returned to resting length between each level of preload. Upon completion of measurements, CFs were lifted out of the solution, which caused the cell or any cell debris to be pulled off the CFs by surface tension. CFs could then be reused.
Data analysis
Initial analysis of the contractility data was performed using IonWizard software (IonOptix LLC) and MATLAB. For each cell, at least three contraction traces per level of preload were analyzed individually, and these triplicate values were then averaged to obtain parameter values for the cell. Mean ± SE was then calculated across all cells for each condition. Forces were calculated from the average stiffness of the two CFs multiplied by CF bending. CF stiffness (0.05−0.20 μN μm−1) was measured using a force transducer system, courtesy of C. Bollensdorf. CF bending is the distance between the CF holders minus the distance between the CF tips. End-diastolic (passive) and end-systolic (total) forces were obtained for each preload level and normalized to the cross-sectional area of each cell. The cardiomyocytes were assumed to be ellipsoidal in cross section, with a short-to-long axis ratio of 1:3 (38). End-diastolic force length relation (EDFLR) and end-systolic force length relation (ESFLR) were obtained by performing linear least squares regressions to the end-diastolic and end-systolic points, respectively. Care was taken not to include points beyond cell lengths at which the force-length relationship was no longer linear. Over the length range studied, the assumption was made that the slopes of EDFLR and ESFLR are independent of the mode of contraction (35).
The maximum rates of force development during contraction (dF/dtmax,C) and force reduction during relaxation (dF/dtmax,R) were similarly normalized to the cross-sectional area of each cell. The preload dependence of these rates was obtained by grouping the data into bins of size ∼5% of ESL0, from which errors in rates as well as normalized effective cell length were calculated. The preload dependence of the time taken to return halfway from systole to diastole during relaxation (t50,R) was analyzed in the same way.
Results
Preload application experimental approach
Fig. 1 illustrates the forces measured during different stages of the loading protocol. Force is calculated as the product of the average stiffness of the two CFs (kave) and the total distance that the CFs are bent. At resting length (no preload) and while relaxed, or in diastole (Fig. 1 a), the distance between the two CFs at the piezo translators (initial carbon fiber distance, CFD0) and at the cell-attached ends (initial end-diastolic length, EDL0) are equal. This distance, EDL0 was kept to 55–60 μm to standardize, as far as possible across cells, the number of sarcomeres between the CFs that contribute to their bending. Initial diastolic force is hence zero. At maximal contraction, or in systole (Fig. 1 b), the distance at the CF tips is reduced to ESL0 (initial end-systolic length), and the maximum force generated by the cardiomyocyte at resting length is F = kave(CFD0-ESL0). To apply preload (Fig. 1, c and d), the piezo translators are moved apart to length CFD1. In diastole, (Fig. 1 c) the stretching of compliant elements such as titin (39,40) within the cardiomyocyte exerts a passive force or tension on the CFs, bending the CFs a total distance of (CFD1-EDL1). The passive force in diastole is thus FP = kave(CFD1-EDL1). In systole, the cardiomyocyte exerts an active contractile force, bringing the CF tips to a distance of ESL1. Here, the total force generated by the cardiomyocyte is FT = kave(CFD1-ESL1). The active force generated by the cardiomyocyte is therefore FA = FT−FP. Increasing preload can be applied by further axial stretch of the cardiomyocyte, with returns to resting length between each level of preload.
Figure 1.
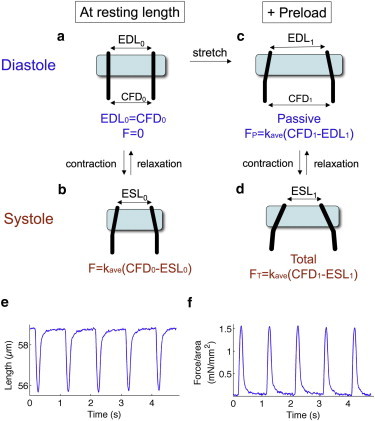
Schematic of forces measured in cardiomyocyte-contractility experiments. (a) At resting length in diastole, the distance between two CFs at piezo translators (CFD0) and at the cell-attached CF tips (initial end-diastolic length, EDL0) are equal. Force is zero. (b) Upon contraction in systole, the distance between CF tips decreases (initial end-systolic length, ESL0). The force is the product of the average stiffness of the CFs (kave) and the distance that the CFs are bent (CFD0-ESL0). (c) Preload is applied by moving the piezo-translator mounted CFs apart to length CFD1. Cell-attached CF tips also move apart but to a lesser extent, to length EDL1. The passive force exerted by the cardiomyocyte is FP = kave(CFD1-EDL1). (d) Contraction under preload moves the CF tips to length ESL1, making the total force exerted FT = kave(CFD1-ESL1). (e) Representative trace of changes in distance between cell-attached CF tips (effective cell length) as the cardiomyocyte contracts at a rate of 1 Hz. (f) Normalization of force to cross-sectional area of the cardiomyocyte gives changes in contractile force per area, from which dynamic parameters can be determined.
