

Abstract
Although the lung expresses procoagulant proteins under inflammatory conditions, underlying mechanisms remain unclear. Here, we addressed lung endothelial expression of tissue factor (TF), which initiates the coagulation cascade and expression of which signifies development of a procoagulant phenotype in the vasculature. To establish the model of acid-induced acute lung injury (ALI), we intranasally instilled anesthetized mice with saline or acid. Then 2 h later, we isolated pulmonary vascular cells for flow cytometry and confocal microscopy to detect the leukocyte antigen, CD45 and the endothelial markers VE-cadherin and von Willebrand factor (vWf). Acid increased both the number of vWf-expressing cells as well as TF and P-selectin expressions on these cells. All of these effects were markedly inhibited by treating mice with antiplatelet serum, suggesting the involvement of platelets. The increased expressions of TF, vWf, and P-selectin in response to acid also occurred in platelets. Moreover, the effects were replicated in endothelial cells derived from isolated, blood-perfused lungs. However, the effect was inhibited completely in lungs perfused with platelet-depleted and, to a lesser extent, with leukocyte-depleted blood. Acid injury increased endothelial expressions of the platelet proteins, CD41 and CD42b, providing evidence that platelet proteins were transferred to the vascular surface. Reactive oxygen species (ROS) were implicated in these responses, in that the endothelial and platelet protein expressions were inhibited. We conclude that acid-induced ALI causes NOX2-mediated ROS generation that activates platelets, which then generate a procoagulant endothelial surface.
Keywords: von Willebrand factor, P-selectin, reactive oxygen species, CD41, gp91phox−/−
although anticoagulant properties of the vascular endothelial lining protect against clot formation, coagulation, and abnormalities of fibrinolysis are features of acute lung injury (ALI) (1, 11, 24, 31). Thus ALI is associated with increases in procoagulant markers in the lung including fibrin deposition and increase of plasminogen activator inhibitor (PAI-1) in the bronchoalveolar lavage (BAL) (1, 24, 31). However, mechanisms initiating these procoagulant responses are not understood.
Critical to these mechanisms are tissue factor (TF), which initiates coagulation, and the lung endothelium, which is a major venue for inflammatory processes leading to ALI. Although TF is expressed in lung endothelium (26), endothelial-specific deletion of the TF gene does not modify plasma coagulant activity in LPS-treated mice (23). However, the possibility needs to be considered that the endothelial surface provides a platform for the effects of TF derived from cells other than endothelial cells.
Here, we consider this issue in the context of acid-induced ALI, which is associated with high morbidity and mortality (16, 17). In animal models of acid-induced ALI, leukocyte-platelet interactions and secretion of inflammatory factors cause pulmonary edema (14, 20, 33). However, the underlying roles of platelet proteins in these responses are not understood (25, 27, 29). Although it is likely that platelet-leukocyte interactions induce activated TF expression on endothelium, this possibility has never been tested.
We address these mechanisms in freshly recovered lung endothelial cells to determine lung responses in situ under inflammatory conditions. We report below that within 2 h of acid instillation, reactive oxygen species (ROS) induce platelet-endothelial interactions leading to the expression of adhesive and procoagulant proteins on lung endothelium. These data provide the first evidence that epithelial injury induces a partnership between platelets and endothelial cells to induce procoagulant mechanisms in lung.
MATERIALS AND METHODS
Reagents and Antibodies
We purchased the following: PBS (Mediatech, Herndon, VA); 0.25% trypsin-EDTA (Sigma, St. Louis, MO); collagenase type 1 (Worthington, Lakewood, NY); rabbit polyclonal anti-von Willebrand factor (wVf) (Dako, Carpinteria, CA); rabbit anti-vWf, rabbit IgG (Abcam, Cambridge, MA); goat anti-mouse IgG-Alexa 488, goat anti-rabbit IgG-Alexa 633, goat anti-rabbit IgG-Alexa 488, and goat anti-rat IgG-Alexa 633 (Life Technologies, Invitrogen, Carlsbad, CA); mouse anti-human TF-FITC (American Diagnostica, Stamford, CT); rat anti-mouse CD144 (VE-cadherin), isotype control rat IgG, mouse IgG-FITC, mouse anti human CD42b-FITC, mouse IgG-FITC, rabbit polyclonal anti-P-selectin, rat anti-mouse P-selectin-FITC, rabbit IgG, mouse IgG, rat IgG-FITC (BD Bioscience, Sparks, MD); rat anti-CD45-APC, rat IgG-APC, rat anti-mouse CD144 (VE-cadherin)-Alexa 488, rat IgG-Alexa 488, rat anti-mouse CD41-FITC, rat IgG-FITC, rat anti-mouse CD45-FITC, rat anti-mouse CD45 PE, rat IgG2b K isotype PE (eBioscience, San Diego, CA); antiplatelet and nonimmune control sera (Accurate Chemical, Westbury, NY).
Animals and Animal Protocols
The Animal Care and Use Committee of Columbia University Medical Center approved all animal procedures. We used Swiss Webster and NOX2−/− mice (gp91phox−/−, strain B6.129S6-Cybbtm1Din/J, C57BL/6 background) as well as age-, sex-, and strain-matched wild-type (WT) mice (Jackson Laboratory, Bar Harbor, ME).
In vivo.
