

Abstract
Super-resolution fluorescence microscopy can achieve resolution beyond the optical diffraction limit, partially closing the gap between conventional optical imaging and electron microscopy for elucidation of subcellular architecture. The centriole, a key component of the cellular control and division machinery, is 250 nm in diameter, a spatial scale where super-resolution methods such as stimulated emission depletion (STED) microscopy can provide previously unobtainable detail. We use STED with a resolution of 60 nm to demonstrate that the centriole distal appendage protein Cep164 localizes in nine clusters spaced around a ring of ∼300 nm in diameter, and quantify the influence of the labeling density in STED immunofluorescence microscopy. We find that the labeling density dramatically influences the observed number, size, and brightness of labeled Cep164 clusters, and estimate the average number of secondary antibody labels per cluster. The arrangements are morphologically similar in centrioles of both proliferating cells and differentiated multiciliated cells, suggesting a relationship of this structure to function. Our STED measurements in single centrioles are consistent with results obtained by electron microscopy, which involve ensemble averaging or very different sample preparation conditions, suggesting that we have arrived at a direct measurement of a centriole protein by careful optimization of the labeling density.
Introduction
Most biological fluorescence microscopic studies at visible wavelengths are restricted to the diffraction limit of resolution (1) of ∼200 nm, a scale that is unable to resolve details of many structures in the cell. In recent years, a variety of techniques have appeared that may be termed super-resolution microscopy, in which various approaches are used to surpass the optical diffraction limit (2,3). Such super-resolution techniques commonly rely on the ability of the experimenter to modulate the fluorescence of the dye labels, thus allowing adjacent emitters more closely spaced than the width of the conventional microscope point spread function (PSF) to be distinguished. Two major approaches have been identified: a), The first set of approaches requires spatial patterning of the illumination and nonlinear processes to force the emitting region to be smaller than the size required by the diffraction limit, such as stimulated emission depletion (STED) microscopy (4) or structured illumination microscopy (5). These methods rely on the ability to modulate the emission of the underlying fluorophores (6), but they do not require single-molecule sensitivity. b), In the (F)PALM/STORM approaches (7–9), sequential widefield single-molecule imaging is used with the critical addition of photoactivation, blinking, or photoswitching of the fluorescent dye labels to maintain the emitting concentration at extremely low levels followed by PSF localization (10) and image reconstruction. Because many physical and chemical processes have been identified for actively controlling the concentration of emitters at very low levels (2,11), this class of super-resolution approaches may be termed single-molecule active control microscopy or SMACM (12). This work provides a specific application of STED microscopy to elucidate and quantify the molecular details of an important cellular structure beyond the optical diffraction limit.
In STED microscopy, two lasers are usually employed in a confocal scanning geometry: the first, called the pump laser, is focused to a diffraction-limited spot inside the sample and pumps the molecules into the electronic excited state. The beam from a second laser at longer wavelengths called the STED laser is carefully prepared in the shape of a doughnut with a dark center, and the STED laser spot is carefully overlapped with the pump laser. Although STED has been implemented with continuous wave beams (13), here we focus on the pulsed version of the technique. The STED laser pulse closely follows the pump laser pulse and stimulates many emitters to produce stimulated light at the STED laser wavelength that is discarded; this brings about depletion of, or the prevention of emission from, the fluorophores not at the center of the pump spot. Therefore, only those fluorophores in a very small region at the center of the pump pulse are allowed to emit normal fluorescence, thus restricting the fluorescence emission to a transverse region much smaller than the optical diffraction limit. Because our setup uses a confocal pinhole, diffraction-limited optical sectioning along the axial direction is also achieved. A complete image is formed by rapid scanning of either the sample or the focused pump and STED spots.
Due to its ability to noninvasively detect specific subcellular objects with optical sectioning to reject out-of-focus background, the confocal STED approach has been successfully employed in early studies for immunofluorescence cell imaging (14), live cell imaging with sectioning of the cell interior (15), and video-rate imaging of cell dynamics (16). In addition, for STED microscopy, the resolution enhancement arises intrinsically from a subdiffraction-sized response to a point emitter such as a single molecule, which we term PSFSM. As a result of the nonlinear response of the fluorophores, the PSFSM has a smaller width than the diffraction-limited PSF because the positions of molecules allowed to emit are restricted to smaller and smaller regions when the intensity of the STED laser is increased. STED images are not reconstructed from images of photoactivated or photoswitched single dye labels; therefore, the STED method can exclude image artifacts such as undersampling of dye labels that may be present in SMACM-type methods (17). At the same time, the high STED laser intensity can lead to excited state processes that produce photobleaching, therefore the fluorophore must be chosen carefully.
