

Abstract
The inhibitor of κ B kinase-ε (IKKε), a breast cancer oncogene, functions as a transforming kinase by activating NF-κB. IKKε is often elevated in breast cancers in the absence of any gene amplification. Because Akt-mediated transformation was shown to require IKKε, we examined if Akt regulates IKKε level in breast cancer cells. Knockdown of Akt2, but not other Akt isoforms, decreased the basal and TNF-induced IKKε protein and mRNA level, and overexpression of Akt2 in MDA-MB-231 cells increased IKKε level. The decrease in IKKε level by Akt2 knockdown was not only restricted to MDA-MB-231 cells but was also observed in several other breast cancer cells, including HCC1937 and MCF-10CA1a cells. Knockdown of p65/RelA subunit of NF-κB decreased IKKε level and attenuated the increase in IKKε caused by Akt2 overexpression, suggesting that Akt2-mediated induction of IKKε involves NF-κB activation. Silencing of IKKε also decreased long-term clonogenic survival of Akt2-overexpressing MDA-MB-231 cells. Taken together, these results demonstrate for the first time that IKKε functions downstream of Akt2 to promote breast cancer cell survival.
Keywords: IKKε, Akt2, NF-κB, TNF, breast cancer
Introduction
The transcription factor nuclear factor-κB (NF-κB) is an important regulator of critical cellular processes such as cell growth, proliferation, and survival.1-3 Aberration in NF-κB–associated pathways in various breast cancer cell lines and primary tumors has been associated with neoplastic transformation.3-5 NF-κB is primarily activated by the inhibitor of the κ B kinase (IKK) family of proteins, which phosphorylate the inhibitor of NF-κB, IκB. IκB is subsequently degraded, releasing NF-κB to translocate into the nucleus to initiate its transcriptional activities.1-3 Apart from the canonical IKKα, IKKβ, and the adaptor protein IKKγ, the noncanonical members of the IKK family, IKKε and TANK-binding kinase-1 (TBK1), also activate NF-κB.6,7 IKKε was reported to function as a transforming kinase by activating NF-κB.8 It was shown to phosphorylate IκBα preferentially at the Ser36 residue, leading to its degradation and NF-κB activation.9,10 It can also contribute to NF-κB activation by phosphorylating different NF-κB subunits.11-14
Integrative genomic approaches established IKBKE as a novel breast cancer oncogene,8 although subsequent studies have demonstrated the oncogenic properties of IKKε in other forms of cancer.15-18 Elevated levels of IKKε have been detected in primary human breast cancer specimens and in a mouse model of breast cancer,19 and suppression of IKKε compromised cell proliferation and viability in MCF-7 and ZR-75-1 cells.8 IKKε was shown to substitute for Akt in inducing transformation. Two recent reports suggested that IKKε directly phosphorylates Akt,20,21 placing it upstream of the Akt signaling pathway. However, Akt-driven transformation required IKKε, indicating that IKKε may act downstream of Akt.8,22
IKKε is frequently elevated in breast cancers in the absence of any increase in gene copy number or somatic mutation.8 It is an inducible kinase and was shown to be induced by lipopolysaccharide in mouse macrophages.10 It is also induced by tumor-promoting phorbol esters as well as cytokines such as interleukin-1 and -6 and tumor necrosis factor-α (TNF).10 Thus, a defect in the signaling pathways that causes an induction of IKKε may also contribute to breast cancer in the absence of any genetic changes in IKKε. Because IKKε was shown to functionally substitute for Akt, we examined if IKKε acts downstream of Akt. We made a novel observation that Akt2, but not Akt1 or Akt3, positively regulates basal and TNF-mediated induction of IKKε in several breast cancer cells, including MDA-MB-231 breast cancer cells. In addition, induction of IKKε by Akt2 involves activation of NF-κB. Moreover, our results demonstrate for the first time that IKKε promotes breast cancer cell survival by acting downstream of Akt2.