In anesthetized mice breathing 100% inspired oxygen (3% isoflurane, ketamine 100 mg/kg, xylazine 10 mg/kg), we instilled 2 ml/kg normal saline or hydrochloride acid (HCl, pH 1.2) intranasally. After 2 h, we excised lungs to obtain vascular cells (3). In one group of mice, we injected antiplatelet serum or control-nonimmune serum (25 μl, diluted with PBS 75 μl iv, tail vein) 2 h before acid instillation.
Isolated lungs.
Using our reported procedures (3), we prepared isolated, blood-perfused (1 ml/min, 37°C, 10% hematocrit) lungs, maintaining pulmonary artery, left atrial, and airway pressures of 12, 3, and 6 cmH2O, respectively. We instilled normal saline or acid intratracheally, obtaining vascular cells after 2 h.
Blood Perfusates
We injected heparin (200 units/kg, intracardiac) into anesthetized animals and withdrew blood. For platelet depletion, as before (32), we centrifuged blood (200 g, 10 min, 4°C) to induce platelet, leukocyte, and red cell separation into layers. For platelet depletion, we discarded platelet-rich supernatants, washing leukocytes and red cells (suspension in 4% albumin/PBS and centrifugation, 4°C × 3) to further remove platelets. In leukocyte depletion, buffy coats were removed and platelet supernatants were added back to the washed red cells. In platelet and leukocyte depletion, we used washed red cells suspended in buffer. As previously described (32), these procedures depleted ∼86 ± 9% of the leukocytes and platelets (Table 1).
Table 1.
Leukocyte and platelet counts in blood perfusates
Values are means ± SE. WB, whole blood; DB, leukocyte- or platelet-depleted blood.
P < 0.05 compared with WB.
Vascular Cell Isolation for Flow Cytometry
Although previously we used immunomagnetic beads to selectively isolate endothelial cells (32), here we avoided the beads to better enable flow cytometry of isolated vascular cells. We perfused the pulmonary artery with ice-cold buffer (4°C, 0.1% albumin/PBS, 5 ml) to clear blood. Then, immersing the lung in cold PBS (4°C), we sequentially perfused the lung vasculature with collagenase (1 ml, 1,000 units/ml, 15 min incubation) and trypsin (0.25%) and EDTA, 5 mmol (3 ml, 30 min incubation). We next perfused buffer (4°C) to collect a left atrial effluent containing dislodged cells. We washed the cells (×3, 1% BSA/PBS, suspension and centrifugation at 2,000 RPM, 10 min). After filtering the cells (100-μm pore, Becton Dickinson Labware, Franklin Lakes, NJ), we removed red blood cells (RBC lysis buffer, 5 min) and washed the cells again (×3). We fluorescently labeled the cells for microscopy and flow cytometry (BD FACScalibur Cell Analyzer), gating against isotype-specific and fluorophore-matched nonimmune IgGs. We acquired a minimum of 20,000 events for each sample.
Platelet Isolation
We heparinized (200 U/kg, intracardiac) anesthetized mice and withdrew blood, which we centrifuged (300 g, 5 min, 4°C) to obtain a platelet-rich supernatant for immunofluorescent labeling.
Immunofluorescent Labeling
We surface labeled freshly isolated cells as specified for TF, vWf, P-selectin, VE-cadherin, the leukocyte antigen, CD45 (19), the platelet integrin α-IIb (CD41) (2) and the platelet glycoprotein Ibα (GP1bα), also known as CD42b (2) using fluorescently tagged antibodies or untagged primary antibodies and appropriately tagged secondary antibodies (10 μg/ml in 1% BSA/PBS, 60 min) at 4°C to avoid antibody internalization. On platelets, we labeled TF, vWf, and P-selectin. The vWf and VE-cadherin labels on isolated cells were used as markers for endothelial phenotype. We labeled duplicate samples with fluorescently labeled nonimmune, isotype-specific IgGs.
Confocal Microscopy
We fixed the labeled cells (2% paraformaldehyde, 10 min, room temperature) for confocal microscopy (LSM 510, Zeiss) and image acquisition. An investigator blinded to the experimental plan digitally quantified fluorescence intensities (MCID-M4, Imaging Research, Canada). We determined the correlation coefficient for colocalization (LSM5 image examiner software).
Statistics
All data are means ± SE. We used standard t-tests for two-group comparisons and in experiments with multiple groups used an ANOVA with post hoc test for multiple comparisons. Statistical significance was accepted at P < 0.05.
RESULTS
Endothelial Cell Characterization
Mice were intranasally instilled with saline or acid. Then 2 h later, the pulmonary artery was buffer perfused to clear the vasculature before infusing collagenase at 4°C to dislodge endothelial cells that were collected in the venous effluent. For flow cytometry (Fig. 1), the collected cells were surface labeled for the leukocyte antigen CD45 and the endothelial cell marker VE-cadherin. Although VE-cadherin is ubiquitously present in endothelial cells (6), a subset of lung endothelial cells also expresses vWf-containing, Weibel-Palade bodies (WPBs). Activation of these endothelial cells causes WPB exocytosis, resulting in release of vWf, which remains adherent to the endothelial surface (30). Since these WPB-containing cells are present only in extra-alveolar vascular segments (4) and, importantly, not in the alveolar capillary segment, we also used a vWf marker to identify endothelial responses of extra-alveolar vascular segments.
Fig. 1.
