

Abstract
The aggregation of alpha-synuclein (α-syn) and huntingtin (htt) into fibrillar assemblies in nerve and glial cells is a molecular hallmark of Parkinson's and Huntington's diseases. Within the aggregation process, prefibrillar and fibrillar oligomeric species form. Prefibrillar assemblies rather than fibrils are nowadays considered cytotoxic. However, recent reports describing spreading of fibrillar assemblies from one cell to another, in cell cultures, animal models, and brains of grafted patients suggest a critical role for fibrillar assemblies in pathogenesis. Here we compare the cytotoxic effect of defined and comparable particle concentrations of on-assembly pathway oligomeric and fibrillar α-syn and Htt fragment corresponding to the first exon of the protein (HttEx1). We show that homogeneous populations of α-syn and HttEx1 fibrils, rather than their precursor on-assembly pathway oligomers, are highly toxic to cultured cells and induce apoptotic cell death. We document the reasons that make fibrils toxic. We show that α-syn and HttEx1 fibrils bind and permeabilize lipid vesicles. We also show that fibrils binding to the plasma membrane in cultured cells alter Ca2+ homeostasis. Overall, our data indicate that fibrillar α-syn and HttEx1, rather than their precursor oligomers, are highly cytotoxic, the toxicity being associated to their ability to bind and permeabilize the cell membranes.
Introduction
Alzheimer's, Parkinson's (PD), and Huntington's disease (HD) are neurological human proteinopathies caused by the accumulation at specific sites inside or outside the cells of specific proteins' high molecular mass, fibrillar, β-sheet-rich, insoluble assemblies termed “amyloids”.
In PD, the intracellular aggregates, which major constituent is aggregated α-syn, are termed “Lewy bodies” and “Lewy neurites” (1,2). Alpha-synuclein (α-syn) is a 140-residue presynaptic protein normally found in both soluble and membrane-associated fractions of the brain (3) that is believed to play an important role in the regulation of synaptic vesicle release and trafficking, fatty acid binding, and neuronal survival (4).
HD is a dominant heritable disorder linked to the presence of an abnormally long polyglutamine (polyQ) repeat (37Q and over) in the N-terminal domain of huntingtin (Htt), a large protein of ∼3144 residues corresponding to the expression of the 67 exons of the HTT gene. Htt is expressed at its highest levels in the brain and plays an essential role in early development, as well as in axonal trafficking, regulation of gene transcription, and cell survival (5). The proteolytic cleavage of disease associated full-length Htt yields polyQ-containing N-terminal Htt fragments corresponding to the exon 1 (HttEx1) (6) that have a high propensity to aggregate and accumulate into cytoplasmic and nuclear inclusions in neurons (7,8).
Most neurological proteinopathies are believed to be due to the formation of folding intermediates that self-associate into nonnative oligomers that can elongate rapidly by incorporation of monomers. Growth is unlimited, because as one monomer is incorporated, a new binding site for yet another monomer is created (9). In vitro, at physiological salt and temperature, α-syn and HttEx1 assemble into fibrillar aggregates that are structurally similar to those found in the inclusions of disease-affected brains. Upon fibril formation, metastable prefibrillar oligomeric intermediates (that are believed to represent precursors of mature fibrils) are populated (10,11). Whether prefibrillar oligomeric and/or fibrillar α-syn and HttEx1 assemblies are toxic to cells is subject to intense debate and conflicting reports. Indeed, although fibrillar α-syn, HttEx1 and other amyloidogenic proteins have been shown to possess a cytotoxic potential (12–15), the oligomers that form during the early stages of assembly are considered most toxic, with the fibrils representing a storage or inert form (16–20). On-assembly pathway oligomers and fibrils cytotoxicities are believed to be the consequence of impairment of membrane integrity and permeability (16,21,22) and the amplification of assemblies by recruitment of homolog endogenous protein within the cytosol upon assemblies crossing the membrane (23–26).
The recent findings reporting spreading of fibrillar assemblies from one cell to another, in cell cultures, animal models, and brains of grafted patients, suggest a role for fibrillar assemblies in pathogenesis (23,24,26,27). This, together with the view that the abovementioned discrepancy most probably originates from comparison of protein particles at different concentrations, prompted us to revisit the respective cytotoxicity of α-syn and HttEx1 assemblies. Indeed, at a given concentration of precursor protein the concentrations of on-assembly pathway oligomers and fibrils diverge by several orders of magnitude, given that the assemblies have sizes, molecular masses, and number of constituting molecules that differ very significantly.
Here, we compare for the first time (to our knowledge) the cytotoxic effect of defined particle concentrations of on-assembly pathway α-syn and HttEx1 oligomers and fibrils of known size and molecular masses. We show that homogeneous populations of α-syn and HttEx1 fibrils, rather than their precursors, are highly toxic to cultured cells and induce apoptotic cell death.
We document the interaction of α-syn and HttEx1 fibrils with synthetic phospholipid vesicles of different composition. We show that α-syn fibrils bind and permeabilize preferentially vesicles made of anionic, rather than neutral, phospholipids, whereas HttEx1 fibrils show no charge selectivity. We assess the interaction of α-syn and HttEx1 fibrils with the plasma membrane in cultured cells and show fibrils-induced alterations in Ca2+ homeostasis.
Overall, our data indicate that fibrillar α-syn and HttEx1, rather than their precursor oligomers, are highly cytotoxic, the toxicity being associated to their ability to bind and permeabilize the cell membranes.
Materials and Methods
Expression and purification of α-syn and HttEx1Q41
The Escherichia coli strain BL21(DE3) (Stratagene, La Jolla, CA) was transformed with the expression vector pRK172 encoding for α-syn. Recombinant human wild-type α-syn was purified as described in Ghee et al. (28). Recombinant HttEx1Q41 was purified from MBP-TEV-HttEx1Q41 as described in the Supporting Material.
Assembly of α-syn and HttEx1Q41 into fibrils
α-Syn and HttEx1Q41 assembly into fibrils was monitored as described in the Supporting Material. To obtain fibril populations that are homogeneous in size, fibrillar samples were sonicated for 20 min on ice in 2-ml Eppendorf tubes in a VialTweeter powered by an ultrasonic processor UIS250v (250 watts, 2 4kHz; Hielscher Ultrasonic, Teltow, Germany) set at 75% amplitude, 0.5 s pulses.
The analytical ultracentrifugation, quantitative electron microscopy, cell viability assay, cell survival assay, caspase-3 activity measurements, preparation of phospholipid unilamellar vesicles Htt and α-syn assemblies membrane association assay, calcein release assay, α-syn and HttEx1Q41 labeling with extrinsic fluorophores, intracellular Ca2+ levels measurements, epifluorescence microscopy imaging, and statistical analysis are detailed in the Supporting Material.
