

Abstract
Keloid scarring is a consequence of aberrant wound healing that leads to expansion of the scar beyond the confines of the skin injury. Keloid scars are characterized by excessive extracellular matrix disposition, prolonged proliferation of fibroblasts, increased angiogenesis, and inflammatory cell infiltration. There is no single satisfactory treatment for keloid, and it can lead to severe disfigurements and bodily dysfunction. Thus, clarification of the mechanisms underlying keloid formation, as well as those that prevent it from behaving as a malignant tumor, has significant consequences not only for treatment of keloid but also for the prevention of malignant tumor formation. Senescence is an irreversible form of growth arrest that has been shown to play a role, both in vitro and in vivo, in preventing malignant tumorigenesis upon oncogenic stress. In this study it is shown that fibroblasts embedded inside keloid scars proliferate at a slower rate compared with either those residing at the proliferative edges of the scar or normal fibroblasts. Likewise it is demonstrated that keloid fibroblasts exhibit a cell-cycle arrest with a G2/M DNA content and a higher rate of senescence. The results also indicate that levels of the tumor suppressor protein PML are higher in the active regions of keloid. The study therefore suggests that senescence is one possible mechanism by which keloid is maintained in a benign state. On this basis, “pro-senescence therapy” should be taken into consideration when designing treatment strategies for keloid.
Keywords: keloid, senescence, PML
Introduction
Dermal injury triggers the complicated process of wound healing, which engages several cellular pathways whose activation leads to repair and rebuilding of the injured site.1 Keloid scarring is an abnormal form of wound healing characterized by excessive disposition of the extracellular matrix, in particular collagen, as well as extended presence of proliferative fibroblasts at the wound site.2,3 The mechanisms involved in keloid formation are only partially understood and are believed to include genetic predisposition, aberrant growth factor regulation, and collagen production as well as dysfunction in the immune system.4 Treatment for keloid remains elusive. Keloid is also described as benign dermal tumors arising from an aberrant wound healing process. The process of wound healing and tumorigenesis share many characteristics; hence, cancer has been described as an “overhealing wound.”1 One of the common aspects shared by keloid and malignant tumors is the capacity of prolonged cellular proliferation.1 After a certain period, however, keloid fibroblasts spontaneously cease to proliferate, and therefore keloid remains benign. Elucidating the molecular mechanisms of this trigger could have therapeutic implications not only for keloid but also for cancer.
Cellular senescence describes an irreversible growth arrest that operates as a failsafe mechanism in response to oncogenic insults.5 Although the ability of cellular senescence to prevent cellular transformation in cultured cells in vitro is well established, several recent studies have also demonstrated a role for senescence in preventing transformation in vivo.6-9 We investigated here a possible role for cellular senescence in halting proliferation of keloid fibroblasts. Our results demonstrate that senescence is indeed one possible mechanism involved in halting proliferation of keloid fibroblasts and that the induction of the tumor suppressor protein PML may participate in this process.
Results and Discussion
Decreased proliferation rate in keloid fibroblasts
Aberrant proliferation is one of the fundamental components underlying tumorigenesis and has been suggested to increase in keloid fibroblasts during keloid formation. To examine the proliferative capacity of keloid fibroblasts in vitro, we isolated fibroblasts from 2 different regions of keloid scars. These included fibroblasts at the edges of keloid scars and those embedded deep in the scars. Fibroblasts isolated from normal skin were included as control. Fibroblasts were cultured and plated with the same density at each passage. At passage 3, the number of cells was counted following plating at the indicated times (Fig. 1). As shown in Fig. 1, fibroblasts from the edges of keloid scars proliferated at a similar rate to normal fibroblasts. Hereafter, we term these fibroblasts proliferative keloid fibroblasts (PKFs). Interestingly, fibroblasts isolated from deep within the keloid scars proliferated at a significantly slower rate (Fig. 1). Hereafter, we term these fibroblasts quiescent keloid fibroblasts (QKFs). These results suggest that keloid fibroblasts lose their proliferative capacity earlier than normal fibroblasts. This finding is in agreement with a previous observation demonstrating that keloid fibroblasts when sparsely plated lose their replicative capacity earlier than their normal counterparts.10,11 Our QKFs were isolated from within the scars where the cell populations were sparse and separated by excessive amount of extracellular matrix. Considering the increased number of fibroblasts at the edges of keloid in vivo, we expected PKFs to show a faster proliferation rate than normal fibroblasts. This, however, was not the case, which could be due to the differences in the surrounding environment of the fibroblasts in vivo and in vitro. Our finding that PKFs show similar growth kinetics to normal fibroblasts is, however, in agreement with previous studies.11
Figure 1.