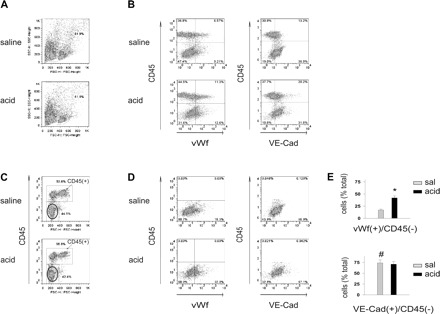
Airway acid increases the vWf+ fraction of cells isolated from the pulmonary vasculature. vWf, von Willebrand factor; VE-Cad, VE-cadherin. Pulmonary vascular cells were freshly isolated from mice subjected to intranasal saline or acid instillation. Dot plots from single experiments show expressions (% of total). The x and y ordinates (gray lines) indicate nonimmune isotype-specific IgG label. A: forward scatter (FSC) and side scatter (SSC) on isolated cells. Particles and granular clumps are outside the boundary line. B: plots show cells labeled on CD45 and vWf (left) and CD45 and VE-cadherin (right). C: CD45− fraction (encircled). D: plots show vWf+ (left) and VE-cadherin+ (right) cells in CD45− fraction. E: group data of vWf+ and VE-cadherin+ cells in CD45− fractions, respectively, n = 4 mice each bar. Means ± SE, *P < 0.05 compared with bar on left, #P < 0.05 comparing saline (sal) instillation data for VE-cadherin and vWf.
Since platelets are a nonendothelial vWf source, vWf+ cells identified by flow cytometry potentially include platelets and endothelial cells. However, platelets are ∼1 μm in diameter, whereas isolated endothelial cells are 10–12 μm. Hence, platelets can be size discriminated from endothelial cells by flow cytometry. Accordingly, our gating conditions excluded not only cell aggregates (high side scatter), but also platelets and debris (low forward scatter) (Fig. 1A). Therefore, the vWf+ cells were predominantly endothelial cells.
The CD45+ and CD45− fractions comprised 55 and 45% of the total cells, respectively. vWf+ and VE-cadherin+ cells were present in both fractions (Fig. 1, B and C). Of the CD45− fraction, which is the fraction encircled in Fig. 1C, ∼70% were endothelial cells, since they were positive for both VE-cadherin and vWf (Fig. 1, D and E). Hence of the ∼2 × 106 cells recovered per collection, ∼6 × 105 were endothelial cells. In control lungs (saline instillation), VE-cadherin+ cells were four times more abundant that vWf+ cells, attesting to the more widespread expression of VE-cadherin in the pulmonary vascular bed. However, acid instillation increased the vWf+ fraction two times above control, but did not change the VE-cadherin+ fraction. These findings suggest that acid instillation increased endothelial vWf expression.
TF and P-Selectin
We determined surface TF expression on the lung vascular isolates to define procoagulant responses in acid injury. The numbers of TF+/vWf+ cells increased markedly after acid instillation (Fig. 2, A and B). Acid instillation also increased the numbers of vWf+ cells that also expressed the leukocyte adhesion receptor, P-selectin (Fig. 2, C and D). These increases in TF, vWf, and P-selectin suggest that acid injury induced a procoagulant, platelet-leukocyte adhesive endothelial surface in the lung.
Fig. 2.
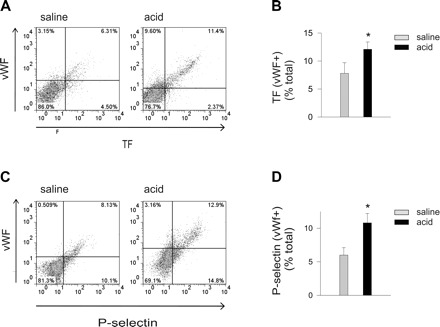
Airway acid increases vWf+ cells expressing TF and P-selectin. Pulmonary vascular cells were freshly isolated from mice subjected to intranasal saline or acid instillation. Dot plots from single experiments show expression (% of total). The x and y ordinates (gray lines) indicate nonimmune isotype specific IgG label. A: TF and vWf. B: group data. C: vWf and P-selectin. D: group data; n = 4 mice each bar. Means ± SE, *P < 0.05 compared with bar on left.
To determine the extent to which the induced expressions of TF, vWf, and P-selectin colocalized on the endothelial cell surface, we imaged fluorescently labeled endothelial cells by confocal microscopy. Although saline treatment caused weak TF and P-selectin expressions in vWf-expressing cells, acid instillation markedly increased the expressions (Fig. 3). Colocalization analyses revealed that the major sites of TF and P-selectin expression were also the major sites of vWf expression (Fig. 3, B and D), indicating that the proteins were expressed in a colocalized manner on the endothelial surface. We did not see platelets adherent to the endothelial cells.
Fig. 3.
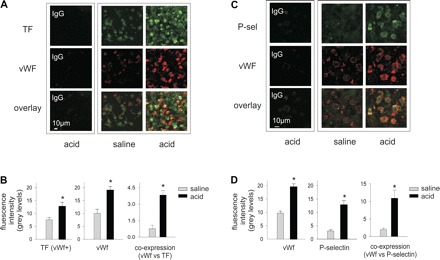
Confocal images of acid instillation-induced TF, vWf, and P-selectin (P-sel) colocalizations on lung endothelium. Images of pulmonary vascular cells freshly isolated from one mouse lung show protein expressions as indicated. Yellow in overlay indicates colocalization. Group data show fluorescence intensity and colocalization (coefficient for colocalization by LSM5 image examiner software). A: TF and vWf. B: group data for fluorescence intensity and colocalization, n = 4 mice each bar, 30 cells each mouse lung. C: vWf and P-selectin. D: group data for fluorescence intensity and colocalization, n = 4 mice each bar, 30 cells each mouse lung. Means ± SE, *P < 0.05 compared with saline.