A representative trace of changes in distance between cell-attached CF tips (hereafter referred to as effective cell length) as the cardiomyocyte contracts at a rate of 1 Hz is shown in Fig. 1 e. To allow comparison across cells and animals, contractile force is normalized to the cross-sectional area of the cardiomyocyte, which is assumed to be ellipsoidal with a short-to-long axis ratio of 1:3 (38). From the dynamic changes in this normalized contractile force (Fig. 1 f), parameters such as the normalized maximum rate of force development during contraction (dF/dtmax,C/area) and the normalized maximum rate of force reduction during relaxation (dF/dtmax,R/area) can be determined.
At resting length, 403/+ cardiomyocytes have slowed relaxation rates compared to +/+ cardiomyocytes
We began by observing the behavior of wild-type (+/+) cardiomyocytes and cardiomyocytes heterozygous for the hypertrophic cardiomyopathy mutation R403Q (403/+) at resting length, before application of preload (Table 1). Resting cell lengths, sarcomere lengths, and cell widths were similar for both groups. Parameters describing cardiomyocyte contraction, namely the time to systole, the normalized systolic force as well as the normalized maximum rate of force development during contraction (dF/dtmax,C/area) did not differ significantly between +/+ and 403/+ cardiomyocytes. However, for cardiomyocyte relaxation parameters, we observed that the time taken to return halfway from systole to diastole during relaxation (t50,R) as well as the normalized maximum rate of force reduction during relaxation (dF/dtmax,R/area) at resting length were significantly slower in 403/+ cardiomyocytes, indicative of diastolic dysfunction.
Table 1.
Cardiomyocyte behavior at resting length
| +/+ | 403/+ | |
|---|---|---|
| n (cells, animals) | 14, 5 | 15, 6 |
| Resting cell length (μm) | 126 ± 6 | 131 ± 6 |
| Resting sarcomere length (μm) | 1.74 ± 0.02 | 1.68 ± 0.02 |
| Cell width (μm) | 20.7 ± 0.6 | 20.1 ± 0.5 |
| Systolic Parameters | ||
| Time to systole (ms) | 112 ± 6 | 117 ± 5 |
| Systolic force/area (mN/mm2) | 1.33 ± 0.15 | 1.33 ± 0.11 |
| dF/dtmax,C/area (mN/[s mm2]) | 25.6 ± 3.4 | 24.9 ± 2.3 |
| Diastolic Parameters | ||
| t50,R (ms) | 38 ± 3∗ | 72 ± 7∗ |
| dF/dtmax,R/area (mN/[s mm2]) | 16.9 ± 1.9∗ | 11.2 ± 1.3∗ |
Data are shown as mean ± SE.
dF/dtmax,C/area: Maximum rate of force development during contraction normalized to cross-sectional area.
t50,R: Time taken to return halfway from systole to diastole during relaxation.
dF/dtmax,R/area: Maximum rate of force reduction during relaxation normalized to cross-sectional area.
Significantly different between +/+ and 403/+ by two-tailed unpaired t-test, p < 0.05.
403/+ cardiomyocytes have higher end-diastolic and end-systolic stiffness compared to +/+ cardiomyocytes
We next studied the response of the cardiomyocytes to preload. Fig. 2 a shows a representative trace of effective cell length changes as increasing levels of preload were applied. Initially, the cardiomyocyte was at resting length, and transitioned from EDL0 in diastole (blue line and dot, time segment R) to ESL0 in systole (red line and dot, time segment R). Increasing levels of preload was applied from time segments P1 to P4, with corresponding end-diastolic lengths shown in blue and end-systolic lengths in red. The increase in the amplitude of contraction (the difference between end-diastolic and end-systolic lengths) demonstrates the Frank-Starling effect at the level of the single cell.
Figure 2.
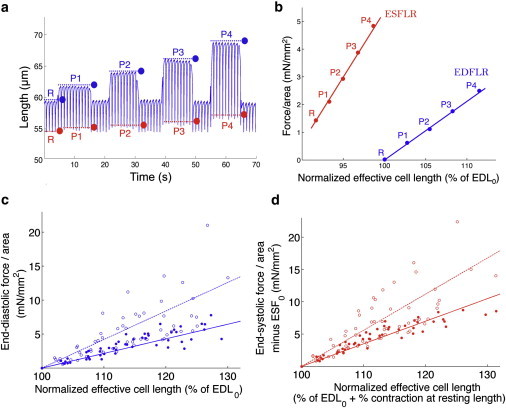
403/+ cardiomyocytes have higher end-diastolic and end-systolic stiffness. (a) Representative data of effective cell length measurements (distance between the CF tips at the cell-attached end) of a cardiomyocyte undergoing voltage stimulation and contraction at 1 Hz, with the application of preload. Initially, the cell is at resting length (time segment R); increasing levels of preload are applied from time segments P1–P4. Dotted lines and dots indicate end-diastolic (blue) and end-systolic (red) lengths, respectively, and correspond to EDL and ESL in Fig. 1. (b) End-diastolic (blue) and end-systolic (red) points at rest (R) and per level of preload (P1–P4) are plotted as normalized force against normalized effective cell length. End-diastolic force-length relation (EDFLR, blue) and end-systolic force-length relation (ESFLR, red) are obtained from linear regressions of end-diastolic and end-systolic points, respectively. (c) EDFLRs of +/+ (solid circles, solid lines) and 403/+ (open circles, dotted lines) cardiomyocytes. The slope of EDFLR for 403/+ cardiomyocytes (0.36 mN/mm2) is steeper than that for +/+ cardiomyocytes (0.22 mN/mm2), indicating that 403/+ cardiomyocytes have higher end-diastolic stiffness. (d) Similarly, ESFLR of 403/+ cardiomyocytes (slope = 0.50 mN/mm2) is steeper than that for +/+ cardiomyocytes (slope = 0.37 mN/mm2), indicating higher end-systolic stiffness.