Results
α-Syn assembly into fibrils, determination of the particle concentration of oligomeric and fibrillar α-syn and characterization of the α-syn toxic species
α-Syn readily assembles in vitro, under physiological conditions, into fibrils, as shown by the time-course measurement of Thioflavin T binding (Fig. 1 a). Transmission electron microscopy (TEM) analysis after negative staining reveals the presence of i), globular oligomers, with an average outer diameter of ∼20 nm (Fig. 1 b) within the lag phase preceding assembly (e.g., 1–6 h after the onset of assembly), and ii), mature fibrils, heterogeneous in length (Fig. 1 c), at steady state (e.g., 40–50 h after the onset of assembly). The length heterogeneity of the fibrils was reduced by sonication (Fig. 1 i, inset).
Figure 1.
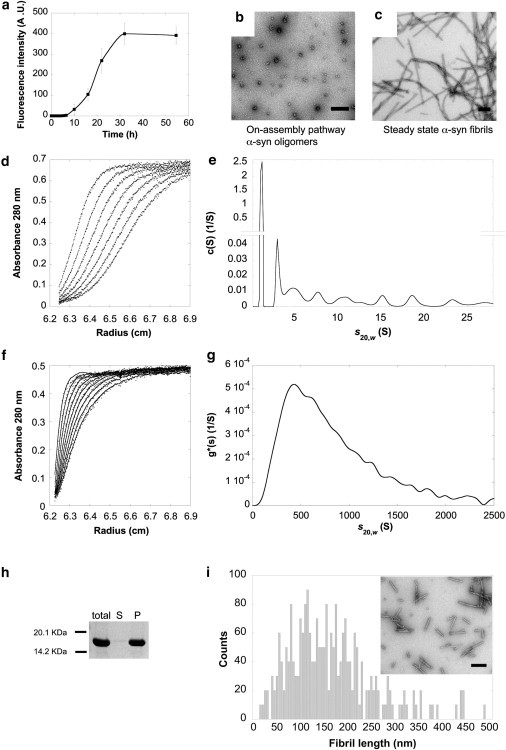
Characterization of on-assembly pathway oligomeric and fibrillar α-syn assemblies. (a) Assembly kinetics of α-syn (200 μM) measured by Thioflavin-T binding assay. Data are mean ± SD. (n = 4). (b and c) Negatively stained TEM of on-assembly pathway α-syn oligomers (b) and of mature α-syn fibrils (c). Scale bars, 200 nm. (d) Sedimentation velocity data for oligomeric α-syn (100-μM soluble protein concentration) at 50,000 rpm and 15°C in assembly buffer. Shown are sedimentation boundaries obtained at intervals of 20 min. (e) Sedimentation coefficient continuous c(s) distribution of oligomeric α-syn calculated from the sedimentation velocity data of panel d and corrected to s20,w. (f) Sedimentation velocity data for sonicated fibrillar α-syn (20-μM soluble protein concentration) at 3000 rpm and 20°C in assembly buffer. Shown are sedimentation boundaries obtained at intervals of 5 min. (g) Sedimentation coefficient g∗(s) distribution of fibrillar α-syn calculated from the sedimentation velocity data of panel f and corrected to s20,w. (h) Sodium-dodecyl-sulfate polyacrylamide-gel electrophoresis analysis of the distribution of fibrillar α-syn in the supernatant and pellet fractions upon centrifugation at 16,100 × g for 15 min at 20°C. (Total) Noncentrifuged α-syn fibrils. (S) Supernatant. (P) Pellet. (i) Length distribution of sonicated α-syn fibrils obtained by measuring the length of ∼2300 fibrils in at least 50 different quantitative negatively stained TEM samples. (Inset) Representative negatively stained TEM of sonicated α-syn fibrils. Scale bar, 200 nm.
A comparison of the toxicity of monomeric, soluble α-syn species that form during the lag phase, i.e., the on-assembly pathway oligomeric (oligomeric α-syn) and fibrillar α-syn based on the input concentration of the soluble protein, does not reflect the specific toxicities. This is due to the different forms of α-syn because the toxicities of assemblies that are not monomeric, and made of very different numbers of molecules, are compared. To estimate the relative toxicities of monomeric, oligomeric, and fibrillar α-syn one needs to determine the average number of α-syn molecules in the two forms of assemblies. This allows calculating the particle concentration of α-syn assemblies for a given concentration of monomeric α-syn.
We used analytical ultracentrifugation and length distribution measurements by quantitative TEM to estimate the average number of monomeric α-syn within the oligomeric and fibrillar assemblies.
The sedimentation velocities of monomeric, oligomeric, and fibrillar α-syn were determined. For monomeric and oligomeric α-syn, no rapidly sedimenting material was detected while the rotor was accelerating to reach its operating speed, ruling out the presence of large aggregates in freshly thawed and oligomeric α-syn samples. The raw data were modeled using the software Sedfit (National Institutes of Health, Bethesda, MD) and the c(s) model. The best fits yielded a single species with sedimentation coefficient (s20,w) of 1.15 S for monomeric α-syn (not shown). The sedimentation boundaries of oligomeric α-syn taken at 20-min intervals are presented in Fig. 1 d. Analysis of the data (Fig. 1 e) reveals the presence of a predominant peak, representing ∼86% of the species, with sedimentation coefficient (s20,w) of 1.15 S, consistent with the sedimentation coefficient of monomeric α-syn (RH = 2.98; f/f0 = 1.85). Nine additional peaks with higher sedimentation coefficients were observed (Fig. 1 e). They represent on-assembly pathway α-syn oligomers that populate the lag phase preceding fibrils formation. The sedimentation coefficient (s20,w), the relative abundance of the different oligomeric species, the average molecular mass, and the corresponding oligomeric state of the species, calculated with the assumption that α-syn oligomers are spherical particles, are given in Table 1. Overall, the oligomeric species account for ∼14% of α-syn in solution. This number varies very little from one measurement to another within a time-frame spanning from 1 to 6 h after the onset of assembly.
Table 1.
Sedimentation coefficients and relative abundances of the different α-syn oligomeric species populating the lag phase preceding assembly (100 μM protein monomer concentration)
| Sedimentation coefficient s20,w (S) | Relative abundance (%) | Estimated MW (KDa) | Estimated oligomeric state |
|---|---|---|---|
| 1.15 | 85.62 | 14.5 | Monomer |
| 3.1 | 2.79 | 34.8 | Dimer |
| 4.8 | 3.01 | 69 | Tetramer |
| ∼7.7–27 | 7.65 | ∼140–900 | ∼10–60 Monomers |
Estimated molecular masses and the corresponding state of oligomerization are also indicated.