Results
Akt2 positively regulates IKKε expression
IKKε is an inducible kinase, but little is known about how its level is regulated. Because Akt-mediated transformation required IKKε in breast cancer cells,8 we wanted to know if IKKε level is regulated by Akt. Although most of the studies have focused on Akt1, there are 3 isoforms of Akt.23 Because MDA-MB-231 cells express high levels of IKKε, we silenced each Akt isoform with the specific siRNA and monitored IKKε level. Figure 1 shows that while Akt1 and Akt3 knockdown had little effect on IKKε, Akt2 knockdown attenuated IKKε level (Fig. 1A). In contrast, knockdown of Akt isoforms had no effect on IKKβ level.
Figure 1.

Knockdown of Akt2 decreased TNF-induced IKKε levels in MDA-MB-231 cells. (A) Cells were transfected with Akt1, Akt2, Akt3, IKKε, or nontargeting SMARTpool siRNA. (B) Cells were transfected with Akt1, Akt2, or nontargeting siRNA and then treated with or without 1 nM TNF, as indicated in the Materials and Methods. Western blot analyses were carried out with indicated antibodies. (C) The intensity of IKKε was determined by densitometry using ImageJ software and normalized with actin. The data represent the fold change in IKKε level compared with control siRNA–transfected cells. Each bar represents the mean ± standard error of 4 independent experiments. The open bar represents the untreated cells, and the hatched bar represents TNF treatment. **P < 0.01 and ***P < 0.005, using paired Student t test.
Because IKKε is induced by cytokines, such as TNF,10,24 we examined if Akt2 regulates induction of IKKε by TNF. As shown in Figure 1B, TNF caused an increase in IKKε but not IKKβ, and knockdown of Akt2, but not Akt1, decreased TNF-induced IKKε level. Based on densitometric scanning of 4 independent experiments, TNF caused a 1.5-fold increase in IKKε level (Fig. 1C). Upon Akt2 depletion, the basal level of IKKε was decreased by 2-fold, and the TNF-induced IKKε level was decreased by 2.2-fold. To determine if Akt2 regulates IKKε level in other cell lines besides MDA-MB-231 cells, we extended our study to include several other breast cancer cell lines. Figure 2 shows that silencing of Akt2 by siRNA also attenuated basal and TNF-induced IKKε level in HCC1937 and MCF-10CA1a cells. Based on the densitometric quantification, Akt1 knockdown had little effect on IKKε level when corrected for loading.
Figure 2.

Knockdown of Akt2 decreased IKKε level in breast cancer cells. HCC1937 (A) or MCF-10CA1a (B) cells were transfected with indicated siRNAs, treated with or without 1 nM TNF, and Western blot analyses were carried out with the indicated antibodies, as described in the Materials and Methods. The results are representative of 2 individual experiments.
To determine if Akt2 affects IKKε expression at the transcriptional level, we depleted Akt2 using siRNA and monitored IKKε mRNA using RT-PCR. As shown in Figure 3A, the knockdown of Akt2 in MDA-MB-231 cells caused a substantial decrease in IKKε mRNA. The densitometric scanning of 3 independent experiments revealed a significant decrease in IKKε mRNA (~2.8 fold) upon Akt2 depletion (Fig. 3B).
Figure 3.
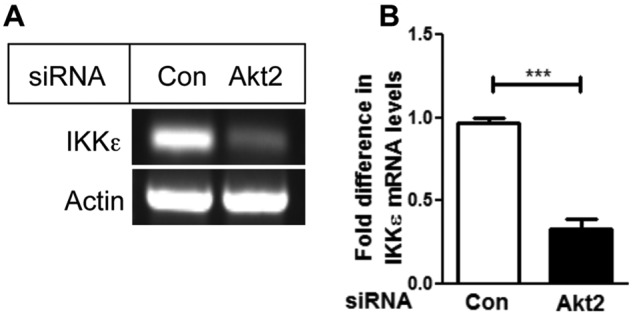
Depletion of Akt2 decreased IKKε mRNA level. (A) MDA-MB-231 cells were transfected with Akt2 siRNA. Twenty-four hours after transfection, RT-PCR analysis was performed to detect IKKε mRNA level. (B) Densitometric quantification of the mRNA from 3 independent experiments normalized with actin. Data are the mean ± standard error. ***P < 0.005, using paired Student t test.