Platelet and Leukocyte Effects in Isolated Lungs
To determine the effects of acid injury on platelet responses, we exposed mice to airway instillations of saline or acid as before, then obtained blood 2 h later to isolate platelets. Flow cytometry indicated that acid injury increased platelet expressions of TF, vWf, and P-selectin (Fig. 4), suggesting that these proteins might be transferred from platelets to the endothelium.
Fig. 4.
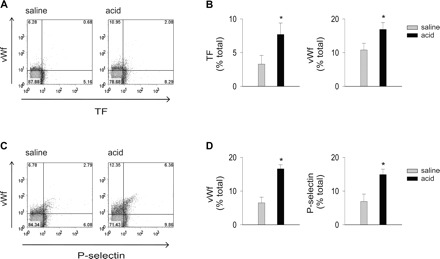
Airway acid induces platelet TF, vWf, and P-selectin expressions. Platelets were isolated from the blood of mice subjected to intranasal saline or acid instillation. Dot plots of single experiments show expression (% of total). The x and y ordinates (gray lines) indicate nonimmune isotype specific IgG label. A: TF and vWf. B: group data, n = 4 mice each bar. C: vWf and P-selectin. D: group data, n = 4 mice each bar. Means ± SE, *P < 0.05 compared with control and saline.
To test this possibility, we perfused isolated lungs with whole blood or platelet-depleted blood and stained cells recovered from the vasculature for TF, vWf, and P-selectin. In the presence of whole blood perfusion, acid instillation increased TF expression in vWf+ cells, as also the numbers of vWf+/P-selectin+ cells. These effects were abrogated in the presence of platelet-free perfusion (Fig. 5). Although perfusion with leukocyte-free blood also decreased the TF and P-selectin expressions, these decreases were smaller than the decreases with platelet-free perfusion. These findings indicate that the endothelial expressions of TF and P-selectin and vWf depended on the presence of platelets in the lung perfusion, and to a lesser extent on that of leukocytes.
Fig. 5.
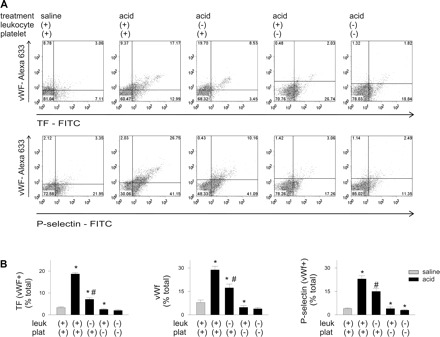
Platelet depletion inhibits airway acid-induced endothelial TF, vWf, and P-selectin expressions. Pulmonary vascular cells were freshly obtained from isolated lungs subjected to airway saline or acid instillation during perfusion with whole blood or blood depleted of leukocytes (leuk), platelets (plat), or both. Dot plots of single experiments show expression (% of total). The x and y ordinates (gray lines) indicate nonimmune isotype specific IgG label. A: TF and vWf (top row), and vWf and P-selectin (bottom row). B: group data, n = 4 mice each bar. Means ± SE, *P < 0.05 compared with bar on left. #P < 0.05, compared with saline.
Transfer of Platelet Proteins
To address the possibility that platelets transferred proteins to endothelium, we determined the expressions of platelet-specific proteins. We labeled the isolated cells for the platelet integrin CD41 (α2b, IIb/IIIa complex), the platelet receptor CD42b (glycoprotein Ibα) and vWf. In control lungs, the numbers of vWf+ cells expressing CD41 and CD42b were small (Fig. 6, A and B). Acid instillation increased both vWf+/CD41+ and vWf+/CD42b+ cells (Fig. 6, C and D). In other experiments, we labeled the cells for CD45, CD41, CD42b, and VE-cadherin. CD45−/VE-cadherin+ cells constituted the endothelial fraction not associated with leukocytes. In control lungs, CD41 was more extensively expressed than CD42b in this endothelial fraction (Fig. 7, A and B). Acid instillation increased both CD41 and CD42b expressions in the fraction, indicating that the platelet proteins were transferred to the endothelium.
Fig. 6.
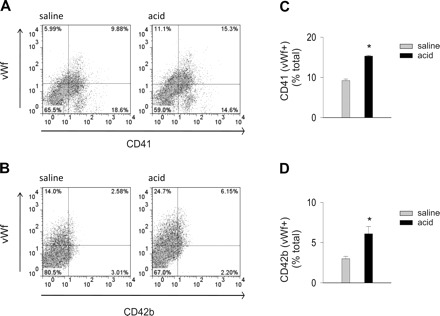
Airway acid induces expression of the platelet proteins, CD41 and CD42b on vWf-expressing cells. CD41, integrin α2b; CD42b, platelet glycoprotein Ibα. Pulmonary vascular cells were freshly isolated from mice subjected to intranasal saline or acid instillation. Dot plots from single experiments show expression (% of total). The x and y ordinates (gray lines) indicate nonimmune isotype specific IgG label. A: CD41 and vWf. B: CD42b and vWf. C: group data for vWf+/CD41+ cells. D: group data for and vWf+/CD42b+ cells. Means ± SE, n = 4 mice each bar, *P < 0.05 compared with bar on left.