For fibrillar α-syn, the sedimentation boundaries (Fig. 1 f) were analyzed with the software Sedfit (National Institutes of Health), using the least-squares boundary modeling ls-g∗(s) that is most suited for heterogeneous mixtures of large particles. This yielded a distribution of particles with sedimentation coefficients ranging from 150 to 1500 S (Fig. 1 g), centered on a species that has a sedimentation coefficient of ∼430 S, corresponding to particles that have a molecular mass of ∼125,000 kDa, e.g., made of ∼8600 α-syn molecules (125,000 kDa/14.5 kDa).
These measurements allowed us to estimate an average particle concentration of α-syn oligomers and of fibrillar α-syn by knowing the initial concentration of the soluble protein. The particle concentration of on-assembly pathway α-syn oligomers was estimated using the equation
| (1) |
where [soluble α-syn] is the initial concentration of the soluble protein, %Ni is the relative abundance of each oligomeric species reported in Table 1, and ni is the average number of monomers composing each oligomeric species.
Thus at the working concentration 20 μM, the overall particle concentration of α-syn oligomers is ∼0.5 μM. Similarly, the particle concentration of fibrillar α-syn is 20 μM/∼8600, i.e., ∼0.002 μM, as 100% of α-syn is in a fibrillar form at steady state. Indeed, 100% of the protein was found in the pellet fraction upon centrifugation at 16,100 × g for 15 min at 20°C of the fibrillar α-syn samples used throughout this work (Fig. 1 h).
We also performed length distribution measurements on the α-syn fibrils used throughout this study using quantitative TEM. The length and width of ∼2300 fibrils were measured using ∼50 quantitative negatively stained electron micrographs. The length distribution (Fig. 1 i) reveals that the fibrils (represented in the inset in Fig. 1 i) are ∼23-nm wide and ∼165-nm long. If the fibrils are assimilated to cylinders, an average fibril volume can be calculated and the number of monomers within a fibril estimated. We used
| (2) |
where Vfib is the volume of a cylinder (× nm long with a y nm diameter), Vaa is the volume on average of an amino acid (1.28 × 10−28 m3), and N is the number of residues within the polypeptide chain (i.e., 140).
Thus, a 165-nm-long and 23-nm-wide fibril contains ∼3800 α-syn monomers. This value is twofold lower than that derived from analytical ultracentrifugation measurements. Such a discrepancy could be due to fibrils dehydration in the electron microscope and/or underestimation of fibrils width and length because of negative staining. It also could be due to analytical ultracentrifugation analysis that yields accurate molecular masses for globular or slightly elongated molecules.
The cytotoxicity of equal particle concentrations of oligomeric and fibrillar α-syn assemblies was assessed on murine endothelioma H-END cells, previously shown to be highly susceptible to the toxicity of different protein assemblies (29) (Fig. 2 a), and human neuroblastoma SH-SY5Y cells (Fig. 2 b) using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Whereas oligomeric α-syn did not affect cell viability (Fig. 2, a and b, open bars) at any concentration we tested, fibrillar α-syn appeared highly toxic to the cells (Fig. 2 a and b, solid bars). Toxicity is apparent at concentrations as low as 0.25 pM. The toxicity we observe is not the consequence of sonication because sonicated and untreated fibrils, which length ranges from 0.16 μm to 2 μm with an average length of 0.6 μm (see Fig. S1 a in the Supporting Material), have similar toxicities (see Fig. S1, b and c).
Figure 2.
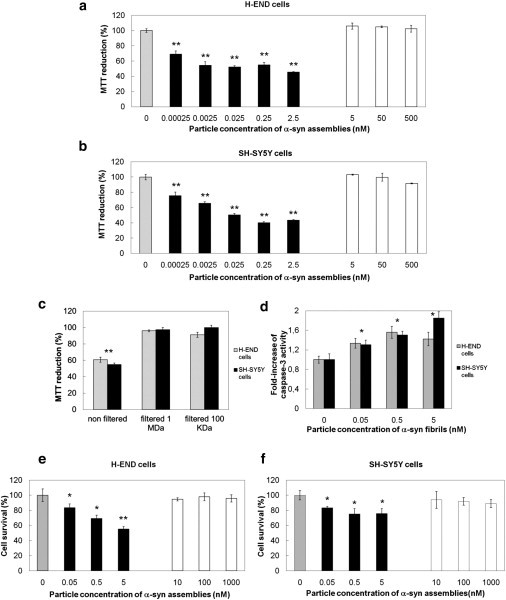
Fibrillar α-syn is cytotoxic. (a and b) Viability of H-END (a) and SH-SY5Y cells (b) treated for 24 h with increasing particle concentrations of on-assembly pathway oligomeric (open bars) or fibrillar α-syn (solid bars), measured by MTT assay. (Shaded bars) control cells treated with assembly buffer. MTT reduction data are mean ± SE (n = 6) expressed as percentage relative to control cells treated with buffer (100%), ∗∗P < 0.01 (Student's t-test). (c) Viability of H-END (shaded bars) and SH-SY5Y cells (solid bars) treated with 0.1 nM fibrillar α-syn preincubated in cell culture medium and ultrafiltered or not through different cutoffs (1 MDa or 100 KDa) before their addition to cell cultures. (d) Caspase-3 activation in H-END (shaded bars) or SH-SY5Y cells (solid bars) treated with increasing particle concentrations of fibrillar α-syn. Data are shown as fold increase relative to the caspase-3 activity measured in control cells treated with assembly buffer. Data are mean ± SE (n = 3), ∗ P < 0.05 (Student's t-test). (e and f) Percentages of H-END (e) or SH-SY5Y (f) cell survival upon treatment for 24 h with increasing particle concentrations of on-assembly pathway oligomeric (open bars) or fibrillar α-syn (solid bars). Data are percentages of the number of the surviving cells in controls treated with assembly buffer (100% survival, shaded bars). Data are mean ± SE (n = 6), ∗P < 0.05; ∗∗P < 0.01 (Student's t-test).
We conclude from these observations that α-syn fibrils, rather than their precursors, are toxic to the cells. As the observed toxicity might be due to the presence of oligomeric species in equilibrium with α-syn fibrils and/or to dissociation of the fibrils upon their dilution in the culture medium, the fibrils were preincubated overnight in the cell culture medium, ultrafiltered through membranes with different molecular-mass cutoffs, and the toxicity of the filtrate assessed using the MTT assay. The α-syn fibrils preincubated in cell culture medium for >48 h retained their fibrillar aspect (see Fig. S1 d) and toxicity. Toxicity was lost when the fibrils were ultrafiltered through 100-KDa or 1-MDa membranes (Fig. 2 c). These observations show that α-syn fibrils (rather than oligomeric species that are neither in equilibrium with the fibrils nor originating from fibrils disassembly) are toxic to cells.