Because knockdown of Akt2 decreased IKKε level, we examined if overexpression of Akt2 increases IKKε level. Figure 4A shows that both basal and TNF-induced IKKε levels are higher in Akt2-overexpressing MDA-MB-231 cells compared to vector-transfected cells (Fig. 4B). Taken together, our results demonstrate that IKKε level is regulated by the Akt2 isoform.
Figure 4.

Overexpression of Akt2 increased IKKε level. (A) MDA-MB-231 cells stably transfected with pBabe or pBabe-Akt2 were treated with or without 1 nM TNF, and Western blot analyses were carried out with the indicated antibodies. (B) Densitometric quantification of IKKε level from 3 independent experiments normalized with actin. The data represent the fold change in IKKε level compared with untreated vector-transfected cells.
Akt2 induces IKKε via activation of NF-κB
Although IKKε is known to activate NF-κB,8-14 it also has a NF-κB binding site in its promoter region.24 To determine if Akt2 induces IKKε via activation of NF-κB, we first examined if Akt2 regulates NF-κB activity in MDA-MB-231 cells. Figure 4A shows that overexpression of Akt2 was associated with an increase in phosphorylation of p65/RelA, indicative of NF-κB activation.25 Figure 5A shows that TNF increased NF-κB activity as determined by the NF-κB luciferase reporter assay. Knockdown of Akt2 decreased, and overexpression of Akt2 increased both basal and TNF-induced NF-κB activity in MDA-MB-231 cells (Fig. 5A and 5B).
Figure 5.

Akt2 induced IKKε by activating NF-κB. (A) MDA-MB-231 cells transfected with Akt2 or control nontargeting siRNA were transfected with 3X-κB construct and Renilla luciferase and treated with or without 1 nM TNF, as described in the Materials and Methods. Luciferase activity was determined, and the relative luciferase units were normalized and represented as fold change. *P < 0.05. The results are representative of 3 independent experiments. (B) The MDA-MB-231/pBabe and MDA-MB-231/Akt2 cells were transfected with 3X-κB construct and Renilla luciferase and treated with or without 1 nM TNF, as indicated in the Materials and Methods. The results are representative of 2 independent experiments. (C) The MDA-MB-231/pBabe and MDA-MB-231/Akt2 cells were transfected with p65/RelA siRNA or nontargeting control siRNA and treated with 1 nM of TNF for 24 hours and processed. Western blot analyses were carried out with the indicated antibodies. The results are representative of 2 independent experiments.
To determine if Akt2 regulates IKKε level via NF-κB, we depleted p65/RelA subunit of NF-κB in cells transfected with an empty vector (pBabe) or a vector containing Akt2 construct (pBabe-Akt2) and monitored IKKε level. Figure 5C shows that knockdown of p65/RelA in cells transfected with the empty vector decreased both basal and TNF-induced IKKε levels. While overexpression of Akt2 enhanced basal and TNF-induced increase in IKKε levels, knockdown of p65/RelA attenuated the increase in IKKε level caused by Akt2 overexpression. These results demonstrate that Akt2 upregulates IKKε via NF-κB.
Akt2 promotes MDA-MB-231 cell survival via IKKε
To determine the functional significance of Akt2-mediated induction of IKKε, we performed a long-term clonogenic cell survival assay. Figure 6A shows that knockdown of either Akt2 or IKKε caused a decrease in the clonogenicity of MDA-MB-231 cells. Figure 6B represents the quantification of the number of colonies. While overexpression of Akt2 enhanced the number of colonies, knockdown of IKKε decreased the number of colonies not only in MDA-MB-231 cells but also in Akt2-overexpressing cells. These results suggest that IKKε acts downstream of Akt2 to promote cell survival.
Figure 6.

Akt2 promotes cell survival via IKKε. (A) MDA-MB-231 cells were transfected with Akt2, IKKε, or nontargeting siRNA, and a clonogenic assay was performed, as detailed in the Materials and Methods. (B) Quantification of the number of colonies as determined by the clonogenic assay in MDA-MB-231 and MDA-MB-231/Akt2 cells transfected with control nontargeting or IKKε siRNA. The results are representative of 3 independent experiments. **P < 0.01 and ***P < 0.005, using paired Student t test.
