

Abstract
In the title compound, C19H13N3O, the dihedral angle between the two quinoline systems is 11.54 (3)°. The molecular conformation is stabilized by intramolecular N—H⋯N and C—H⋯O hydrogen bonds, with N—H⋯N being bifurcated towards the two N atoms of the two quinoline rings. In the crystal, there are weak intermolecular π–π interactions present involving the quinoline rings [centroid–centroid distance 3.7351 (14) Å].
Related literature
For the synthesis of the title compound and related structures, see: Kim et al. (2009 ▶). For applications of the title compound and background to the synthesis, see: Wang et al. (2011 ▶).
Experimental
Crystal data
C19H13N3O
M r = 299.32
Orthorhombic,
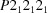
a = 6.3651 (13) Å
b = 11.475 (2) Å
c = 19.861 (4) Å
V = 1450.6 (5) Å3
Z = 4
Mo Kα radiation
μ = 0.09 mm−1
T = 173 K
0.25 × 0.15 × 0.15 mm
Data collection
Rigaku Saturn 724 CCD diffractometer
Absorption correction: multi-scan (ABSCOR; Higashi, 1995 ▶) T min = 0.978, T max = 0.987
6769 measured reflections
1553 independent reflections
1442 reflections with I > 2σ(I)
R int = 0.036
Refinement
R[F 2 > 2σ(F 2)] = 0.035
wR(F 2) = 0.082
S = 1.06
1553 reflections
208 parameters
H-atom parameters constrained
Δρmax = 0.10 e Å−3
Δρmin = −0.13 e Å−3
Data collection: CrystalClear (Rigaku, 2008 ▶); cell refinement: CrystalClear; data reduction: CrystalClear; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: DIAMOND (Brandenburg, 2006 ▶); software used to prepare material for publication: SHELXTL (Sheldrick, 2008 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812020144/zl2476sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812020144/zl2476Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812020144/zl2476Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2—H2A⋯N1 | 0.88 | 2.27 | 2.693 (2) | 109 |
| N2—H2A⋯N3 | 0.88 | 2.27 | 2.684 (2) | 109 |
| C12—H12⋯O1 | 0.95 | 2.25 | 2.867 (2) | 122 |
Acknowledgments
The authors thank the National Natural Science Foundation of China for financial support (grant No. 51072071).
supplementary crystallographic information
Comment
The title compound (Hqcq) can act as a tridentate ligand, and has been incorporated into the cyanometalate building block [Fe(qcq)(CN)3]- {qcq = 8-(2-quinolinecarboxamido)quinoline anion}, in which the FeIII ion is coordinated by three carbon atoms of cyanide groups and three N-donors from the qcq ligand in a mer-arrangement (Kim et al., 2009). Through replacement of the cyanide ligands the Fe(qcq) fragment can coordinate to transition metal ions to form various polynuclear and one-dimensional structures with fascinating magnetic properties such as single molecular magnets and single-chain magnets (Kim et al., 2009; Wang et al., 2011). Herein, the crystal structure of the tridentate ligand of Hqcq is presented.
The molecular structure of the title compound is shown in Fig. 1. The quinoline rings are essentially planar, with a maximum deviation of 0.046 (1) Å for atom C8 in the (N1/C1-C9) ring and 0.016 (1) Å for atom C14 in the (N3/C11-C19) ring. The dihedral angle between the two quinoline rings is 11.54 (3)°. The amide (N2/C10/O1) plane forms dihedral angles of 14.1 (1)° and 4.2 (1)° with the quinoline rings of (N1/C1-C9) and (N3/C11-C19), respectively. The bond lengths of the title molecule are slightly different from those reported for [Fe(qcq)(CN)3]- (Kim et al., 2009), probably owing to the coordination effect to the tridentate ligand. There are intramolecular hydrogen-bonding interactions between the amido N atom and the N atoms of the quinoline rings, and between the O atom of amide group and the C atom of the quinoline ring. The amido N atom (N2) forms bifurcated hydrogen bonds towards the two N atoms (N1, N3) of the two quinoline rings (Table 1).