Fig. 7.
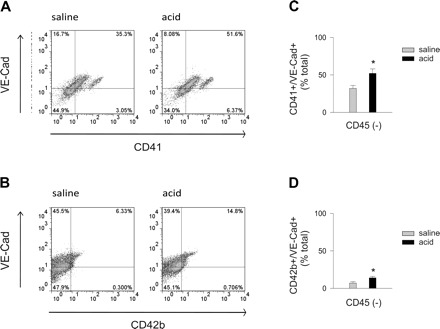
Airway acid induces expression of the platelet proteins, CD41 and CD42b on CD45− cells expressing VE-cadherin. Pulmonary vascular cells were freshly isolated from mice subjected to intranasal saline or acid instillation. Dot plots from single experiments show expression (% of total) in isolated CD45− cells. The x and y ordinates (gray lines) indicate nonimmune isotype specific IgG label. A: CD41 and VE-cadherin. B: CD42b and VE-cadherin. C: group data for VE-cadherin+/CD41+ cells. D: group data for and VE-cadherin+/CD42b+ cells. Means ± SE, n = 4 mice each bar, *P < 0.05 compared with bar on left.
To further determine the effects of platelets on the endothelium, we intravenously injected antiplatelet serum to deplete blood platelets. For control, we injected nonimmune serum. Antiplatelet serum given 2 h before acid instillation depleted platelets by 86 ± 2% of control (P < 0.05, n = 4), but it did not deplete leukocytes. Under these platelet-depleted conditions, acid injury markedly reduced TF and P-selectin expression in vWf+ cells (Fig. 8). Control serum had no effects. These responses support the notion that, in acid injury, platelets enhance TF expression on the lung endothelium.
Fig. 8.
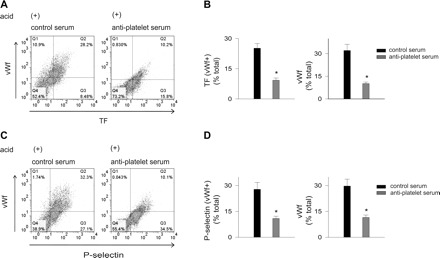
Platelet depletion in vivo decreases acid-induced lung endothelial TF, vWf, and P-selectin expressions. vWf, von Willebrand factor; TF, tissue factor. Mice were injected intravenously with nonimmune control serum or antiplatelet serum. After 2 h, they were subjected to intranasal saline or acid instillation. Pulmonary vascular cells were freshly isolated 2 h after instillation. Dot plots from single experiments show expression (% of total). The x and y ordinates (gray lines) indicate nonimmune isotype specific IgG label. A: TF and vWf. B: group data, n = 4 mice each bar. Means ± SE, *P < 0.05 compared with saline. C: vWf and P-selectin. D: group data, n = 3 mice each bar. Means ± SE, *P < 0.05 compared with saline.
ROS
To define the role of ROS, we determined expressions of coagulant proteins on cells collected from NOX2-deficient, gp91phox−/− mice and strain-matched WT mice. For WT mice, acid instillation increased TF+, vWf+, and P-selectin+ cells (Fig. 9). However, these responses were markedly reduced for gp91phox−/− mice, indicating that the increased protein expressions were due to ROS. To define the ROS source, we perfused acid-instilled isolated lungs of WT mice with WT or gp91phox−/− blood. Perfusion with gp91 phox−/− blood reduced TF+, vWf+, and P-selectin+ cells (Fig. 10), indicating that the responses resulted from ROS production by leukocytes and platelets. Platelet expressions of TF, vWf, and P-selectin were also abrogated in blood from gp91phox−/− but not in blood of WT mice (Fig. 11), indicating that the expressions were determined by ROS from leukocytes and platelets.
Fig. 9.
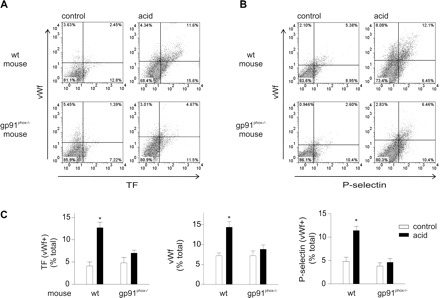
Airway acid-induced lung endothelial TF, vWf, and P-selectin expressions are inhibited in gp91phox−/− mouse. WT, wild-type; control, untreated. Pulmonary vascular cells were freshly isolated from gp91phox−/−, NOX2-deficient mice subjected to intranasal acid instillation and from strain-matched WT untreated mice. Dot plots (single experiments), top (WT) and bottom (gp91phox−/−) show expression (% of total). The x and y ordinates (gray lines) indicate nonimmune isotype specific IgG label. A: TF and vWf. B: vWf and P-selectin. C: paired group data showing WT and gp91phox−/− responses, n = 3 mice each bar. Means ± SE. *P < 0.05 compared with control.
Fig. 10.