End-diastolic and end-systolic forces were calculated as described in Fig. 1, normalized to the cardiomyocyte cross-sectional area and plotted against effective cell length normalized to EDL0, yielding the data points in Fig. 2 b (labels in Fig. 2 b correspond to those in Fig. 2 a). The end-diastolic points (blue circles) describe a linear dependence of passive force on cell length, a dependence known as the end-diastolic force-length relation (EDFLR, blue line). The slope of EDFLR is a measure of the passive stiffness or elasticity of the cardiomyocyte (end-diastolic stiffness), a property likely conferred by the presence of compliant elements in the cell that resist stretching. A linear dependence is similarly described by the end-systolic points (red circles), giving the end-systolic force-length relation (ESFLR, red line), which indicates the total force the cardiomyocyte is able to produce at each level of preload. The slope of ESFLR is a measure of the stiffness of the cardiomyocyte at maximal contraction (end-systolic stiffness).
The end-diastolic points and average EDFLR slopes (Fig. 2 c, Table 2) indicate that 403/+ cardiomyocytes (open circles, dotted line; slope = 0.36 mN/mm2) have significantly higher end-diastolic stiffness compared to +/+ cardiomyocytes (solid circles, solid line; slope = 0.22 mN/mm2). The same trend is seen for end-systolic points and average ESFLR slopes (Fig. 2 d, Table 2; 403/+ slope = 0.50 mN/mm2; +/+ slope = 0.37 mN/mm2), indicating higher end-systolic stiffness of 403/+ cardiomyocytes.
Table 2.
Measures of passive, total, and active force
| +/+ | 403/+ | |
|---|---|---|
| Passive force, FP | ||
| EDFLR slope (mN/mm2) | 0.22 ± 0.02∗ | 0.36 ± 0.04∗ |
| Total force, FT | ||
| ESFLR slope (mN/mm2) | 0.37 ± 0.03∗ | 0.50 ± 0.05∗ |
| Active force, FA | ||
| ESFLR – EDFLR slope (mN/mm2) | 0.15 ± 0.05 | 0.14 ± 0.09 |
| FSG Index | 1.61 ± 0.09 | 1.45 ± 0.06 |
Data are shown as mean ± SE.
Significantly different between +/+ and 403/+ by two-tailed unpaired t-test, p < 0.05.
Active force generation of 403/+ cardiomyocytes is the same as +/+ cardiomyocytes
It is important to note that ESFLR describes preload dependence of total force, which consists of two components—passive force and active force. As mentioned before, passive force is primarily a result of resistance from compliant cellular elements being stretched. Active force, on the other hand, is primarily generated by the Ca2+-regulated and energy-dependent sliding of actin filaments along myosin in the cardiomyocyte sarcomeres, resulting in contraction. It is the increase in amplitude of this active force component with preload that is the Frank-Starling effect (41), and as described earlier is seen in the increase in amplitude of contraction with cell stretch (Fig. 2 a). The contribution of the active force to the total force of contraction may be teased apart in two ways. We can look at the difference in slopes between ESFLR and EDFLR, which gives the dependence of the normalized active force component on preload. This value is 0.15 mN/mm2 for +/+ cardiomyocytes and 0.14 mN/mm2 for 403/+ cardiomyocytes (Table 2). Alternatively, the use of the Frank-Starling Gain (FSG) index, defined as the ratio of the slopes of ESFLR and EDFLR, has been proposed to be useful as a dimensionless descriptor of active force recruitability (41). This index basically expresses the active force component as a multiple of the passive force at each preload. The FSG Index is 1.61 for +/+ cardiomyocytes and 1.45 for 403/+ cardiomyocytes (Table 2). Neither the difference in slopes between ESFLR and EDFLR nor the FSG Index is significantly different between the two groups of cardiomyocytes. Hence, it appears that although 403/+ cardiomyocytes have higher end-diastolic and end-systolic stiffness and greater overall force production, active force generation is unchanged.