Discussion
IKKε is an inducible kinase and is amplified and overexpressed in breast cancers,8 but little is known about how its level is upregulated in these breast cancers. The results of our present study demonstrate for the first time that IKKε level is regulated by the Akt signaling pathway in highly aggressive breast cancer cells and that Akt2, but not the Akt1 or Akt3 isoform, is responsible for IKKε induction. In addition, Akt2 enhanced IKKε expression by activating the transcription factor NF-κB. Moreover, IKKε functions downstream of Akt2 to promote the survival of MDA-MB-231 breast cancer cells.
The functional relationship between Akt and IKKε was explored by Boehm et al.8 IKKε was shown to substitute for Akt in promoting transformation.8 Because Akt-driven transformation required IKKε, it was proposed that IKKε functions downstream of the Akt signaling pathway.8,22 Two recent studies, however, demonstrated that IKKε could directly phosphorylate Akt in vitro.20,21 While Xie et al. suggested that PI3K-dependent membrane association of Akt is required for its phosphorylation by IKKε,20 Guo et al. showed PI3K-independent phosphorylation of Akt by IKKε.21 However, these studies do not explain how IKKε level is elevated in breast cancers.
Although IKKε is often amplified or overexpressed in breast cancers, there was no evidence of somatic mutations.8 It was speculated that IKKε is required for the proliferation and survival of cells in which IKKε is amplified.8 These studies were conducted in MCF-7 and ZR-75-1 cells, which harbored 5 and 10 copies of IKKε, respectively. However, IKKε was also required for the anchorage-independent growth of breast cancer Hs578t cells19 and for the survival of cervical cancer HeLa cells,13 which overexpressed IKKε without any gene amplification, suggesting additional mechanisms leading to IKKε upregulation.8 Because IKKε is an inducible kinase, an elevation in IKKε level could be caused by a deregulation in the signaling pathway that leads to IKKε induction.
Our observation that the Akt signaling pathway is responsible for the induction of IKKε is extremely important because the Akt signaling pathway is frequently deregulated in breast cancer and plays a central role in the development and progression of breast cancer. We have utilized both a loss-of-function and gain-of-function approach to demonstrate that Akt2 positively regulates IKKε level. It was reported that Akt2 phosphorylates and interacts with IKKα in ovarian cancer and HEK293 cells in which myr-Akt2 was overexpressed.26 We, however, found that silencing of endogenous Akt2, but not Akt1 or Akt3, by siRNA decreased, whereas ectopic expression of Akt2 increased both the basal and TNF-induced IKKε levels in MDA-MB-231 breast cancer cells. The regulation of IKKε level by Akt2 was not unique to MDA-MB-231 cells because knockdown of Akt2 also attenuated IKKε levels in HCC1937, MCF-10CA1a, HCC1395, and MCF-10CA1d cells (Fig. 2 and data not shown). However, knockdown of Akt2 did not decrease IKKε levels in MCF-7 and ZR-75-1 cells in which IKKε was amplified (data not shown). It was reported that overexpression of the protein kinase CK2 increased IKKε level in murine fibroblasts and MCF-10F breast epithelial cells and that pharmacological inhibition of CK2 or ectopic expression of kinase-inactive CK2 decreased IKKε expression in MDA-MB-231 and Hs578t cells, respectively.19 We also found that the CK2 inhibitor DMAT (2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole) inhibited Akt2 level in MDA-MB-231 cells (data not shown). Because CK2 was shown to phosphorylate Akt,27 it is conceivable that CK2 regulates IKKε expression via Akt2.
We also explored the mechanism by which Akt2 upregulates IKKε in breast cancer cells. Depletion of Akt2 decreased IKKε not only at the protein level but also at the mRNA level, suggesting that Akt2 transcriptionally regulates IKKε expression. The promoter region of IKKε and the transcription factors required for IKKε induction were extensively characterized by Wang et al., who demonstrated that IKKε is transcriptionally regulated by p65/RelA upon induction by TNF.24 Our results show that Akt2 regulates IKKε expression via NF-κB in MDA-MB-231 breast cancer cells. First, knockdown of Akt2 decreased both basal and TNF-induced NF-κB activity. Second, overexpression of Akt2 enhanced constitutive and TNF-induced activation of NF-κB. Third, knockdown of p65 decreased IKKε level. Finally, knockdown of p65/RelA attenuated the ability of Akt2 to induce IKKε level. Although Akt1 is well known to activate NF-κB,28-30 reports of Akt2 activating NF-κB have been emerging.26,31,32 Knockdown of Akt2 was shown to inhibit NF-κB activity by decreasing the phosphorylation of p65/RelA at the Ser536 residue in PC3 prostate cancer cells.31 These results are consistent with our observation that Akt2 activates NF-κB in MDA-MB-231 cells.