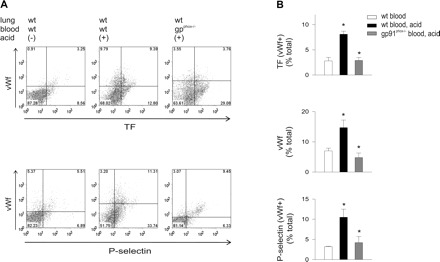
Airway acid-induced endothelial TF, vWf, and P-selectin expressions are inhibited in WT lungs perfused with gp91phox−/− blood. Pulmonary vascular cells were freshly isolated from WT untreated lungs perfused with WT blood or from WT lungs that were acid instilled and either perfused with WT blood or blood from gp91phox−/− mice. Dot plots of single experiments show expression (% of total). The x and y ordinates (gray lines) indicate nonimmune isotype specific IgG label. A: top row, TF and vWf and bottom row, vWf, and P-selectin. B: group data, n = 4 mice each bar. Data are means ± SE. *P < 0.05 compared with bar on left.
Fig. 11.
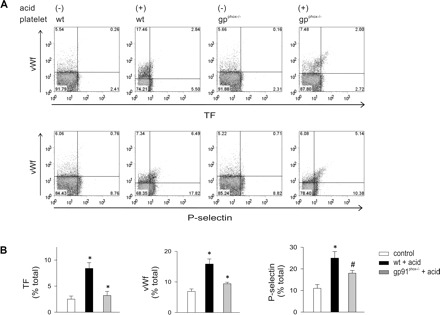
Airway acid-induced TF, vWf, and P-selectin expressions are inhibited on platelets of gp91phox−/− mouse. Control, untreated. Platelets were isolated from WT and gp91phox−/− blood in untreated or acid instillation conditions. Dot plots of single experiments show expression (% of total). The x and y ordinates (gray lines) indicate nonimmune isotype specific IgG label. A: TF and vWf (top row), and vWf and P-selectin (bottom row). B: group data show paired responses on platelets from WT and gp91phox−/−, respectively, n = 4 mice each bar. Data are means ± SE. *P < 0.05 compared with bar on left. #P < 0.05 compared with untreated control.
DISCUSSION
We report here a flow cytometry approach for defining procoagulant mechanisms in endothelium of acutely injured lungs. We defined the endothelial cell fraction as the platelet-free fraction that was CD45− and vWf+ or VE-cadherin+. By these criteria, we estimate that we collected 6 × 105 endothelial cells, a number that compares well with our previously reported isolations using immunomagnetic beads (32). We conclude that flow cytometry of mixed cell populations provides a good approach for global analyses of lung endothelial cells.
Using vWf as an endothelial marker, we detected expression of procoagulant proteins. Our major finding was that airway acid instillation increased expression of TF and P-selectin on vWf+ cells suggesting that acid injury induces a procoagulant endothelial response. vWf and another endothelial marker, VE-cadherin together labeled a majority of the leukocyte-free (CD45−) fraction, indicating that endothelial cell recovery was substantial. In the leukocyte-free fraction, acid instillation increased vWf+, but not VE-cadherin+ cells, suggesting that this form of lung injury increases endothelial vWf expression.
Since both endothelial cells and platelets produce vWf, the increase in vWf+ cells could be due to either cell type. To define the platelet role, we depleted blood platelets by 85% using antiplatelet serum. This treatment blocked the increase in vWf+ cells, as well as the increased TF and P-selectin expressions in the vWf+ fraction, implicating platelets in these responses. Similarly, perfusing isolated lungs with platelet-depleted blood also reduced the protein expressions in acid injury. Platelet priming for the procoagulant response was evident in that TF, vWf, and P-selectin expressions were higher in platelets isolated directly from blood.
These findings suggest that platelets were responsible for the procoagulant protein expression on endothelial cells. This possibility is further supported in that acid injury increased the expressions of the platelet proteins CD41 and CD42b in VE-cadherin+ cells of the CD45− fraction, indicating that platelets transferred proteins to the endothelium. The possibility that these protein expressions were due to free platelets in the cell samples was ruled out by size criteria of flow cytometry that eliminated free platelets from the analyses, as well as by direct confocal microscopy of the endothelial cells. Taking these findings together we interpret that the endothelial procoagulant responses were due to protein transfer from platelets. To our knowledge, this is the first evidence that an inflammatory injury in the lung induces a procoagulant phenotype in the lung vasculature by deposition of platelet-derived proteins on the endothelial surface.
Our findings provide novel insight regarding the mechanism by which TF appears on the endothelial surface. The bulk of this understanding is derived from the systemic circulation and rests on the notion that TF is constitutively expressed in fibroblasts, vascular smooth muscle cells, and pericytes (22). Under pathological conditions such as sepsis, TF is expressed in monocytes and human platelets (8, 12, 23). However, uncertainty surrounds the understanding of how the TF from these cell types becomes displayed on the endothelial surface to initiate coagulation. This uncertainty is also evident in lung literature in that in ALI several coagulation relevant proteins are reported in the BAL, including TF, PAI-1, thrombin-anti-thrombin complexes, and fibrin and its degradation products (1, 11, 24, 31). However, the TF source is not known.
Here we show for the first time that in the setting of acid injury the TF expressed on the endothelial surface comes not from vascular smooth muscle, as in systemic coagulation, but almost entirely from platelets. These inferences arise from our studies with platelet-depleted blood perfusion and with antiplatelet serum. Our novel finding that acid injury induces CD41 and CD42b expressions in endothelium suggest the feasibility of protein transfers from platelets to endothelium in vivo. This transfer might occur as a consequence of the continuous attachment and detachment of flowing platelets with the endothelium (2). The presence of the platelet proteins on vWf-expressing cells also suggests that the interactions occur in the extra-alveolar vascular segment. Furthermore, acid injury-induced enhancement in expressions of these platelet proteins on VE-cadherin constitutes evidence of platelet-endothelial interactions over an extensive area of the lung vascular surface. Since activated platelets release CD41+ and CD42b+ microparticles (28), it is possible that endothelial uptake of platelet particles plays a role in the protein transfer. To a lesser extent, leukocytes also contributed to the TF expressions on endothelium. Taken together, our findings indicate that under injury conditions, activated platelets condition the vascular endothelium to establish the proinflammatory-procoagulant phenotype. The role of platelets in lung injury has generally been considered in the context of leukocyte activation (27, 29, 33). Our findings enlarge this role in that we show that in lung injury, platelets are initiators of endothelial TF display.