In the crystal structure, no significant intermolecular hydrogen bonds are observed. The crystal structure features intermolecular π-π interactions between different types of quinoline rings with a distance of ca. 3.735 Å between the centroids of the respective rings (Fig. 2), and the adjacent rings tilted against each other.
Experimental
The compound of 8-(2-quinolinecarboxamido)quinoline (Hqcq) was preparecd according to a literature method (Kim et al., 2009). Then, 0.3 mmol of Hqcq was added to MeCN (20 mL) with stirring. The resulting solution was filtered and the filtrate was left for slow evaporation in the dark at room temperature. Yellow block-shaped crystals of the title compound suitable for single-crystal X-ray diffraction were obtained after two weeks. Melting point = 429.6-430.5 K. IR (KBr, cm-1): 3314(s), 3044(m), 1678(vs), 1523(vs), 1488(s), 1427(s), 1325(s), 1126(m), 913(s), 834(s), 764(vs), 611(m), 588(m).
Refinement
All non-H atoms were refined with anisotropic thermal parameters. The C- and N-bound H atoms were calculated in idealized positions and included in the refinement in a riding mode (C-H = 0.95 Å, N-H = 0.88 Å) with Uiso for H assigned as 1.2 times Ueq of the attached atoms. In the absence of atoms heavier than Si and with Mo Kα radiation used Friedel-pair reflections have been merged (using a MERG 3 command) during the refinement. Assignment of the absolute structure is arbitrary.
Figures
Fig. 1.

ORTEP diagram of the title compound with displacement ellipsoids drawn at the 30% probability level.
Fig. 2.

The crystal packing diagram of the title compound showing the intermolecular π-π interactions.The distances shown are between the centroids of the respective rings, and the symmetry operator codes for generating adjacent aromatic rings are -1+x, y, z and 1+x, y, z.
Crystal data
| C19H13N3O | F(000) = 624 |
| Mr = 299.32 | Dx = 1.371 Mg m−3 |
| Orthorhombic, P212121 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P2ac2ab | Cell parameters from 6054 reflections |
| a = 6.3651 (13) Å | θ = 3.4–29.0° |
| b = 11.475 (2) Å | µ = 0.09 mm−1 |
| c = 19.861 (4) Å | T = 173 K |
| V = 1450.6 (5) Å3 | Block, yellow |
| Z = 4 | 0.25 × 0.15 × 0.15 mm |
Data collection
| Rigaku Saturn 724 CCD diffractometer | 1553 independent reflections |
| Radiation source: Rotating Anode | 1442 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.036 |
| φ and ω scans | θmax = 25.3°, θmin = 3.4° |
| Absorption correction: multi-scan (ABSCOR; Higashi, 1995) | h = −7→6 |
| Tmin = 0.978, Tmax = 0.987 | k = −13→13 |