It has been proposed that IKKε is required for breast cancer cell proliferation and survival.8 Although the Akt signaling pathway is also believed to promote breast cancer cell proliferation and survival,33 a recent study demonstrated that introduction of myristoylated Akt1 and Akt2 inhibited proliferation of MDA-MB-231 cells.34 In contrast, Santi et al. used siRNA-mediated silencing of Akt2 to demonstrate that Akt2 facilitates cell cycle progression and cell proliferation in these cells.35 We have used both ectopic expression and siRNA-mediated knockdown of Akt2 and a long-term clonogenic assay to establish the importance of Akt2 in mediating the survival of MDA-MB-231 cells. In addition, we show that IKKε functions downstream of Akt2 to promote the survival of MDA-MB-231 cells.
In summary, we made a novel observation that the Akt2 isoform positively regulates IKKε expression via NF-κB to promote the survival of MDA-MB-231 cells. Because the Akt signaling pathway is activated in more than 50% of breast cancers, and there is a close correlation between IKKε upregulation and pAkt in breast cancer tissue specimens,21 this study underscores the importance of targeting the Akt2/IKKε axis for the treatment of breast cancer.
Materials and Methods
Materials
Rabbit monoclonal antibodies to IKKε, IKKβ, Akt1, Akt2, Akt3, and S536-p65 were obtained from Cell Signaling Technology (Danvers, MA). Mouse monoclonal antibodies to p65 and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and Sigma-Aldrich (St. Louis, MO), respectively. The Dual-Luciferase Reporter Assay System was purchased from Promega (Madison, WI). Horseradish peroxidase–conjugated goat anti-mouse and donkey anti-rabbit antibodies were obtained from Jackson ImmunoResearch Laboratories (Bar Harbor, ME). Control nontargeting siRNA and siRNA SMARTpool specific to AKT1, AKT2, AKT3, IKBKE, and RELA were purchased from Dharmacon (Lafayette, CO). The polyvinylidene fluoride membrane was from Millipore (Billerica, MA), and the enhanced chemiluminescence detection kit was from Amersham (Piscataway, NJ). Puromycin was obtained from Sigma-Aldrich. The 3X-κB construct was kindly provided by Dr. Zhijian Chen (University of Texas Southwestern Medical Center, Dallas, TX). pBabe-Akt2 (plasmid 14551) was obtained from Addgene (Cambridge, MA).36
Cell culture and transfection
MDA-MB-231 and HCC1937 cells were maintained in RPMI medium supplemented with 10% FBS and 2 mmol/L glutamine. MCF-10CA1a cells were maintained in DMEM-F12 medium supplemented with 5% horse serum. Cells were kept in a humidified incubator at 37°C with 95% air and 5% CO2. The cells were authenticated by DNA fingerprinting at the Department of Forensic Genetics at the University of North Texas Health Science Center. siRNA was transfected using Lipofectamine RNAiMax according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA).
Immunoblot analysis
Equivalent amounts of total cellular extracts were electrophoresed by SDS-PAGE and transferred electrophoretically to the polyvinylidene fluoride membrane. Immunoblot analyses were carried out as described before.37
RT-PCR analysis
Total RNA was isolated using TRI reagent RT from Molecular Research Center (Cincinnati, OH) as per the manufacturer’s protocol and subjected to RT reaction using reverse transcriptase enzyme from Promega. PCR amplifications were performed using the following primers: IKBKE: forward, 5′-GAA GTT CGT CTC GGT CTA TG-3′; reverse, 5′-CGG TAC ATG ATC TCC TTG TT-3′; and ACTB: forward, 5′-TAC AAT GAG CTG CGT GTG GCT-3′; reverse, 5′-ATC CAC ATC TGC TGG AAG GTG GA-3′. Cycle conditions for all PCRs were set up for 35 cycles as follows: denaturation at 94°C for 1 minute, annealing at 55°C for 1 minute, extension at 72°C for 1 minute, and final extension at 72°C for 10 minutes. The products were resolved on 1% agarose gel containing ethidium bromide.