Slowed relaxation of 403/+ cardiomyocytes compared to +/+ cardiomyocytes is consistent across preloads studied
We analyzed the dynamics of contraction and relaxation across different preloads. The normalized maximum rate of force development during contraction of 403/+ cardiomyocytes (Fig. 3 a, open circles, dotted line), while possibly reduced by ∼10%, was similar to +/+ cardiomyocytes (Fig. 3 a, solid circles, solid line) across preloads studied. The normalized maximum rate of force reduction during relaxation, however, was consistently slower in 403/+ cardiomyocytes by a significant amount (Fig. 3 b). This slowed relaxation was also evident in a considerably longer t50,R, which is the time taken to return halfway from systole to diastole (Fig. 3 c), for 403/+ cardiomyocytes across studied preloads.
Figure 3.
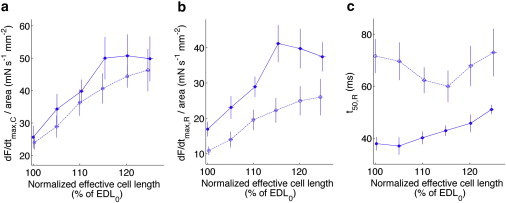
403/+ cardiomyocytes have slowed rates of relaxation compared to +/+ cardiomyocytes across all preloads studied. (a) Normalized maximum rates force development during contraction, dF/dtmax,C/area for +/+ (solid circles, solid lines) and 403/+ (open circles, dotted lines) cardiomyocytes are similar across all preloads. (b) Normalized maximum rates force reduction during relaxation, dF/dtmax,R/area are consistently slower for 403/+ compared to +/+ cardiomyocytes across all preloads. (c) The time taken to return halfway from systole to diastole, t50,R is greater for 403/+ cardiomyocytes than +/+ cardiomyocytes across all preloads.
Discussion
In this work, we used a dual carbon nanofiber technique to investigate the effects of the R403Q familial hypertrophic cardiomyopathy mutation at the single cardiomyocyte level. The use of isolated cardiomyocytes has allowed us to identify the effects of hypertrophic cardiomyopathy that are inherent in the cardiomyocyte itself, as contributions from extracellular fibrosis or neurohormonal influences are absent. To our knowledge, this is the first study to examine in detail the preload-dependent contractile properties of single cardiomyocytes bearing a HCM mutation. Importantly, the application of preload has allowed us to measure the end-diastolic and end-systolic stiffness of cardiomyocytes, and also investigate the Frank-Starling effect at the level of single cells (35,41).
Investigating the effects of the mutation at the single cardiomyocyte level has allowed us to isolate and identify the cell-intrinsic contributions of intracellular factors to disease, which cannot easily be done at the tissue or whole heart level because these are complicated by effects from extracellular factors such as fibrosis and hormones. Studies done on whole mouse hearts either in vivo (23,24,42) or in isolated heart preparations (25) have shown increased passive elastance and end-systolic elastance in 403/+ mice compared to +/+ mice. Similarly, at the tissue level, isolated papillary muscle strips (20) and myocardial strips (22) from 403/+ mice demonstrated increased passive stiffness and increased maximum total force respectively. The increased end-diastolic and end-systolic stiffness that we see in 403/+ single cardiomyocytes are consistent with the characteristics seen at the whole heart and tissue level. Additionally, diastolic dysfunction manifest as slowed maximal rates of pressure reduction and increased time to peak filling is observed in whole 403/+ hearts (23,24,42). The slowed relaxation dynamics that we observe for isolated 403/+ cardiomyocytes are again in agreement with the properties of the whole heart in 403/+ mice. This suggests that these hallmarks of human HCM, namely increased passive and end-systolic chamber elastance and slowed cardiac relaxation (3,43), do not require the presence of extra-cardiomyocyte factors to explain them, and may be cell-intrinsic and inherent in the cardiomyocytes. Cardiac morphology and histological changes that occur with disease progression such as chamber wall thickening and interstitial fibrosis (44) most likely exacerbate these phenotypes.
Multiple studies have been done with rodent and human cardiac myosin containing the R403Q mutation expressed in and purified from a variety of sources such as insect cells (17,18), mammalian cells (16), transgenic mouse models (10,13,14,19), and human soleus and cardiac biopsies (9,15). Here, we consider only the most relevant studies done with α-cardiac myosin purified from R403Q mouse hearts (10,13,14,19). At the single molecule level, the average unitary displacement and force of R403Q myosin purified from homozygous 403/403 mouse hearts are unchanged (19) compared to myosin purified from +/+ mouse hearts. However, the duty ratio of R403Q myosin is increased as a result of ∼1.4–2.2× increased Vmax rates in actin-activated ATPase assays (14,19), and 1.25–1.50× increased actin translocation velocities (vactin) measured by in vitro motility assays (10,13,19). This increased duty ratio is consistent with a ∼2.2× increased average isometric force generation seen in ensemble myosin assays (10,19).