| 6769 measured reflections | l = −23→18 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.035 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.082 | H-atom parameters constrained |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0494P)2] where P = (Fo2 + 2Fc2)/3 |
| 1553 reflections | (Δ/σ)max < 0.001 |
| 208 parameters | Δρmax = 0.10 e Å−3 |
| 0 restraints | Δρmin = −0.13 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | −0.0149 (2) | 0.41328 (13) | 0.73413 (7) | 0.0458 (4) | |
| N1 | −0.1752 (2) | 0.46907 (14) | 0.56729 (8) | 0.0328 (4) | |
| C1 | −0.3327 (3) | 0.44137 (16) | 0.52317 (10) | 0.0309 (4) | |
| N2 | 0.1160 (2) | 0.54135 (14) | 0.65635 (8) | 0.0336 (4) | |
| H2A | 0.0898 | 0.5707 | 0.6163 | 0.040* | |
| C2 | −0.3099 (3) | 0.47410 (18) | 0.45477 (10) | 0.0367 (5) | |
| H2 | −0.1874 | 0.5146 | 0.4407 | 0.044* | |
| N3 | 0.3570 (3) | 0.69181 (14) | 0.58750 (8) | 0.0385 (4) | |
| C3 | −0.4630 (3) | 0.44780 (19) | 0.40896 (10) | 0.0405 (5) | |
| H3 | −0.4453 | 0.4691 | 0.3631 | 0.049* | |
| C4 | −0.6456 (3) | 0.38961 (18) | 0.42926 (11) | 0.0416 (5) | |
| H4 | −0.7514 | 0.3724 | 0.3970 | 0.050* | |
| C5 | −0.6737 (3) | 0.35718 (18) | 0.49498 (10) | 0.0390 (5) | |
| H5 | −0.7993 | 0.3187 | 0.5082 | 0.047* | |
| C6 | −0.5164 (3) | 0.38082 (16) | 0.54318 (10) | 0.0324 (5) | |
| C7 | −0.5302 (3) | 0.34377 (17) | 0.61049 (10) | 0.0364 (5) | |
| H7 | −0.6505 | 0.3024 | 0.6257 | 0.044* | |
| C8 | −0.3693 (3) | 0.36763 (17) | 0.65394 (10) | 0.0358 (5) | |
| H8 | −0.3739 | 0.3413 | 0.6993 | 0.043* | |
| C9 | −0.1960 (3) | 0.43222 (16) | 0.62995 (10) | 0.0317 (5) | |
| C10 | −0.0232 (3) | 0.46113 (17) | 0.67895 (10) | 0.0329 (4) | |
| C11 | 0.2965 (3) | 0.58357 (17) | 0.68860 (10) | 0.0322 (5) | |
| C12 | 0.3571 (3) | 0.55062 (19) | 0.75238 (9) | 0.0362 (5) | |
| H12 | 0.2716 | 0.4994 | 0.7781 | 0.043* | |
| C13 | 0.5465 (3) | 0.59343 (19) | 0.77909 (10) | 0.0409 (6) | |
| H13 | 0.5880 | 0.5697 | 0.8229 | 0.049* | |
| C14 | 0.6721 (3) | 0.66767 (18) | 0.74419 (10) | 0.0388 (5) | |
| H14 | 0.7999 | 0.6945 | 0.7635 | 0.047* | |
| C15 | 0.6127 (3) | 0.70472 (17) | 0.67950 (10) | 0.0341 (5) | |
| C16 | 0.7326 (3) | 0.78261 (18) | 0.64020 (11) | 0.0404 (5) | |
| H16 | 0.8604 | 0.8134 | 0.6572 | 0.049* | |
| C17 | 0.6643 (4) | 0.81326 (18) | 0.57795 (11) | 0.0423 (5) | |
| H17 | 0.7423 | 0.8667 | 0.5513 | 0.051* | |
| C18 | 0.4767 (4) | 0.76476 (18) | 0.55348 (10) | 0.0436 (6) | |
| H18 | 0.4330 | 0.7860 | 0.5094 | 0.052* | |
| C19 | 0.4227 (3) | 0.66226 (17) | 0.65098 (10) | 0.0315 (4) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0481 (9) | 0.0504 (9) | 0.0389 (9) | −0.0030 (8) | −0.0031 (8) | 0.0133 (7) |
| N1 | 0.0336 (9) | 0.0318 (9) | 0.0328 (9) | 0.0016 (8) | 0.0003 (8) | −0.0013 (7) |