NF-κB luciferase reporter assay
Cells were plated or transfected with the siRNA using RNAiMax reagent per the manufacturer’s protocol (Invitrogen). After 24 hours, the cells were transfected with the 3X-κB construct and Renilla luciferase construct using Lipofectamine 2000 (Invitrogen). Cells were harvested after 48 hours of transfection and lysed in the passive lysis buffer (Promega). Luciferase assay was measured with a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA), and Renilla luciferase was measured using the Stop n’ Glo reagent (Promega).
Clonogenic assay
Cells transfected with or without control nontargeting, Akt2, or IKKε siRNA were cultured at 37°C in a humidified incubator with 5% CO2 until there were at least 50 cells per colony. At the end of the incubation, the cells were washed with PBS and incubated with 0.25% crystal violet solution for 15 minutes. Colonies were counted using ImageJ software (http://rsbweb.nih.gov/ij/), and the plate was photographed using the BioChemi System (BioImaging System, UVP, Upland, CA).
Statistical analysis
Statistical significance was determined by a paired Student t test using the GraphPad Prism software (La Jolla, CA). P < 0.05 was considered statistically significant.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the National Institutes of Health/National Cancer Institute (grant CA071727 to A. Basu) and by the Department of Defense/Breast Cancer Research Program (predoctoral fellowship BC083099 to S. Krishnamurthy).
References
- 1. Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5(4):297-309 [DOI] [PubMed] [Google Scholar]
- 2. Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8(1):33-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haffner MC, Berlato C, Doppler W. Exploiting our knowledge of NF-kappaB signaling for the treatment of mammary cancer. J Mammary Gland Biol Neoplasia. 2006;11(1):63-73 [DOI] [PubMed] [Google Scholar]
- 4. Wu JT, Kral JG. The NF-kappaB/IkappaB signaling system: a molecular target in breast cancer therapy. J Surg Res. 2005;123(1):158-69 [DOI] [PubMed] [Google Scholar]
- 5. Biswas DK, Singh S, Shi Q, Pardee AB, Iglehart JD. Crossroads of estrogen receptor and NF-kappaB signaling. Sci STKE. 2005;(288):pe27. [DOI] [PubMed] [Google Scholar]
- 6. Clement JF, Meloche S, Servant MJ. The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 2008;18(9):889-99 [DOI] [PubMed] [Google Scholar]
- 7. Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30(6):631-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boehm JS, Zhao JJ, Yao J, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129(6):1065-79 [DOI] [PubMed] [Google Scholar]
- 9. Peters RT, Liao SM, Maniatis T. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol Cell. 2000;5(3):513-22 [DOI] [PubMed] [Google Scholar]
- 10. Shimada T, Kawai T, Takeda K, et al. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int Immunol. 1999;11(8):1357-62 [DOI] [PubMed] [Google Scholar]
- 11. Buss H, Dorrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem. 2004;279(53):55633-43 [DOI] [PubMed] [Google Scholar]
- 12. Wietek C, Cleaver CS, Ludbrook V, et al. IkappaB kinase epsilon interacts with p52 and promotes transactivation via p65. J Biol Chem. 2006;281(46):34973-81 [DOI] [PubMed] [Google Scholar]
- 13. Adli M, Baldwin AS. IKK-i/IKKepsilon controls constitutive, cancer cell-associated NF-kappaB activity via regulation of Ser-536 p65/RelA phosphorylation. J Biol Chem. 2006;81(37):26976-84 [DOI] [PubMed] [Google Scholar]
- 14. Harris J, Oliere S, Sharma S, et al. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. J Immunol. 2006;177(4):2527-35 [DOI] [PubMed] [Google Scholar]
- 15. Peant B, Forest V, Trudeau V, Latour M, Mes-Masson AM, Saad F. IkappaB-kinase-epsilon (IKKepsilon/IKKi/IkappaBKepsilon) expression and localization in prostate cancer tissues. Prostate. 2011;71(10):1131-8 [DOI] [PubMed] [Google Scholar]