Cell death and caspase-3 activation by fibrillar α-syn
We next investigated whether the metabolic impairment we observed using the MTT assay leads to cell death. Cells lifted by EDTA were incubated in the presence of increasing concentrations of oligomeric or fibrillar α-syn, replated, and counted after 24 h in the presence of Trypan blue to quantify the surviving cells.
H-END and SH-SY5Y cell survival was not affected by treatment with oligomeric α-syn at a particle concentration ranging from 0.01 to 1 μM (Fig. 2, e and f, open bars). In contrast, exposure of the cells to fibrillar α-syn resulted in a dose-dependent marked decrease of surviving cells after 24 h (Fig. 2, e and f, solid bars). Indeed, 55 ± 4% and 76 ± 7% H-END and SH-SY5Y cells, respectively, survived to exposure to 5 nM α-syn fibrils (Fig. 2, e and f, respectively) as compared to ∼100% survival for cells that were exposed to oligomeric α-syn (open bars in Fig. 2, e and f) or that were not exposed to α-syn (shaded bars in Fig. 2, e and f).
We then measured the activation of the effector caspase-3 in H-END or SH-SY5Y cells exposed for 24 h to increasing concentrations of fibrillar α-syn. The fibrils induced the activation of caspase-3 in a concentration-dependent manner (Fig. 2 d). No activation of caspase-3 could be observed when cells were exposed to oligomeric α-syn (not shown). We conclude from these observations that apoptotic cell death is triggered by fibrillar α-syn.
Interaction of α-syn assemblies with synthetic membranes
The interaction of α-syn assemblies with the membranes and its consequences, e.g., perturbation of the membranes, in particular permeabilization, may account for the toxicity we observe. Whereas soluble monomeric α-syn is known to interact with a variety of phospholipids (30,31) and oligomeric species of α-syn have been shown to disrupt negatively charged synthetic vesicles (32), the interaction of on-assembly pathway oligomeric and fibrillar α-syn with membranes and lipid vesicles of known composition has not been yet well characterized. We therefore set up an ultracentrifugation floatation assay to assess, within a density gradient, the interaction of protein assemblies with lipid vesicles.
Both oligomeric and fibrillar α-syn (50 nM) were found in the bottom fractions of the sucrose gradient (fractions 6–9) in the absence of lipid vesicles (see Fig. S2 a and Fig. S3 a). In contrast, α-syn assemblies were found in the top fractions of the gradient (fractions 1–4) when mixed with 6.25 mM vesicles made of 1,2-dioleoyl phosphatidylcholine DOPC (see Fig. S2 c and Fig. S3 c), 1,2-dioleoyl phosphatidylserine (DOPS; see Fig. S2 d and Fig. S3 d), 1,2-dioleoyl phosphatidylglycerol (DOPG; see Fig. S2 e and Fig. S3 e), and lipids extracted from porcine brain (see Fig. S2 f and Fig. S3 f). This is indicative of α-syn assemblies-lipid vesicle interaction. No interaction was observed with 1-palmitoyl, 2-oleoyl phosphatidylethanolamine (POPE) lipid vesicles (see Fig. S2 b and Fig. S3 b). The proportion of oligomeric and fibrillar α-syn found in each fraction of the gradient was determined. The amount of protein in fractions 1–4, i.e., interacting with the lipid vesicles, from three independent experiments was summed up and is presented in Fig. 3, a and b. Higher amounts of α-syn assemblies bound to DOPS and DOPG vesicles compared to POPE and DOPC vesicles. The highest amounts of α-syn assemblies bound to vesicles were prepared from brain lipid extracts (see Fig. S2 f and Fig. S3 f). We then assessed the effect of adding cholesterol (cholesterol/total lipids molar ratio 1:4) to lipid vesicles on the interaction of α-syn assemblies with the membranes. Fig. 3, a and b, Fig. S2, g and h, and Fig. S3, g and h, show that cholesterol did not affect significantly the interaction. Interestingly, oligomeric and fibrillar α-syn showed similar abilities to bind lipid vesicles.
Figure 3.
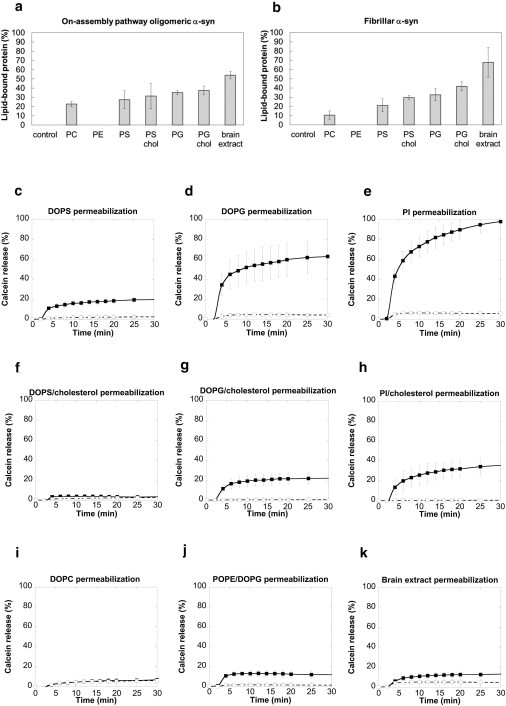
Interaction of α-syn assemblies with synthetic membranes and their permeabilization. (a and b) Binding of 50 nM on-assembly pathway oligomeric (a) or fibrillar (b) α-syn to lipid unilamellar vesicles with different phospholipid composition (6.25 mM lipids) measured by floatation. Membrane-bound α-syn was separated from free α-syn by density gradient ultracentrifugation. Data are the sum of the percentages of α-syn detected in the top fractions 1–4 of the gradient (see Fig. S2 and Fig. S3) in three independent experiments (data are mean ± SE). (c–k) Calcein release from lipid unilamellar vesicles with different phospholipid composition, induced by 0.2 nM oligomeric (open squares) or fibrillar α-syn (solid squares). The α-syn assemblies were added to calcein-loaded vesicles and the fluorescence measured for 30 min. The data are obtained by subtracting the percentage of spontaneous calcein release measured in control vesicles treated with identical volumes of assembly buffer. Data are mean ± SE (n = 3).
We conclude from these observations that α-syn assemblies bind preferentially to negatively charged lipid vesicles and complex lipid mixtures. We also conclude that cholesterol does not affect binding significantly.
α-Syn-mediated permeabilization of synthetic membranes
To assess the consequences of the interaction of α-syn assemblies with lipid vesicles, we monitored membranes' integrity and permeability using a calcein release assay. Unilamellar vesicles with different compositions were prepared in the presence of calcein at a concentration (60 mM) where fluorescence is self-quenched. 0.2 nM α-syn assemblies were then added to the vesicles and the time course of calcein release from the vesicles recorded.