| C1 | 0.0303 (10) | 0.0286 (10) | 0.0337 (10) | 0.0012 (9) | 0.0019 (9) | −0.0059 (8) |
| N2 | 0.0334 (9) | 0.0372 (9) | 0.0301 (8) | −0.0003 (8) | −0.0032 (8) | 0.0028 (7) |
| C2 | 0.0339 (11) | 0.0405 (12) | 0.0356 (11) | −0.0021 (11) | 0.0030 (9) | −0.0031 (9) |
| N3 | 0.0466 (10) | 0.0354 (9) | 0.0335 (9) | −0.0013 (9) | −0.0026 (9) | 0.0011 (7) |
| C3 | 0.0426 (12) | 0.0425 (12) | 0.0366 (11) | 0.0039 (11) | −0.0025 (10) | −0.0047 (10) |
| C4 | 0.0383 (12) | 0.0374 (12) | 0.0492 (14) | 0.0031 (11) | −0.0089 (11) | −0.0080 (10) |
| C5 | 0.0342 (11) | 0.0317 (11) | 0.0512 (14) | −0.0025 (11) | −0.0032 (10) | −0.0044 (10) |
| C6 | 0.0317 (11) | 0.0257 (10) | 0.0397 (11) | 0.0023 (9) | 0.0021 (9) | −0.0040 (8) |
| C7 | 0.0357 (11) | 0.0287 (10) | 0.0448 (12) | 0.0019 (10) | 0.0075 (10) | −0.0004 (9) |
| C8 | 0.0393 (11) | 0.0314 (10) | 0.0367 (11) | 0.0020 (10) | 0.0046 (10) | 0.0003 (9) |
| C9 | 0.0343 (11) | 0.0276 (10) | 0.0332 (11) | 0.0046 (9) | 0.0032 (9) | −0.0015 (8) |
| C10 | 0.0328 (11) | 0.0327 (10) | 0.0332 (10) | 0.0056 (10) | 0.0031 (9) | −0.0010 (9) |
| C11 | 0.0324 (11) | 0.0337 (10) | 0.0307 (11) | 0.0043 (10) | −0.0002 (9) | −0.0053 (8) |
| C12 | 0.0385 (11) | 0.0417 (12) | 0.0283 (10) | 0.0031 (11) | 0.0018 (10) | −0.0015 (9) |
| C13 | 0.0418 (12) | 0.0510 (14) | 0.0300 (11) | 0.0074 (12) | −0.0052 (10) | −0.0054 (9) |
| C14 | 0.0352 (11) | 0.0462 (13) | 0.0350 (12) | 0.0071 (11) | −0.0055 (10) | −0.0104 (10) |
| C15 | 0.0360 (11) | 0.0332 (10) | 0.0330 (10) | 0.0030 (10) | 0.0024 (9) | −0.0092 (9) |
| C16 | 0.0402 (12) | 0.0358 (11) | 0.0454 (13) | −0.0030 (11) | 0.0036 (10) | −0.0139 (10) |
| C17 | 0.0496 (14) | 0.0350 (11) | 0.0423 (13) | −0.0087 (11) | 0.0051 (11) | −0.0044 (9) |
| C18 | 0.0563 (14) | 0.0372 (12) | 0.0374 (11) | −0.0050 (12) | −0.0009 (11) | 0.0031 (9) |
| C19 | 0.0336 (10) | 0.0313 (10) | 0.0295 (10) | 0.0053 (9) | 0.0008 (9) | −0.0052 (8) |
Geometric parameters (Å, º)
| O1—C10 | 1.227 (2) | C7—H7 | 0.9500 |
| N1—C9 | 1.321 (2) | C8—C9 | 1.412 (3) |
| N1—C1 | 1.369 (2) | C8—H8 | 0.9500 |
| C1—C2 | 1.417 (3) | C9—C10 | 1.505 (3) |
| C1—C6 | 1.417 (3) | C11—C12 | 1.377 (3) |
| N2—C10 | 1.354 (3) | C11—C19 | 1.421 (3) |
| N2—C11 | 1.402 (2) | C12—C13 | 1.406 (3) |
| N2—H2A | 0.8800 | C12—H12 | 0.9500 |
| C2—C3 | 1.367 (3) | C13—C14 | 1.358 (3) |
| C2—H2 | 0.9500 | C13—H13 | 0.9500 |
| N3—C18 | 1.318 (3) | C14—C15 | 1.405 (3) |
| N3—C19 | 1.371 (2) | C14—H14 | 0.9500 |
| C3—C4 | 1.400 (3) | C15—C16 | 1.411 (3) |
| C3—H3 | 0.9500 | C15—C19 | 1.421 (3) |
| C4—C5 | 1.369 (3) | C16—C17 | 1.357 (3) |
| C4—H4 | 0.9500 | C16—H16 | 0.9500 |
| C5—C6 | 1.412 (3) | C17—C18 | 1.405 (3) |