The increase in duty ratio of R403Q myosin would predict that both the active force and total force produced by 403/+ cardiomyocytes are increased. Although we observe an increased total force production, this is due primarily to the increased passive force. No significant increase in preload-dependent active force generation, or the Frank-Starling effect, is seen in 403/+ cardiomyocytes. A reason for this discrepancy may be that the expected increase in active force is balanced by a reduced efficiency of force production due to myofibrillar disarray (27), disorder at the myosin-actin interface (8), or disrupted coordination between the two myosin heads (19) as a result of the R403Q mutation. Although the 403/+ cardiomyocytes used in our assays do not show gross myofibrillar disarray by eye, it is likely that some disorganization is present at the molecular level.
Myofibrillar disarray (27) could also account for the increased cardiomyocyte passive stiffness that we observe in 403/+ cardiomyocytes. The disruption of the regular parallel arrangement and crystalline organization of myofilaments could increase the resistance of compliant elements within the disorganized myofibrils to stretching. An alternative explanation could be the decreased elastance of titin, the primary determinant of passive stiffness, in 403/+ cardiomyocytes. Protein kinase C-α (PKC-α) has been implicated in and shown to be upregulated in hypertrophy (45–48), and phosphorylation of titin's PEVK spring element by PKC-α has been reported to increase titin-based passive tension in myocardial tissue (49,50). Although neither phenomenon has been directly demonstrated in HCM, this PKC-α-dependent increase in titin-based passive tension is a probable mechanism given the myocardial and cardiomyocyte hypertrophy evident in HCM. The exact molecular mechanism by which the R403Q mutation causes an increase in cardiomyocyte passive stiffness remains to be uncovered.
A previous study on the contractile properties of 403/+ cardiomyocytes reported a reduced maximum rate of decrease in cell length during contraction (27) compared to +/+ cardiomyocytes. Although a slight impairment of contraction dynamics in 403/+ cardiomyocytes is observed in our study (Fig. 3 a), it is to a lesser extent than previously reported. It is possible that frictional or weak adhesion effects from the cover slip surface in the earlier study or differences in data normalization could account for the discrepancy. Another possible reason could be the selection of primarily rod-shaped, type I cardiomyocytes for our study, as contraction velocity was less impaired in rod-shaped cardiomyocytes compared to other populations of cardiomyocytes (27).
The diastolic dysfunction that we observe in 403/+ cardiomyocytes (confirming a previous report (27)) may be rationalized by changes in affinity of R403Q cardiac myosin for regulated actin filaments. Although in vitro ATPase and motility assays generally show increased Km and hence decreased affinity of R403Q myosin for unregulated purified actin filaments (19), the use of regulated tropomyosin-troponin-actin filament complexes, better reflecting conditions in the cardiomyocyte, instead gives a decrease in Km and increased apparent affinity of R403Q myosin for these regulated actin filaments (14). This increased affinity of R403Q myosin for regulated actin, in addition to its increased duty ratio, may cause the R403Q myosin population in 403/+ cardiomyocytes to exert a greater drag on the actin filaments during relaxation, resulting in the observed diastolic dysfunction.
Increased expression of the fetal cardiac myosin isoform has been shown to occur in cardiac hypertrophy in both humans and rodents (51). In 403/+ mice, this increase in β-cardiac myosin proportion occurs no earlier than 20 weeks of age, as the myosin isoform profiles of 10–20 week old +/+ and 403/+ mice have been shown to be similar (22). Additionally, at the level of the whole heart, increased chamber elastance and diastolic dysfunction is already evident in young, 5–6-week-old 403/+ mice (23,26), an age at which any increase in β-cardiac myosin proportion or significant fibrosis is unlikely to have occurred. These functional effects are hence likely due to the presence of R403Q α-cardiac myosin. At older ages, such as in our study, the cell-intrinsic functional effects seen in 403/+ cardiomyocytes are similarly due at least partially, if not primarily to R403Q α-cardiac myosin.
Although the HCM-causing mutation being studied is in a sarcomeric protein, we do not rule out the possibility that the phenotypes observed are caused by secondary effects on Ca2+ handling in the 403/+ cardiomyocyte. Cardiomyocyte relaxation kinetics is importantly affected by the rate of Ca2+ reuptake into the sarcoplasmic reticulum (SR). 403/+ cardiomyocytes have been shown to exhibit delayed rates of Ca2+ reuptake into the SR (27), which may be a result of reduced levels of the SR Ca2+-binding protein calsequestrin (42). Additionally, reduced levels (42) of SR Ca2+ but normal cytosolic Ca2+ levels (52) in 403/+ cardiomyocytes at rest suggest that the mutant cardiomyocytes may display enhanced Ca2+ retention in the sarcomere. These observations are all consistent with defects in Ca2+ handling in 403/+ cardiomyocytes that may result in diastolic dysfunction.