Oligomeric α-syn induced very little permeabilization of lipid vesicles of any composition, as judged from the negligible fluorescence increase we recorded (Fig. 3, c–k, open squares). In contrast, fibrillar α-syn (0.2 nM) permeabilized anionic DOPS, DOPG, and soy L-α-phosphatidylinositol (PI) vesicles (Fig. 3, c–e, solid squares). Whereas the extent of DOPS vesicles' permeabilization was modest (∼20% calcein release after 30-min incubation), it was significant and extreme for DOPG or PI vesicles (60 and 100% calcein release after 30-min incubation, respectively). Finally, negligible amounts of calcein were released from vesicles made of neutral phospholipids (DOPC or POPE/DOPG molar ratio 1:1) upon incubation with fibrillar α-syn (Fig. 3, i and j). The observation that fibrillar α-syn permeabilizes negatively charged, but not neutral, vesicles is consistent with the binding data (Fig. 3, a and b, and Fig. S3).
Interestingly, when DOPS, DOPG, or PI vesicles were enriched in cholesterol (cholesterol/total lipids molar ratio 1:4), the extent of permeabilization by fibrillar α-syn strongly diminished (Fig. 3, f–h), although fibrils binding to the vesicles did not decrease (Fig. 3, a and b and Fig. S3). Similarly, although we found the fibrils to bind efficiently to vesicles prepared from brain lipid extracts (Fig. 3, a and b, and Fig. S3), a negligible level of fibrils-mediated calcein release was observed for the latter vesicles upon exposure to fibrillar α-syn (Fig. 3 k).
The interaction of fibrillar α-syn with the cell membrane induces an increase in intracellular calcium levels
Fluorescently labeled (Alexa Fluor 488; Invitrogen, Carlsbad, CA) fibrillar α-syn binds efficiently to the plasma membranes of cultured H-END and SH-SY5Y cells (Fig. 4, a and b) whereas oligomeric α-syn does not (not shown).
Figure 4.
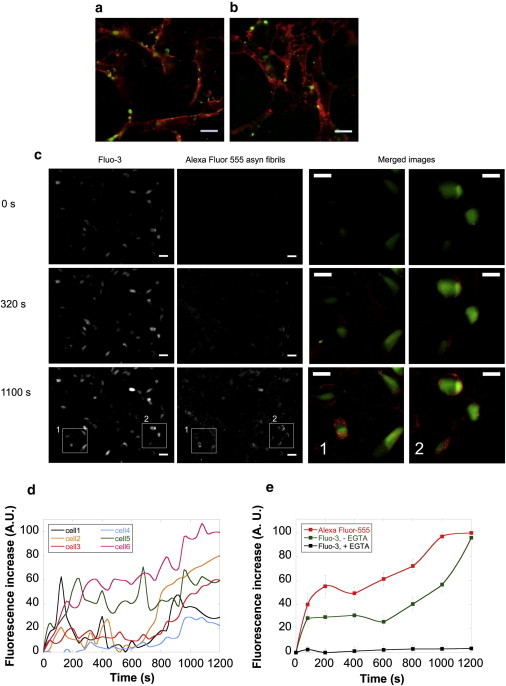
Interaction of fibrillar α-syn with the cell membrane induces an increase in intracellular calcium levels. (a and b) Binding of 0.1 nM Alexa Fluor 488-labeled α-syn fibrils to H-END (a) or SH-SY5Y cells (b) imaged by epifluorescence microscopy. Red fluorescence, wheat germ agglutinin, tetramethylrhodamine conjugate cell membrane staining. Green fluorescence, Alexa Fluor 488-labeled α-syn fibrils. Scale bars, 13 μm. (c) Imaging of intracellular free Ca2+ alterations and α-syn fibrils-membrane interaction at three different incubation stages of SH-SY5Y cells with 5 nM Alexa Fluor 555-labeled α-syn fibrils. Cells were loaded with Fluo-3-AM. (First column) Fluo-3 fluorescence (green). (Second column) Alexa Fluor 555 fluorescence (red). Scale bars, 30 μm. (Third and fourth column) Higher magnification view of merged images for the areas 1 and 2, respectively. Scale bars, 15 μm. (d) Quantification of the intracellular free Ca2+ increase measured over time for all the duration of the experiment in six representative cells. (e) Kinetics of the intracellular free Ca2+ variation in the absence (green curve) or the presence of 5 mM EGTA (solid curve). Quantification of Alexa Fluor 555-labeled α-syn fibrils binding to the cells (red curve). Data are mean ± SE of the fluorescence measured in 30 different cellular areas of 20 μm2 each.
We investigated the consequences of the interaction of fibrillar α-syn with the cell membranes. We monitored first the intracellular free Ca2+ levels in real-time. To this aim, H-END and SH-SY5Y cells loaded with Fluo-3-acetoxymethyl ester (Fluo-3-AM) were incubated with 5 nM Alexa Fluor 555-labeled fibrillar α-syn, and the fluorescence imaged over time for 20 min to monitor simultaneously intracellular Ca2+ variations (green fluorescence) and fibril binding (red fluorescence).
A progressive and remarkable rise of intracellular green fluorescence due to intracellular free Ca2+ increase was observed upon fibrils addition (Fig. 4 c, first column). This increase is concomitant to the accumulation of fibrillar α-syn on the cell surface (Fig. 4 c, second column). Merged images at higher magnification in two representative areas (Fig. 4 c, area 1 and area 2) are shown in Fig. 4 c, third and fourth columns, respectively.
The time courses of intracellular free Ca2+ increase in six different randomly chosen cells are presented in Fig. 4 d. This graph reveals cell-to-cell variability in the intracellular free Ca2+ increase we report. Some cells exhibited rapid bursts of intracellular free Ca2+ increase upon incubation with fibrillar α-syn, whereas a slow and progressive increase of intracellular free Ca2+ was observed in other cells. The extent of free intracellular Ca2+ increase also varied significantly. Overall, the intracellular free Ca2+ levels increased significantly as revealed by the signals recorded from 30 different cells, presented in Fig. 4 e (green curve). The red curve in Fig. 4 e represents the increase in Alexa Fluor 555 fluorescence due to α-syn fibrils binding to the cells. Interestingly, the rates of intracellular Ca2+ increase and fibrils binding have similar slopes, indicating that the two events are related.
The observed alterations in intracellular free Ca2+ levels induced by fibrillar α-syn might be due either to influx of extracellular Ca2+ through the plasma membrane or to Ca2+ release from the intracellular compartments where it is stored. To identify Ca2+ origin, the culture medium was supplemented with 5 mM EGTA and the measurements repeated. No fibrillar α-syn-induced intracellular free Ca2+ increase was observed under such experimental conditions (Fig. 4 e, solid curve). This suggests that the free Ca2+ influxes originate from the extracellular medium.