| C5—H5 | 0.9500 | C17—H17 | 0.9500 |
| C6—C7 | 1.405 (3) | C18—H18 | 0.9500 |
| C7—C8 | 1.367 (3) | ||
| C9—N1—C1 | 117.09 (17) | C8—C9—C10 | 117.92 (17) |
| N1—C1—C2 | 118.50 (18) | O1—C10—N2 | 124.89 (19) |
| N1—C1—C6 | 122.59 (17) | O1—C10—C9 | 120.68 (19) |
| C2—C1—C6 | 118.90 (18) | N2—C10—C9 | 114.42 (16) |
| C10—N2—C11 | 128.30 (17) | C12—C11—N2 | 123.71 (19) |
| C10—N2—H2A | 115.8 | C12—C11—C19 | 120.01 (19) |
| C11—N2—H2A | 115.8 | N2—C11—C19 | 116.26 (17) |
| C3—C2—C1 | 120.5 (2) | C11—C12—C13 | 119.4 (2) |
| C3—C2—H2 | 119.8 | C11—C12—H12 | 120.3 |
| C1—C2—H2 | 119.8 | C13—C12—H12 | 120.3 |
| C18—N3—C19 | 116.89 (19) | C14—C13—C12 | 122.1 (2) |
| C2—C3—C4 | 120.37 (19) | C14—C13—H13 | 118.9 |
| C2—C3—H3 | 119.8 | C12—C13—H13 | 118.9 |
| C4—C3—H3 | 119.8 | C13—C14—C15 | 119.8 (2) |
| C5—C4—C3 | 120.8 (2) | C13—C14—H14 | 120.1 |
| C5—C4—H4 | 119.6 | C15—C14—H14 | 120.1 |
| C3—C4—H4 | 119.6 | C14—C15—C16 | 123.5 (2) |
| C4—C5—C6 | 120.1 (2) | C14—C15—C19 | 119.3 (2) |
| C4—C5—H5 | 119.9 | C16—C15—C19 | 117.19 (18) |
| C6—C5—H5 | 119.9 | C17—C16—C15 | 119.7 (2) |
| C7—C6—C5 | 122.86 (19) | C17—C16—H16 | 120.2 |
| C7—C6—C1 | 117.81 (18) | C15—C16—H16 | 120.2 |
| C5—C6—C1 | 119.30 (18) | C16—C17—C18 | 119.0 (2) |
| C8—C7—C6 | 119.55 (19) | C16—C17—H17 | 120.5 |
| C8—C7—H7 | 120.2 | C18—C17—H17 | 120.5 |
| C6—C7—H7 | 120.2 | N3—C18—C17 | 124.4 (2) |
| C7—C8—C9 | 118.52 (19) | N3—C18—H18 | 117.8 |
| C7—C8—H8 | 120.7 | C17—C18—H18 | 117.8 |
| C9—C8—H8 | 120.7 | N3—C19—C11 | 117.95 (18) |
| N1—C9—C8 | 124.35 (19) | N3—C19—C15 | 122.76 (19) |
| N1—C9—C10 | 117.73 (18) | C11—C19—C15 | 119.28 (18) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2A···N1 | 0.88 | 2.27 | 2.693 (2) | 109 |
| N2—H2A···N3 | 0.88 | 2.27 | 2.684 (2) | 109 |
| C12—H12···O1 | 0.95 | 2.25 | 2.867 (2) | 122 |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: ZL2476).
References
- Brandenburg, K. (2006). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Higashi, T. (1995). ABSCOR Rigaku Corporation, Tokyo, Japan.
- Kim II, J., Kwak, H. Y., Yoon, J. H., Ryu, D. W., Yoo, I. Y., Yang, N., Cho, B. K., Park, J. G., Lee, H. & Hong, C. S. (2009). Inorg. Chem. 48, 2956–2966. [DOI] [PubMed]
- Rigaku (2008). CrystalClear Rigaku Corporation, Tokyo, Japan.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wang, S., Ding, X. H., Zuo, J. L., You, X. Z. & Huang, W. (2011). Coord. Chem. Rev. 256, 1713–1732.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812020144/zl2476sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812020144/zl2476Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812020144/zl2476Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report