In conclusion, we have demonstrated the use of the dual carbon nanofiber technique and application of preload in probing sarcomeric functions that may be altered as a result of mutations. Much work remains to be done in elucidating the exact mechanisms by which the R403Q mutation results in the phenotypes observed. Additionally, the cell-autonomy of these hallmark HCM phenotypes may be a general phenomenon in HCM, a testable hypothesis with multiple animal models of HCM available (53–57). Potential pharmacological agents for HCM may be tested using CF techniques similar to the one described here because it is likely that therapies that alleviate these cell-intrinsic sarcomeric dysfunctions at the cardiomyocyte level will also have positive effects in vivo. Finally, as advances in generating well-differentiated human cardiomyocytes in vitro are underway, it may soon be possible to explore the functional effects of HCM mutations on human cardiomyocytes.
Acknowledgments
The authors are grateful to P. Kohl and C. Bollensdorf for the generous gift of carbon fibers, C. Bollensdorff, C. Woods, K. Ruppel, D. Charo, P. Lee, M. Helmes,, and J. Soughayer for technical assistance and useful scientific discussions, and T. Finsterbach, J. Yang, and S. Bartholomew for help with mice.
P.C. is supported by the Agency for Science, Technology and Research, Singapore, S.S., E.A.A., and J.A.S. are supported by a Stanford Cardiovascular Institute seed grant, E.A.A. is supported by National Institutes of Health Director's New Innovator Award OD004613, and J.A.S is supported by National Institutes of Health grant GM33289.
References
- 1.Maron B.J., Gardin J.M., Bild D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 2.Maron B.J. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 3.Maron B.J., Spirito P., Arce J. Noninvasive assessment of left ventricular diastolic function by pulsed Doppler echocardiography in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1987;10:733–742. doi: 10.1016/s0735-1097(87)80264-4. [DOI] [PubMed] [Google Scholar]
- 4.Epstein N.D., Cohn G.M., Fananapazir L. Differences in clinical expression of hypertrophic cardiomyopathy associated with two distinct mutations in the beta-myosin heavy chain gene. A 908-Leu----Val mutation and a 403-Arg----Gln mutation. Circulation. 1992;86:345–352. doi: 10.1161/01.cir.86.2.345. [DOI] [PubMed] [Google Scholar]
- 5.Geisterfer-Lowrance A.A., Kass S., Seidman J.G. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 6.Wheeler M., Pavlovic A., Ashley E.A. A new era in clinical genetic testing for hypertrophic cardiomyopathy. J. Cardiovasc. Transl. Res. 2009;2:381–391. doi: 10.1007/s12265-009-9139-0. [DOI] [PubMed] [Google Scholar]
- 7.Becker K.D., Gottshall K.R., Chien K.R. Point mutations in human beta cardiac myosin heavy chain have differential effects on sarcomeric structure and assembly: an ATP binding site change disrupts both thick and thin filaments, whereas hypertrophic cardiomyopathy mutations display normal assembly. J. Cell Biol. 1997;137:131–140. doi: 10.1083/jcb.137.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Volkmann N., Lui H., Hanein D. The R403Q myosin mutation implicated in familial hypertrophic cardiomyopathy causes disorder at the actomyosin interface. PLoS ONE. 2007;2:e1123. doi: 10.1371/journal.pone.0001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuda G., Fananapazir L., Sellers J.R. The in vitro motility activity of beta-cardiac myosin depends on the nature of the beta-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. J. Muscle Res. Cell Motil. 1997;18:275–283. doi: 10.1023/a:1018613907574. [DOI] [PubMed] [Google Scholar]
- 10.Debold E.P., Schmitt J.P., Warshaw D.M. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H284–H291. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 11.Fujita H., Sugiura S., Sutoh K. Characterization of mutant myosins of Dictyostelium discoideum equivalent to human familial hypertrophic cardiomyopathy mutants. Molecular force level of mutant myosins may have a prognostic implication. J. Clin. Invest. 1997;99:1010–1015. doi: 10.1172/JCI119228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris D.E., Work S.S., Warshaw D.M. Smooth, cardiac and skeletal muscle myosin force and motion generation assessed by cross-bridge mechanical interactions in vitro. J. Muscle Res. Cell Motil. 1994;15:11–19. doi: 10.1007/BF00123828. [DOI] [PubMed] [Google Scholar]
- 13.Lowey S., Lesko L.M., Robbins J. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. J. Biol. Chem. 2008;283:20579–20589. doi: 10.1074/jbc.M800554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer B.M., Fishbaugher D.E., Maughan D.W. Differential cross-bridge kinetics of FHC myosin mutations R403Q and R453C in heterozygous mouse myocardium. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H91–H99. doi: 10.1152/ajpheart.01015.2003. [DOI] [PubMed] [Google Scholar]