Cytotoxicity, interaction with synthetic and cellular membranes and consequences of the interaction of huntingtin Exon 1 assemblies
To determine whether what we report for α-syn assemblies is generic and illustrative of the consequences of the interaction of amyloidogenic protein assemblies with cellular membranes, we extended our study to HttEx1Q41.
Under physiological conditions, HttEx1Q41 readily assembles into fibrils. At time 0, no fibrils were observed by TEM (Fig. 5 a) whereas, at steady state, mature fibrils with an average length of 136 ± 3 nm and a diameter of ∼15 nm were present (Fig. 5 b).
Figure 5.
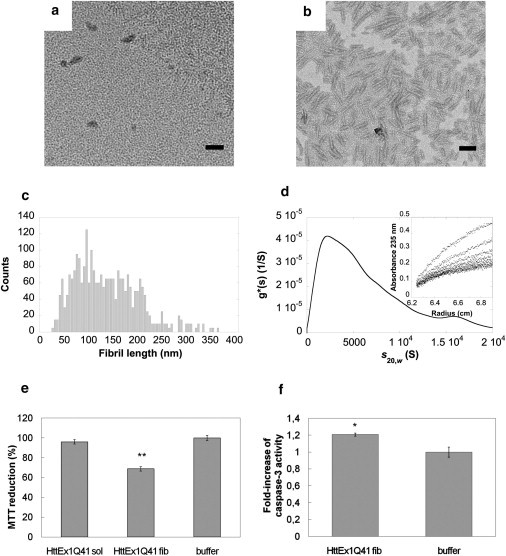
Characterization of fibrillar HttEx1Q41 assemblies and their toxicity. (a and b) Negatively stained TEM samples of soluble HttEx1Q41 at time 0 of assembly (a), and of mature HttEx1Q41 fibrils (b). Scale bars, 200 nm. (c) Length distribution of HttEx1Q41 fibrils obtained by measuring the length of ∼1700 fibrils in at least 30 different quantitative negatively stained electron micrographs. (d) Sedimentation coefficient g∗(s) distribution of HttEx1Q41 fibrils (25 μM protein monomer concentration) obtained at 2000 rpm and 20°C in 50 mM HEPES-KOH pH 7.5, 150 mM NaCl and corrected to s20,w. (Inset) Corresponding sedimentation boundaries, obtained at intervals of 20 min. (e) Viability of SH-SY5Y cells treated for 24 h with 10 μM soluble HttEx1Q41 or 6 nM HttEx1Q41 fibrils, measured by MTT assay. Data are mean ± SE (n = 6) expressed as percentages relative to control cells treated with buffer (∗∗P < 0.01 Student's t-test). (f) Caspase-3 activation in SH-SY5Y cells treated for 24 h with 6 nM HttEx1Q41 fibrils. Data are expressed as fold increase compared to the caspase activity measured in control cells treated with identical volumes of buffer. Data are mean ± SE (n = 3), ∗P < 0.05 (Student's t-test).
TEM imaging and analytical ultracentrifugation measurements allowed us to determine the size distribution of HttEx1Q41 fibrils and to estimate average particle concentrations for the different HttEx1Q41 assemblies.
Based on the length distribution (Fig. 5 c) of sonicated fibrillar HttEx1Q41, we estimated the number of HttEx1Q41 monomers within the fibrils to ∼1600 (see Eq. 2). Fibrillar HttEx1Q41 was also subjected to analytical ultracentrifugation measurements. The sedimentation boundaries (Fig. 5 d, inset) were analyzed as for fibrillar α-syn and yielded a broad distribution of particles centered on a species that has a sedimentation coefficient of ∼2200 S (Fig. 5 d), consistent with particles that have a molecular mass of ∼700,000 kDa, e.g., made of ∼52,000 HttEx1Q41 monomers. This value is ∼30-fold larger than that derived from length-distribution measurements. The discrepancy between the measurements may be the consequence of fibril-fibril association and extensive tangling (33). Because the length-distribution analysis is based on single particles, we considered the number derived from this measurement more reliable than that from analytical ultracentrifugation measurements. The number of HttEx1Q41 monomers within the fibrils is, therefore, on average ∼1600, and the particle concentration of fibrillar HttEx1Q41 at the working concentration 10 μM is 10 μM/∼1600, i.e., ∼0.006 μM. The calculation mode we opted for overestimates the concentration of fibrillar HttEx1Q41. This number would be 10 μM/∼52,000, i.e., ∼0.2 nM if the average molecular mass derived from analytical ultracentrifugation measurements is used instead.
We next investigated the cytotoxicity of HttEx1Q41 fibrils using the MTT assay. The viability of SH-SY5Y cells was unaffected by up to 10-μM soluble, freshly thawed HttEx1Q41, whereas a 31 ± 3% inhibition of MTT reduction was measured for cells exposed to 6-nM HttEx1Q41 fibrils for 24 h (Fig. 5 e). We also observed a significant (20%) caspase-3 activation upon exposure of the cells to 6-nM fibrillar HttEx1Q41 (Fig. 5 f). Thus, although fibrillar HttEx1Q41 concentration is overestimated by a factor 30, fibrillar HttEx1Q41 is approximately three orders-of-magnitude more toxic than soluble on-assembly pathway HttEx1Q41 oligomers.
The interaction of fibrillar HttEx1Q41 with synthetic vesicles and cell membranes was also investigated. HttEx1Q41 fibrils interact with both neutral DOPC and negatively charged DOPS or DOPG synthetic lipid vesicles (Fig. 6 a). In agreement with that, fibrillar HttEx1Q41 permeabilized to similar extents the different types of neutral and anionic vesicles (Fig. 6 b). Finally, fibrillar HttEx1Q41 bound to SH-SY5Y cells (Fig. 6 c) and induced a time-dependent increase in the intracellular free Ca2+ levels within the cells in a manner similar to what we observed for α-syn fibrils (Fig. 6, d and e).
Figure 6.