Since mouse platelets do not synthesize TF (23), although they express vWf and P-selectin in cytoplasmic granules (2), it is important to consider how the platelets might have acquired TF. Under inflammatory conditions, secretion of the granules causes expression of these adhesive proteins on the platelet surface, enabling platelets to adhere to monocytes (7, 29). TF is then transferred from monocytes to platelets (7, 21). Evidence that these events might have occurred in our studies is indicated in that, within 2 h of acid injury, mouse platelets expressed TF, although the expression was negligible in platelets from untreated mice. Furthermore, platelet vWf and P-selectin also increased, suggesting that these adherence mechanisms mediated TF transfer from monocytes to platelets (7, 21).
The fact that the acid-induced endothelial expressions of P-selectin and vWf were also platelet dependent further implicates platelets in a subset of the lung's inflammatory response that has traditionally been interpreted as largely endothelial in origin. The mechanistic importance of P-selectin and vWf in the lung injury is understood in the context of mechano-oxidative coupling during pressure stress-induced leukocyte adherence to lung endothelium (9, 13). Lung injury is blocked in mice given intravenous infusion of anti-P-selectin antibody (14). Chimeric mice containing P-selectin-deficient platelets fail to develop acid injury (33). vWf is a platelet-adhesive molecule (25) and thus could augment injury due to platelet-leukocyte interactions. However, mechanisms underlying P-selectin and vWf expression on lung endothelium are not clear. Our findings address this issue.
Both vWf and P-selectin are expressed in endothelial WPBs (18). One mechanism by which the proteins might be expressed on the endothelial surface could relate to calcium-induced WPB exocytosis. These mechanisms are likely to be important under conditions such as mechanical stress, which causes prolonged calcium oscillations in the lung endothelium (9). Here, however, the exocytosis mechanism was probably not operative, since no expression of P-selectin or vWf was evident in the absence of platelets. The present colocalized expressions of vWf and P-selectin suggest that, under injury conditions, activated platelets are critical for the endothelial display of the proteins and that they establish sites on the endothelial surface for promoting platelet and leukocyte aggregation (33).
The endothelial TF, vWf, and P-selectin expressions were blocked in NOX2-lacking mice subjected to acid injury, indicating ROS induction was crucial. Blunted expressions of these proteins were evident in endothelium of WT lungs, selectively perfused with NOX2-lacking blood. In this perfusion condition, the failure of platelets to increase TF, vWf, and P-selectin expressions indicates that platelet ROS were critical for the endothelial responses. These findings provide the first evidence that platelet NOX2 determine the expression of leukocyte adhesion proteins in platelets.
From the methodological standpoint, we describe a new flow cytometry-based method to define lung endothelial responses in a segment-specific manner. Although the bulk of the lung's endothelium resides in the alveolar capillary segment, it is increasingly understood that the extra-alveolar segments might be the focus of proinflammatory responses (10) and that these segments may be more permeable (5). We took advantage of the fact that the extra-alveolar endothelium specifically expresses WPBs that secrete vWf. Hence, by a gating protocol that excludes platelets, the other vWf source in the pulmonary circulation, it was possible to address responses in the vWf+ endothelial fraction that likely originates from the extra-alveolar segment. The 4:1 ratio of VE-cadherin to vWf-expressing cells is consistent with the higher alveolar than extra-alveolar vessel distribution (4). We suggest that flow cytometry analyses on primary isolates of lung endothelial cells might provide a novel approach to understanding disease responses in different pulmonary segments.
In conclusion, our findings indicate a new platelet-driven mechanism in the induction of a procoagulant lung endothelial surface in acid-induced ALI. Since the effects are ROS dependent, the procoagulant state requires ROS production at the site of injury. The significance of these procoagulant responses needs understanding, not necessarily as an inherent property of the endothelium, but as a set of events that follow platelet activation. Importantly, the roles of the cell adhesion molecules in platelet and leukocyte adhesion and that of TF need to be understood further. Our findings suggest that the extra-alveolar vascular segment might be a focus of TF expression, hence of procoagulant events. Further studies are needed to determine whether the procoagulant effect of TF leads to thrombin generation, hence the hyperpermeability in lung injury.
GRANTS
This study was supported by National Institutes of Health Grant HL36024 (to J. Bhattacharya).
ACKNOWLEDGMENTS
Tara Guclu helped with manuscript preparation.