- 15.Palmiter K.A., Tyska M.J., Warshaw D.M. R403Q and L908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J. Muscle Res. Cell Motil. 2000;21:609–620. doi: 10.1023/a:1005678905119. [DOI] [PubMed] [Google Scholar]
- 16.Roopnarine O., Leinwand L.A. Functional analysis of myosin mutations that cause familial hypertrophic cardiomyopathy. Biophys. J. 1998;75:3023–3030. doi: 10.1016/S0006-3495(98)77743-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sata M., Ikebe M. Functional analysis of the mutations in the human cardiac beta-myosin that are responsible for familial hypertrophic cardiomyopathy. Implication for the clinical outcome. J. Clin. Invest. 1996;98:2866–2873. doi: 10.1172/JCI119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sweeney H.L., Straceski A.J., Faust L. Heterologous expression of a cardiomyopathic myosin that is defective in its actin interaction. J. Biol. Chem. 1994;269:1603–1605. [PubMed] [Google Scholar]
- 19.Tyska M.J., Hayes E., Warshaw D.M. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 2000;86:737–744. doi: 10.1161/01.res.86.7.737. [DOI] [PubMed] [Google Scholar]
- 20.Blanchard E., Seidman C., Maughan D. Altered crossbridge kinetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 1999;84:475–483. doi: 10.1161/01.res.84.4.475. [DOI] [PubMed] [Google Scholar]
- 21.Lankford E.B., Epstein N.D., Sweeney H.L. Abnormal contractile properties of muscle fibers expressing beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J. Clin. Invest. 1995;95:1409–1414. doi: 10.1172/JCI117795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer B.M., Wang Y., Maughan D.W. Myofilament mechanical performance is enhanced by R403Q myosin in mouse myocardium independent of sex. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H1939–H1947. doi: 10.1152/ajpheart.00644.2007. [DOI] [PubMed] [Google Scholar]
- 23.Georgakopoulos D., Christe M.E., Kass D.A. The pathogenesis of familial hypertrophic cardiomyopathy: early and evolving effects from an alpha-cardiac myosin heavy chain missense mutation. Nat. Med. 1999;5:327–330. doi: 10.1038/6549. [DOI] [PubMed] [Google Scholar]
- 24.McConnell B.K., Fatkin D., Seidman J.G. Comparison of two murine models of familial hypertrophic cardiomyopathy. Circ. Res. 2001;88:383–389. doi: 10.1161/01.res.88.4.383. [DOI] [PubMed] [Google Scholar]
- 25.Spindler M., Saupe K.W., Ingwall J.S. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J. Clin. Invest. 1998;101:1775–1783. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geisterfer-Lowrance A.A., Christe M., Seidman J.G. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–734. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]
- 27.Kim S.J., Iizuka K., Vatner S.F. An alpha-cardiac myosin heavy chain gene mutation impairs contraction and relaxation function of cardiac myocytes. Am. J. Physiol. 1999;276:H1780–H1787. doi: 10.1152/ajpheart.1999.276.5.H1780. [DOI] [PubMed] [Google Scholar]
- 28.Sivaramakrishnan S., Ashley E., Spudich J.A. Insights into human beta-cardiac myosin function from single molecule and single cell studies. J. Cardiovasc. Transl. Res. 2009;2:426–440. doi: 10.1007/s12265-009-9129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bird S.D., Doevendans P.A., Mummery C.L. The human adult cardiomyocyte phenotype. Cardiovasc. Res. 2003;58:423–434. doi: 10.1016/s0008-6363(03)00253-0. [DOI] [PubMed] [Google Scholar]
- 30.Peeters G.A., Sanguinetti M.C., Barry W.H. Method for isolation of human ventricular myocytes from single endocardial and epicardial biopsies. Am. J. Physiol. 1995;268:H1757–H1764. doi: 10.1152/ajpheart.1995.268.4.H1757. [DOI] [PubMed] [Google Scholar]
- 31.Kehat I., Kenyagin-Karsenti D., Gepstein L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J. Clin. Invest. 2001;108:407–414. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujiwara M., Yan P., Yamashita J.K. Induction and enhancement of cardiac cell differentiation from mouse and human induced pluripotent stem cells with cyclosporin-A. PLoS ONE. 2011;6:e16734. doi: 10.1371/journal.pone.0016734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi K., Tanabe K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 34.Josowitz R., Carvajal-Vergara X., Gelb B.D. Induced pluripotent stem cell-derived cardiomyocytes as models for genetic cardiovascular disorders. Curr. Opin. Cardiol. 2011;26:223–229. doi: 10.1097/HCO.0b013e32834598ad. [DOI] [PubMed] [Google Scholar]
- 35.Iribe G., Helmes M., Kohl P. Force-length relations in isolated intact cardiomyocytes subjected to dynamic changes in mechanical load. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1487–H1497. doi: 10.1152/ajpheart.00909.2006. [DOI] [PubMed] [Google Scholar]
- 36.Katz A.M. Ernest Henry Starling, his predecessors, and the “Law of the Heart”. Circulation. 2002;106:2986–2992. doi: 10.1161/01.cir.0000040594.96123.55. [DOI] [PubMed] [Google Scholar]