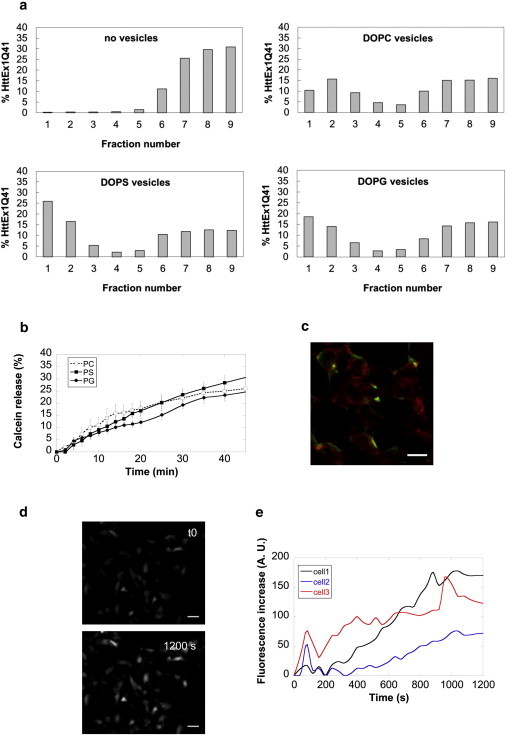
Fibrillar HttEx1Q41 binds and permeabilizes synthetic and cellular membranes. (a) Binding of 0.35 nM Alexa Fluor 488-labeled HttEx1Q41 fibrils to unilamellar vesicles with different phospholipid composition (6.25 mM lipids) measured by floatation. Membrane-bound HttEx1Q41 fibrils were separated from free fibrils by density-gradient ultracentrifugation. The distribution of HttEx1Q41 fibrils in the different fractions of the density gradient in the absence or the presence of vesicles, determined by fluorescence measurements, is shown in the graphs. (b) Calcein release from small unilamellar DOPC (open squares), DOPS (solid squares), or DOPG vesicles (solid circles), induced by incubation with 1.2 nM HttEx1Q41 fibrils for 50 min. The data represent net values obtained by subtracting the percentage of spontaneous calcein release measured in control vesicles treated with buffer. Data are mean ± SE (n = 3). (c) Binding of Alexa Fluor 488-labeled HttEx1Q41fibrils (green) to SH-SY5Y cells labeled with wheat germ agglutinin, tetramethylrhodamine conjugate (red) imaged by epifluorescence microscopy. Scale bar, 15 μm. (d) Imaging of intracellular free Ca2+ alterations induced by 6 nM HttEx1Q41 fibrils in SH-SY5Y cells loaded with Fluo-3-AM. (Upper panel) time 0. (Lower panel) 1200 s of incubation. Scale bars, 30 μm. (e) Quantification of the intracellular free Ca2+ increase measured over time for all the duration of the experiment in three representative cells.
Discussion
The pathogenesis of PD and HD is strongly associated with the aggregation of α-syn and HttEx1 containing polyQ tracts of 37Q and over into high molecular mass assemblies. In vitro, α-syn and HttEx1Q ≥ 37 assemble into fibrils. The role of the latter and the numerous oligomeric precursors that are populated during assembly in PD and HD onset has been actively investigated. Nonetheless, no clear consensus has been reached so far concerning the species that is most toxic to the cells and therefore likely to cause neurodegeneration. Indeed, although some reports suggest that, in a manner similar to polyQ, fibrillar assemblies of α-syn and HttEx1 are most toxic (11–14), others support the view that on-assembly pathway soluble oligomeric species are responsible for disease (17–20). The weakness of all these reports resides in the comparison of assemblies concentrations that differ by several orders of magnitude. Indeed, the concentrations of insoluble fibrils and soluble oligomeric species that are usually compared are expressed as a function of the initial soluble protein concentration. Such a comparison is erroneous as the concentration of an oligomeric species is the initial concentration divided by the number of monomers that constitute the oligomeric species. Thus, the concentration of oligomeric assemblies and fibrils can differ by a factor 1000 and over, because the oligomeric assemblies are made of ∼4–10 monomers whereas fibrils are made of thousands of molecules.
The importance of assessing the respective toxicities of the different oligomeric species has become urgent with the recent findings describing spreading of fibrillar assemblies from one cell to another in cell cultures, animal models, and brains of grafted patients, suggesting a critical role for fibrillar assemblies in pathogenesis.
We document here for the first time (to our knowledge) the respective toxicities of α-syn and pathologic HttEx1 assemblies at defined particle concentrations of on-assembly pathway oligomeric species and fibrils. The molecular mass of the oligomeric species and that of fibrils of defined length was determined (Figs. 1 and 5, Table 1), as was the concentration of particles in solution. Comparison of the relative toxicity of the oligomeric species and the fibrils shows undoubtedly that the fibrils are toxic to the cells at picomolar concentrations whereas micromolar amounts of oligomeric species are not (Figs. 2 and 5). The observed fibrils-associated toxicity is neither the consequence of their dissociation/alteration in fibrils structure nor to oligomeric species in equilibrium with the fibrils, as the fibrils persist upon dilution in the cell growth media (see Fig. S1 d) and toxicity is lost upon filtration of the media through a membrane that has a cutoff of 1 MDa (Fig. 2).
We show that fibrillar α-syn and pathologic HttEx1 trigger apoptotic cell death through the activation of caspase-3 (Figs. 2 and 5). It is now established that cells release soluble and fibrillar α-syn in their culture media. Soluble α-syn is certainly released by exocytosis, whereas the fibrillar form of the protein is most likely released upon cell death after the accumulation of aggregated α-syn within the cell. We and others have shown that released α-syn and HttEx1 assemblies bind to neighbor cultured cells and propagate to healthy neuronal grafts within the brain (24–26). To better understand how the assemblies traffic between cells, also why they are deleterious, we documented their interaction with lipid vesicles of known composition and cells and the consequences, if any, of the interaction. We show here using a floatation assay that fibrillar HttEx1 displays no charge selectivity and interacts with any kind of lipid vesicles (Fig. 6). This contrasts with the behavior of fibrillar α-syn that exhibits, in agreement with observations made on soluble α-syn (30–32), some specificity for anionic lipid membranes (Fig. 3, a and b, and see Fig. S2 and Fig. S3).
This may be due to the net isoelectric points and/or charge distributions within α-syn and HttEx1 (see Fig. S4). The interaction of fibrillar α-syn and HttEx1 with lipid vesicles leads invariably to permeabilization of the membranes (Fig. 3, c–k, and Fig. 6). Soluble α-syn has been reported to bind preferentially to the cholesterol enriched lipid rafts within the membranes (34) and cholesterol has been reported to protect against membrane permeabilization upon binding of Aβ1-42 assemblies (35). We therefore assessed the effect of cholesterol presence within lipid vesicles on fibrillar α-syn and HttEx1 binding to and permeabilization of the vesicles. We show here that although cholesterol has a limited effect on α-syn and HttEx1 binding to the vesicles (Fig. 3, a and b, and data not shown), its presence reduces significantly α-syn and HttEx1-mediated permeabilization (Fig. 3, f–h, and not shown). This is in perfect agreement with an efficient binding of fibrillar α-syn and HttEx1 to lipid vesicles made of complex phospholipid mixtures (e.g., lipid extracts from brain) where binding is accompanied by limited permeabilization. In conclusion, cholesterol appears to protect the membrane from disruption possibly by increasing membrane stability/rigidity rather than altering their ability to bind fibrillar α-syn and HttEx1.