REFERENCES
- 1. Allen GB, Cloutier ME, Larrabee YC, Tetenev K, Smiley ST, Bates JH. Neither fibrin nor plasminogen activator inhibitor-1 deficiency protects lung function in a mouse model of acute lung injury. Am J Physiol Lung Cell Mol Physiol 296: L277– L285, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andrews RK, Berndt MC. Platelet adhesion: a game of catch and release. J Clin Invest 118: 3009– 3011, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhattacharya S, Sen N, Yiming MT, Patel R, Parthasarathi K, Quadri S, Issekutz AC, Bhattacharya J. High tidal volume ventilation induces proinflammatory signaling in rat lung endothelium. Am J Respir Cell Mol Biol 28: 218– 224, 2003 [DOI] [PubMed] [Google Scholar]
- 4. Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev 83: 309– 336, 2003 [DOI] [PubMed] [Google Scholar]
- 5. Chetham PM, Babal P, Bridges JP, Moore TM, Stevens T. Segmental regulation of pulmonary vascular permeability by store-operated Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 276: L41– L50, 1999 [DOI] [PubMed] [Google Scholar]
- 6. Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro A, Ruco L, McDonald DM, Ward PA, Dejana E. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci USA 96: 9815– 9820, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 106: 1604– 1611, 2005 [DOI] [PubMed] [Google Scholar]
- 8. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med 359: 938– 949, 2008 [DOI] [PubMed] [Google Scholar]
- 9. Ichimura H, Parthasarathi K, Issekutz AC, Bhattacharya J. Pressure-induced leukocyte margination in lung postcapillary venules. Am J Physiol Lung Cell Mol Physiol 289: L407– L412, 2005 [DOI] [PubMed] [Google Scholar]
- 10. Ichimura H, Parthasarathi K, Quadri S, Issekutz AC, Bhattacharya J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J Clin Invest 111: 691– 699, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Idell S, Peters J, James KK, Fair DS, Coalson JJ. Local abnormalities of coagulation and fibrinolytic pathways that promote alveolar fibrin deposition in the lungs of baboons with diffuse alveolar damage. J Clin Invest 84: 181– 193, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kretz CA, Vaezzadeh N, Gross PL. Tissue factor and thrombosis models. Arterioscler Thromb Vasc Biol 30: 900– 908, 2010 [DOI] [PubMed] [Google Scholar]
- 13. Kuebler WM, Ying X, Singh B, Issekutz AC, Bhattacharya J. Pressure is proinflammatory in lung venular capillaries. J Clin Invest 104: 495– 502, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kyriakides C, Austen W, Jr, Wang Y, Favuzza J, Moore FD, Jr, Hechtman HB. Endothelial selectin blockade attenuates lung permeability of experimental acid aspiration. Surgery 128: 327– 331, 2000 [DOI] [PubMed] [Google Scholar]
- 15. Lu Q, Malinauskas RA. Comparison of two platelet activation markers using flow cytometry after in vitro shear stress exposure of whole human blood. Artif Organs 35: 137– 144, 2011 [DOI] [PubMed] [Google Scholar]
- 16. Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med 344: 665– 671, 2001 [DOI] [PubMed] [Google Scholar]
- 17. Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295: L379– L399, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McEver RP, Beckstead JH, Moore KL, Marshall-Carlson L, Bainton DF. GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J Clin Invest 84: 92– 99, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mustelin T, Vang T, Bottini N. Protein tyrosine phosphatases and the immune response. Nat Rev Immunol 5: 43– 57, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Nagase T, Ishii S, Kume K, Uozumi N, Izumi T, Ouchi Y, Shimizu T. Platelet-activating factor mediates acid-induced lung injury in genetically engineered mice. J Clin Invest 104: 1071– 1076, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Osterud B. Tissue factor expression in blood cells. Thromb Res 125, Suppl 1: S31– S34, 2010 [DOI] [PubMed] [Google Scholar]
- 22. Owens AP, 3rd, Mackman N. Tissue factor and thrombosis: the clot starts here. Thromb Haemost 104: 432– 439, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pawlinski R, Wang JG, Owens AP, 3rd, Williams J, Antoniak S, Tencati M, Luther T, Rowley JW, Low EN, Weyrich AS, Mackman N. Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood 116: 806– 814, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prabhakaran P, Ware LB, White KE, Cross MT, Matthay MA, Olman MA. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol 285: L20– L28, 2003 [DOI] [PubMed] [Google Scholar]
- 25. Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res 100: 1673– 1685, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Solovey A, Kollander R, Shet A, Milbauer LC, Choong S, Panoskaltsis-Mortari A, Blazar BR, Kelm RJ, Jr, Hebbel RP. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood 104: 840– 846, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Tabuchi A, Kuebler WM. Endothelium-platelet interactions in inflammatory lung disease. Vascul Pharmacol 49: 141– 150, 2008 [DOI] [PubMed] [Google Scholar]
- 28. Terrisse AD, Puech N, Allart S, Gourdy P, Xuereb JM, Payrastre B, Sie P. Internalization of microparticles by endothelial cells promotes platelet/endothelial cell interaction under flow. J Thromb Haemost 8: 2810– 2819, 2010 [DOI] [PubMed] [Google Scholar]
- 29. Totani L, Evangelista V. Platelet-leukocyte interactions in cardiovascular disease and beyond. Arterioscler Thromb Vasc Biol 30: 2357– 2361, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood 117: 5033– 5043, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van der Poll T. Tissue factor as an initiator of coagulation and inflammation in the lung. Crit Care 12, Suppl 6: S3, 1– 9, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yiming MT, Lederer DJ, Sun L, Huertas A, Issekutz AC, Bhattacharya S. Platelets enhance endothelial adhesiveness in high tidal volume ventilation. Am J Respir Cell Mol Biol 39: 569– 575, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest 116: 3211– 3219, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]