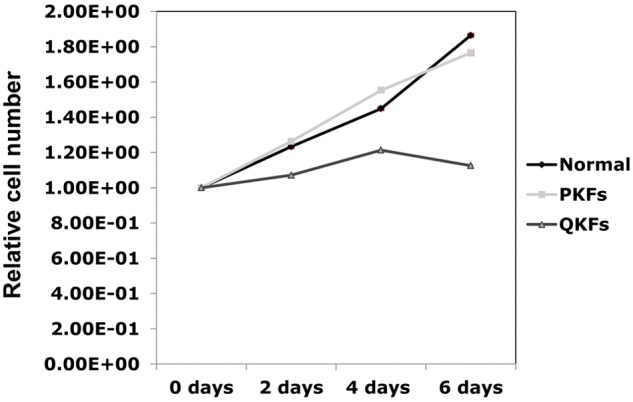
Representative growth curves corresponding to the indicated fibroblasts isolated from keloid scars and normal skin. Fibroblasts isolated from quiescent parts (QKFs) of keloid scars proliferate slower than those isolated from proliferative regions (PKFs) of keloid and normal skin. The experiments were performed using fibroblasts at passage number 3. The results shown are representative of those from 3 independent patients. All standard deviations were negligible (all SD <7.6 × 10−3) and hence barely visible in the figure.
Keloid fibroblasts arrest with a 4N DNA content
Next we examined the cell-cycle profile of the keloid fibroblasts and their normal counterparts (Fig. 2). Fibroblasts at passage 2, 5, and 8 were harvested 24 hours after plating and their cell-cycle profiles were examined. Figure 2 is representative for 3 patients. PKFs showed the same cell-cycle profile as their normal control at all examined passage numbers (Fig. 2). QKFs exhibited the same cell-cycle profile as PKFs and normal fibroblasts at passage 2 (Fig. 2). QKFs, however, accumulated with a 4N DNA content at passage 5 (Fig. 2). The number of QKFs with a 4N DNA content had increased at passage 8 (Fig. 2). These results indicate that the loss in proliferative capacity in keloid fibroblasts is a consequence of cell cycle arrest at the G2/M phase of the cell cycle.
Figure 2.

Comparison of the cell-cycle profiles of keloid and normal skin fibroblasts. Quiescent keloid fibroblasts (QKFs) arrest with a 4N DNA content at passage numbers 5 and 8 when compared with proliferative keloid (PKFs) and normal skin fibroblasts. Bar graphs are quantifications of cell numbers in different phases of the cell cycle (lower panels). Shown results are representative of those obtained from analyzing fibroblasts originated from 3 independent patients. P indicates passage number.
Senescence rate is higher in keloid fibroblasts
Slower proliferation rate and cell-cycle arrest observed in QKFs prompted us to investigate the status of senescence in keloid fibroblasts. We compared the senescence status in the indicated fibroblasts from 2 patients at passage 2 and 5 using the senescence-associated β-galactosidase activity analysis (Fig. 3). Percentage of β-galactosidase positive cells as a measure of occurrence of senescence was slightly higher in PKFs compared with normal fibroblasts as early as passage number 2 (Fig. 3). This percentage increased to approximately twice of that observed in normal fibroblasts in both patients (from about 7% to 15%) at passage number 5 (Fig. 3). Consistent with the above results, QKFs showed higher percentage of β-galactosidase positive cells at passage 2 compared with PKFs and normal fibroblasts (between 15% and 20% compared with 5% and 8%) (Fig. 3). The percentage of β-galactosidase positive QKFs increased to 25% to 30% at passage 5 (Fig. 3). These results indicate that keloid fibroblasts commit to senescence earlier than their normal counterparts in vitro. Thus, cellular senescence and ensuing cell cycle arrest underlie keloid evolution and could explain why these lesions eventually stop growing and do not evolve to malignancy.
Figure 3.