Finally, we show here that the intracellular free Ca2+ levels increase upon exposure of cells to fibrillar α-syn and HttEx1. The changes we measured are concomitant to fibrils binding to the cell surfaces (Figs. 4 and 6) and are suppressed when extracellular Ca2+ is sequestered (Fig. 4 e). This suggests that the free Ca2+ influxes originate from the extracellular medium and that they are the consequence of plasma membrane permeabilization. Oscillations in intracellular free Ca2+ increase and variations between individual cells were also observed (Figs. 4 and 6). This may either reflect the time course of fibrillar α-syn and HttEx1 binding to the cells or differences in the ability of different cells to cope with either the fibrillar insult or ion homeostasis regulation.
Our observations reveal similarities and differences among fibrillar assemblies associated to neurodegenerative disease. Overall, fibrillar assemblies rather than on-assembly pathway oligomeric species are toxic to the cells. This generic property of fibrils appears to be the consequence, at least in part, of the binding of the fibrillar assemblies to the cell membrane lipid bilayers and its permeabilization. Studies aimed at assessing the contribution of membrane proteins to fibrillar assemblies binding to the cell will provide additional insights into the molecular events underlying fibrils-associated pathogenesis, and will allow defining of the respective contributions of lipids and proteins in fibrillar α-syn and HttEx binding to the cells.
Acknowledgments
We thank Yannick Sourigues for technical assistance in the production of recombinant Ex1HttQ41.
This research was supported by the Era-Net Neuron (MIPROTRAN), the Human Frontier Science Program, the Agence Nationale de la Recherche (ANR-08-NEUR-001-01 and ANR-09-MNPS-013-01), a “Coup d'Élan à la Recherche Française” award from the Bettencourt Schueller Foundation, and the Centre National de la Recherche Scientifique.
Supporting Material
References
- 1.Spillantini M.G., Crowther R.A., Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 3.Lee H.J., Choi C., Lee S.J. Membrane-bound α-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 2002;277:671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- 4.Uversky V.N., Eliezer D. Biophysics of Parkinson's disease: structure and aggregation of α-synuclein. Curr. Protein Pept. Sci. 2009;10:483–499. doi: 10.2174/138920309789351921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulte J., Littleton J.T. The biological function of the Huntingtin protein and its relevance to Huntington's Disease pathology. Curr. Trends Neurol. 2011;5:65–78. [PMC free article] [PubMed] [Google Scholar]
- 6.Graham R.K., Deng Y., Hayden M.R. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 7.Sieradzan K.A., Mechan A.O., Mann D.M. Huntington's disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp. Neurol. 1999;156:92–99. doi: 10.1006/exnr.1998.7005. [DOI] [PubMed] [Google Scholar]
- 8.DiFiglia M., Sapp E., Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 9.Brundin P., Melki R., Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010;11:301–307. doi: 10.1038/nrm2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg M.S., Lansbury P.T., Jr. Is there a cause-and-effect relationship between α-synuclein fibrilization and Parkinson's disease? Nat. Cell Biol. 2000;2:E115–E119. doi: 10.1038/35017124. [DOI] [PubMed] [Google Scholar]
- 11.Poirier M.A., Li H., Ross C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 2002;277:41032–41037. doi: 10.1074/jbc.M205809200. [DOI] [PubMed] [Google Scholar]
- 12.El-Agnaf O.M., Jakes R., Wallace A. Aggregates from mutant and wild-type α-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of β-sheet and amyloid-like filaments. FEBS Lett. 1998;440:71–75. doi: 10.1016/s0014-5793(98)01418-5. [DOI] [PubMed] [Google Scholar]
- 13.Yang W., Dunlap J.R., Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 2002;11:2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 14.Nekooki-Machida Y., Kurosawa M., Tanaka M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. USA. 2009;106:9679–9684. doi: 10.1073/pnas.0812083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Novitskaya V., Bocharova O.V., Baskakov I.V. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J. Biol. Chem. 2006;281:13828–13836. doi: 10.1074/jbc.M511174200. [DOI] [PubMed] [Google Scholar]
- 16.Caughey B., Lansbury P.T., Jr. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 17.Winner B., Jappelli R., Riek R. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arrasate M., Mitra S., Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi T., Kikuchi S., Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 2008;17:345–356. doi: 10.1093/hmg/ddm311. [DOI] [PubMed] [Google Scholar]
- 20.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 21.Danzer K.M., Haasen D., Kostka M. Different species of α-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kagan B.L., Hirakura Y., Azimova R. The channel hypothesis of Huntington's disease. Brain Res. Bull. 2001;56:281–284. doi: 10.1016/s0361-9230(01)00654-2. [DOI] [PubMed] [Google Scholar]
- 23.Desplats P., Lee H.J., Lee S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen C., Angot E., Brundin P. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 2011;121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Volpicelli-Daley L.A., Luk K.C., Lee V.M. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ren P.H., Lauckner J.E., Kopito R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J.Y., Englund E., Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 28.Ghee M., Melki R., Mallet J. PA700, the regulatory complex of the 26S proteasome, interferes with α-synuclein assembly. FEBS J. 2005;272:4023–4033. doi: 10.1111/j.1742-4658.2005.04776.x. [DOI] [PubMed] [Google Scholar]
- 29.Pieri L., Bucciantini M., Stefani M. Synthetic lipid vesicles recruit native-like aggregates and affect the aggregation process of the prion Ure2p: insights on vesicle permeabilization and charge selectivity. Biophys. J. 2009;96:3319–3330. doi: 10.1016/j.bpj.2008.12.3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartels T., Ahlstrom L.S., Beyer K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 2010;99:2116–2124. doi: 10.1016/j.bpj.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jo E., McLaurin J., Fraser P.E. α-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 32.Giehm L., Svergun D.I., Vestergaard B. Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation. Proc. Natl. Acad. Sci. USA. 2011;108:3246–3251. doi: 10.1073/pnas.1013225108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacRaild C.A., Hatters D.M., Howlett G.J. Sedimentation velocity analysis of flexible macromolecules: self-association and tangling of amyloid fibrils. Biophys. J. 2003;84:2562–2569. doi: 10.1016/S0006-3495(03)75061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park J.Y., Kim K.S., Park S.M. On the mechanism of internalization of α-synuclein into microglia: roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009;110:400–411. doi: 10.1111/j.1471-4159.2009.06150.x. [DOI] [PubMed] [Google Scholar]
- 35.Arispe N., Doh M. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer's disease AβP (1-40) and (1-42) peptides. FASEB J. 2002;16:1526–1536. doi: 10.1096/fj.02-0829com. [DOI] [PubMed] [Google Scholar]
- 36.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuck P., Perugini M.A., Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.