- 16. Guo JP, Shu SK, He L, et al. Deregulation of IKBKE is associated with tumor progression, poor prognosis, and cisplatin resistance in ovarian cancer. Am J Pathol. 2009;175(1):324-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng A, Guo J, Henderson-Jackson E, Kim D, Malafa M, Coppola D. IkappaB kinase epsilon expression in pancreatic ductal adenocarcinoma. Am J Clin Pathol. 2011;136(1):60-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guan H, Zhang H, Cai J, et al. IKBKE is over-expressed in glioma and contributes to resistance of glioma cells to apoptosis via activating NF-kappaB. J Pathol. 2011;223(3):436-45 [DOI] [PubMed] [Google Scholar]
- 19. Eddy SF, Guo S, Demicco EG, et al. Inducible IkappaB kinase/IkappaB kinase epsilon expression is induced by CK2 and promotes aberrant nuclear factor-kappaB activation in breast cancer cells. Cancer Res. 2005;65(24):11375-83 [DOI] [PubMed] [Google Scholar]
- 20. Xie X, Zhang D, Zhao B, et al. I{kappa}B kinase {epsilon} and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci U S A. 2011;108(16):6474-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo JP, Coppola D, Cheng JQ. IKBKE activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and PH domain to sustain malignant transformation. J Biol Chem. 2011;286(43):37389-98 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Agami R. All roads lead to IKKepsilon. Cell. 2007;129(6):1043-5 [DOI] [PubMed] [Google Scholar]
- 23. Masure S, Haefner B, Wesselink JJ, et al. Molecular cloning, expression and characterization of the human serine/threonine kinase Akt-3. Eur J Biochem. 1999;265(1):353-60 [DOI] [PubMed] [Google Scholar]
- 24. Wang N, Ahmed S, Haqqi TM. Genomic structure and functional characterization of the promoter region of human IkappaB kinase-related kinase IKKi/IKKvarepsilon gene. Gene. 2005;353(1):118-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30(1):43-52 [DOI] [PubMed] [Google Scholar]
- 26. Yuan ZQ, Feldman RI, Sun M, et al. Inhibition of JNK by cellular stress- and tumor necrosis factor alpha-induced AKT2 through activation of the NF kappa B pathway in human epithelial cells. J Biol Chem. 2002;277(33):29973-82 [DOI] [PubMed] [Google Scholar]
- 27. Di Maira G, Salvi M, Arrigoni G, et al. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005;12(6):668-77 [DOI] [PubMed] [Google Scholar]
- 28. Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 1999;9(11):601-4 [DOI] [PubMed] [Google Scholar]
- 29. Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401(6748):86-90 [DOI] [PubMed] [Google Scholar]
- 30. Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401(6748):82-5 [DOI] [PubMed] [Google Scholar]
- 31. Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22(11):1490-500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sugatani T, Hruska KA. Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas Akt is dispensable for cell survival in isolated osteoclast precursors. J Biol Chem. 2005;280(5):3583-9 [DOI] [PubMed] [Google Scholar]
- 33. Liu W, Bagaitkar J, Watabe K. Roles of AKT signal in breast cancer. Front Biosci. 2007;12:4011-9 [DOI] [PubMed] [Google Scholar]
- 34. Yang W, Ju JH, Lee KM, Shin I. Akt isoform-specific inhibition of MDA-MB-231 cell proliferation. Cell Signal. 2011;23(1):19-26 [DOI] [PubMed] [Google Scholar]
- 35. Santi SA, Lee H. Ablation of Akt2 induces autophagy through cell cycle arrest, the downregulation of p70S6K, and the deregulation of mitochondria in MDA-MB231 cells. PLoS One. 2011;6(1):e14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Irie HY, Pearline RV, Grueneberg D, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171(6):1023-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Basu A, Akkaraju GR. Regulation of caspase activation and cis-diamminedichloroplatinum(II)-induced cell death by protein kinase C. Biochemistry. 1999;38(14):4245-51 [DOI] [PubMed] [Google Scholar]