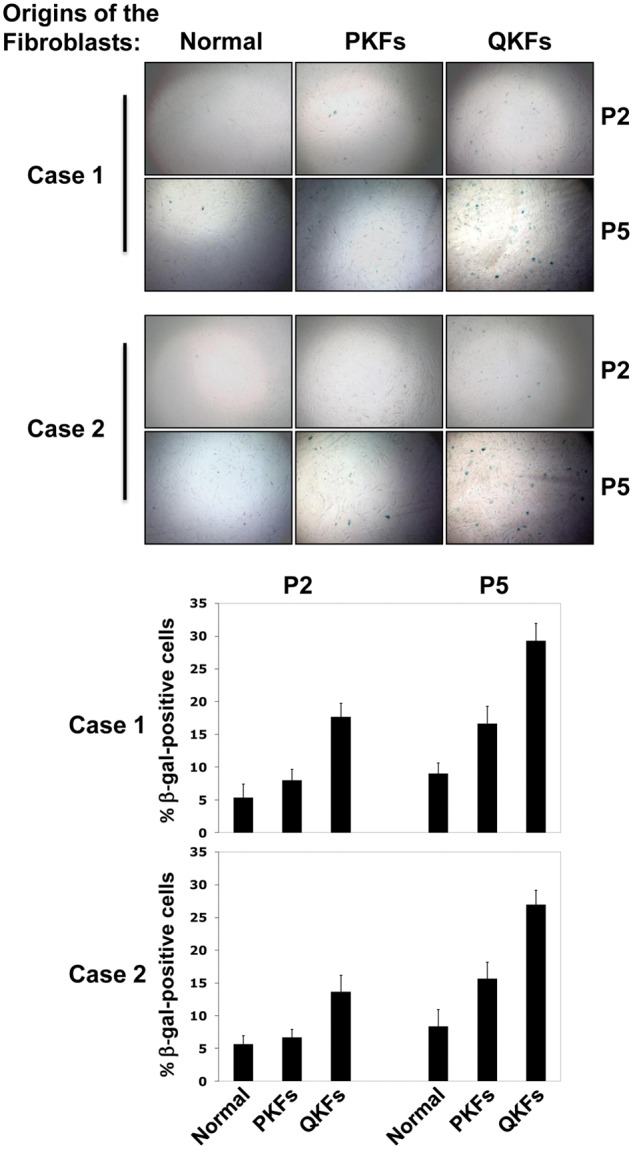
Keloid fibroblasts senesce earlier than normal skin fibroblasts. Fibroblasts with the indicated origins and passage numbers (P2 and 5) were fixed and stained for β-galactosidase (β-gal) activity (upper panels). Bar graphs show quantifications of β-gal positive cells (lower panels). Results from 2 patients are shown.
Increased PML levels in proliferative regions of keloid
The PML tumor suppressor has been shown to be a potent inducer of cellular senescence in fibroblasts.12-14 Higher occurrence of senescence in keloid fibroblasts prompted us to perform immunohistochemistry analysis to investigate the status of PML levels in keloid scars in vivo (Fig. 4). Interestingly, PML levels appeared to be elevated in proliferative regions of keloid scars (characterized by a higher number of cells) when compared with both quiescent parts (regions with sparse cell populations) and normal skin (Fig. 4). Results from 5 independent keloid scars from 5 patients are shown (Fig. 4). These results suggest that PML is a likely inducer of senescence in keloid scars. It’s conceivable that an abnormal increase in proliferation or enhanced inflammatory responses in proliferative fibroblasts residing in proliferative keloid regions triggers an accumulation of PML that in turn activates the senescence response. This in turn may lead to the conversion of proliferative fibroblasts to more quiescent ones embedded in the scars and thus a gradual inhibition of keloid progression. It would be interesting to examine the status and timing of formation of PML nuclear bodies in normal scar tissues. Future experiments to clarify the possible role of PML bodies in normal wound healing and scar formation are warranted. Intriguingly, CCN1/CYR61, a matricellular protein, has been shown to exert antifibrotic effects in normal wound healing process by inducing senescence.15 It would be interesting to examine the status of CCN1/CYR61 and its possible functional cross-talk with PML in the process of keloid formation as well as normal wound healing.
Figure 4.

Enhanced levels of PML expression in proliferative regions of keloid lesions. Immunohistochemistry analysis using PML antibodies shows that PML levels are increased in fibroblasts in proliferative regions (PKFs) of keloid lesions as compared with those in quiescent regions (QKFs) or normal skin. Results from keloid lesions from 5 patients are shown. Magnification 60× of Case 5 is shown as an example to demonstrate nuclear bodies containing PML. The arrows point to examples of nuclear bodies containing PML.