- 37.Campbell K.S. Impact of myocyte strain on cardiac myofilament activation. Pflugers Arch. 2011;462:3–14. doi: 10.1007/s00424-011-0952-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishimura S., Yasuda S., Sugiura S. Single cell mechanics of rat cardiomyocytes under isometric, unloaded, and physiologically loaded conditions. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H196–H202. doi: 10.1152/ajpheart.00948.2003. [DOI] [PubMed] [Google Scholar]
- 39.Fukuda N., Terui T., Kurihara S. Titin-based regulations of diastolic and systolic functions of mammalian cardiac muscle. J. Mol. Cell. Cardiol. 2010;48:876–881. doi: 10.1016/j.yjmcc.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 40.Fukuda N., Terui T., Kurihara S. Titin and troponin: central players in the frank-starling mechanism of the heart. Curr. Cardiol. Rev. 2009;5:119–124. doi: 10.2174/157340309788166714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bollensdorff C., Lookin O., Kohl P. Assessment of contractility in intact ventricular cardiomyocytes using the dimensionless ‘Frank-Starling Gain’ index. Pflugers Arch. 2011;462:39–48. doi: 10.1007/s00424-011-0964-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semsarian C., Ahmad I., Seidman J.G. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J. Clin. Invest. 2002;109:1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pak P.H., Maughan L., Kass D.A. Marked discordance between dynamic and passive diastolic pressure-volume relations in idiopathic hypertrophic cardiomyopathy. Circulation. 1996;94:52–60. doi: 10.1161/01.cir.94.1.52. [DOI] [PubMed] [Google Scholar]
- 44.Braunwald E. Hypertrophic cardiomyopathy: the early years. J. Cardiovasc. Transl. Res. 2009;2:341–348. doi: 10.1007/s12265-009-9128-3. [DOI] [PubMed] [Google Scholar]
- 45.Simonis G., Briem S.K., Strasser R.H. Protein kinase C in the human heart: differential regulation of the isoforms in aortic stenosis or dilated cardiomyopathy. Mol. Cell. Biochem. 2007;305:103–111. doi: 10.1007/s11010-007-9533-3. [DOI] [PubMed] [Google Scholar]
- 46.Muth J.N., Bodi I., Schwartz A. A Ca(2+)-dependent transgenic model of cardiac hypertrophy: a role for protein kinase Calpha. Circulation. 2001;103:140–147. doi: 10.1161/01.cir.103.1.140. [DOI] [PubMed] [Google Scholar]
- 47.Braz J.C., Bueno O.F., Molkentin J.D. PKC alpha regulates the hypertrophic growth of cardiomyocytes through extracellular signal-regulated kinase1/2 (ERK1/2) J. Cell Biol. 2002;156:905–919. doi: 10.1083/jcb.200108062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shubeita H.E., Martinson E.A., Brown J.H. Transcriptional activation of the cardiac myosin light chain 2 and atrial natriuretic factor genes by protein kinase C in neonatal rat ventricular myocytes. Proc. Natl. Acad. Sci. USA. 1992;89:1305–1309. doi: 10.1073/pnas.89.4.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hidalgo C., Hudson B., Bogomolovas J., Zhu Y., Anderson B., Greaser M., Labeit S., Granzier H. PKC phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ. Res. 2009;105:631–638. doi: 10.1161/CIRCRESAHA.109.198465. 617 p following 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hudson B.D., Hidalgo C.G., Granzier H.L. Excision of titin's cardiac PEVK spring element abolishes PKCalpha-induced increases in myocardial stiffness. J. Mol. Cell. Cardiol. 2010;48:972–978. doi: 10.1016/j.yjmcc.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gupta M.P. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2007;43:388–403. doi: 10.1016/j.yjmcc.2007.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fatkin D., McConnell B.K., Seidman J.G. An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J. Clin. Invest. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frey N., Franz W.M., Katus H.A. Transgenic rat hearts expressing a human cardiac troponin T deletion reveal diastolic dysfunction and ventricular arrhythmias. Cardiovasc. Res. 2000;47:254–264. doi: 10.1016/s0008-6363(00)00114-0. [DOI] [PubMed] [Google Scholar]
- 54.Maass A., Leinwand L.A. Animal models of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2000;15:189–196. doi: 10.1097/00001573-200005000-00012. [DOI] [PubMed] [Google Scholar]
- 55.Muthuchamy M., Pieples K., Wieczorek D.F. Mouse model of a familial hypertrophic cardiomyopathy mutation in alpha-tropomyosin manifests cardiac dysfunction. Circ. Res. 1999;85:47–56. doi: 10.1161/01.res.85.1.47. [DOI] [PubMed] [Google Scholar]
- 56.Prabhakar R., Petrashevskaya N., Wieczorek D.F. A mouse model of familial hypertrophic cardiomyopathy caused by a alpha-tropomyosin mutation. Mol. Cell. Biochem. 2003;251:33–42. [PubMed] [Google Scholar]
- 57.Tardiff J.C., Hewett T.E., Leinwand L.A. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J. Clin. Invest. 1999;104:469–481. doi: 10.1172/JCI6067. [DOI] [PMC free article] [PubMed] [Google Scholar]