In summary, our study highlights the importance of cellular senescence as a mechanism of preventing abnormal proliferation in keloid. On this basis we propose that PML-enhancing drugs (e.g., interferon), and “pro-senescence” therapeutic approaches could be used to control keloid onset and recurrence.16,17
Materials and Methods
Generation of primary cultures of fibroblasts isolated from keloid and normal skin tissue
All keloid tissue and normal skin were collected and stored in DMEM (supplemented with 2 mmol/L L-glutamine, 100 U/mL penicillin, 100 U/mL streptomycin, 10% heat-inactivated FCS [PAA, Germany], and 1% nonessential amino acids [Sigma-Aldrich, UK]); they were washed thoroughly in PBS (1×) before being minced and incubated in collagenase A (10 mg/mL) for 4 hours at 37°C. The suspensions were passed through a 100 µm filter to remove any remaining tissue residuals. Cells were then washed once with PBS (PAA, UK) and plated in T25 Cell-Bind flasks (Nunc, Life Technologies Ltd., Germany) as passage 1-3 (p 1-3) cells. Monolayer cultures were maintained in complete DMEM medium. Cells were incubated at 37°C in a 5% (v/v) CO2 humidified atmosphere.
Flow cytometry
Cells were fixed in ice-cold 70% ethanol followed by rinsing with PBS (Invitrogen, Carlsbad, CA). DNA was stained with propidium iodide (20 µg/mL, Sigma) in the presence of 1 mg/mL Ribonuclease A (Sigma, St. Louis, MO). Cell cycle analyses were performed on FACSCalibur, and data were analyzed using CellQuest.
Senescence-associated β-galactosidase analysis
To perform senescence analysis, fibroblasts were plated at 104 cells per well of a 6-well plate in triplicate. After 3 days, senescence-associated β-galactosidase activity was measured following manufacturer’s instructions using a senescence detection kit (Calbiochem/Millipore, Billerica, MA) and quantified including more than 200 cells per sample.
Immunohistochemistry analysis
Paraffin-embedded tissue sections were stained with PML antibodies H-238, Santa Cruz Biotechnology, Santa Cruz, CA) following the manufacturer’s instructions.
Acknowledgments
We thank current members of the Pandolfi lab for critical discussions and Thomas Garvey for insightful editing. This work has been supported by a generous philanthropic donation to P.P.P. and A.B. to foster collaborative research between Harvard University and the University of Manchester.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9(8):628-38 [DOI] [PubMed] [Google Scholar]
- 2. Syed F, Ahmadi E, Iqbal SA, Singh S, McGrouther DA, Bayat A. Fibroblasts from the growing margin of Keloid scars produce higher levels of collagen I and III compared with intralesional and extralesional sites: clinical implications for lesional site-directed therapy. Br J Dermatol. 2010;164:83-96 [DOI] [PubMed] [Google Scholar]
- 3. Viera MH, Caperton CV, Berman B. Advances in the treatment of keloids. J Drugs Dermatol. 2011;10(5):468-80 [PubMed] [Google Scholar]
- 4. Shih B, Bayat A. Genetics of keloid scarring. Arch Dermatol Res. 2010;302:319-39 [DOI] [PubMed] [Google Scholar]
- 5. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729-40 [DOI] [PubMed] [Google Scholar]
- 6. Braig M, Lee S, Loddenkemper C, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436(7051):660-5 [DOI] [PubMed] [Google Scholar]
- 7. Chen Z, Trotman LC, Shaffer D. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642. [DOI] [PubMed] [Google Scholar]
- 9. Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436(7051):720-4 [DOI] [PubMed] [Google Scholar]
- 10. Diegelmann RF, Cohen IK, McCoy BJ. Growth kinetics and collagen synthesis of normal skin, normal scar and keloid fibroblasts in vitro. J Cell Physiol. 1979;98(2):341-6 [DOI] [PubMed] [Google Scholar]
- 11. McCoy BJ, Galdun J, Cohen IK. Effects of density and cellular aging on collagen synthesis and growth kinetics in keloid and normal skin fibroblasts. In Vitro. 1982;18(1):79-86 [DOI] [PubMed] [Google Scholar]
- 12. Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14(16):2015-27 [PMC free article] [PubMed] [Google Scholar]
- 13. Mark Pearson RC, Sebastiani C, Cioce M, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406(13):207-10 [DOI] [PubMed] [Google Scholar]
- 14. Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108(2):165-70 [DOI] [PubMed] [Google Scholar]
- 15. Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12(7):676-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alimonti A, Nardella C, Chen Z, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest. 2010;120(3):681-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011;11(7)503-11 [DOI] [PubMed] [Google Scholar]