Here, we report the application of STED imaging to centrioles, a subcellular structure of animal cells critical for cell signaling (18–20), schematized in Fig. 1 a. Centrioles nucleate the formation of the primary cilium, a cellular signaling organelle, and also serve as a nucleus for the centrosome, the main microtubule organizer in animal cells. The centrioles are cylindrical organelles made of triplets of microtubules and typically present as a pair in cells. The triplet microtubules of the centriole are arranged with 9-fold symmetry around the central lumen; this symmetry was established by electron microscopy (EM) (21) and is a highly conserved feature of centrioles with a known structural basis (22,23). The centriole duplication cycle results in each centriole pair consisting of an older mother centriole and a newer daughter centriole (24). In most animal cells, only the mother centriole is competent to form a basal body that nucleates cilium formation. The mother centriole, our primary interest, is distinguished morphologically from the daughter centriole by the presence of distal and subdistal appendages near the end of the structure, which also have a 9-fold symmetric arrangement around the nine triplets of microtubules (21,25), and are thought to be required for cilia formation. Based on immuno-EM with Au particles, the Cep164 protein was found to be a component of the distal appendages (26). However, it is unclear from these results whether Cep164 is present on each of the distal appendage structures on a given centriole, and how the appendage structures of centrioles that had been observed in the centrosomes of dividing cells relate to those of specialized epithelial cells that make hundreds of centrioles and cilia (27). In other words, the fine details of Cep164 arrangement, which we refer to as the Cep164 superstructure, are unknown. To address these issues, we have used our custom-built STED microscope to image Cep164 protein superstructure in centrioles both in proliferating and in multiciliated cells with optical resolution down to ∼60 nm full width at half-maximum (FWHM), four to five times beyond the optical diffraction limit, for direct comparison with tomographic EM images as illustrated in Fig. 1 a. Extending our prior preliminary work on this system (28), we have quantitatively characterized the shapes, sizes, and numbers of Cep164 clusters, and we have also validated our immunofluorescence labeling over a range of antibody concentrations. Our results provide additional detail and quantification of the number of Cep164 clusters beyond the recently published PALM/STORM imaging of these proteins (29). This work demonstrates that STED microscopy can be used for precise subdiffraction imaging of centriolar protein assemblies in cells when care is taken to avoid the detrimental effects of overlabeling or underlabeling.
Figure 1.
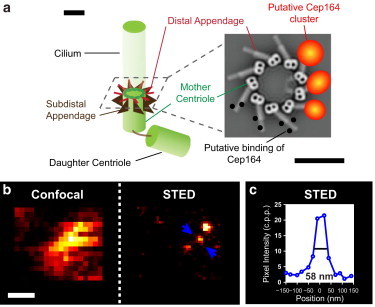
Cep164 model and PSFSM. (a) Schematic of the centriole pair showing 9-fold symmetry of the distal and subdistal appendages on the mother centriole. (Inset) Top view of putative locations of Cep164 clusters on the distal appendage, where the Cep164 immunostained clusters are overlaid on a cross section of the mother centriole from an electron tomographic reconstruction (reprinted with permission from (21)). Scale bars: 200 nm. (b) Confocal and STED images of single molecules of ATTO647N in Mowiol+DABCO+PPD showing the PSF in both cases. (c) Intensity profile along the line demarcated by the arrows in b shows the PSFSM width of FWHM = 58 ± 2 nm or equivalently, width of standard deviation σ = 25 nm.
Materials and Methods
Cell samples
The organic dye-conjugated antibody ATTO647N goat antirabbit IgG (Active Motif, Carlsbad, CA) was used as the secondary antibody. IMCD3 cells and mouse tracheal epithelial cells (MTEC) were cultured and labeled using standard indirect immunostaining protocols (described in the Supporting Material).
Measurements
Images were acquired with a noncommercial pulsed STED microscope fabricated in our laboratory ((28) and the Supporting Material). The quantitative analysis methods including the Cep164 cluster fitting, estimation of Cep164 ring diameter, and estimation of bound antibody labels are described in the Supporting Material.
Results
STED microscopy probes centriole architecture in the centrosome of proliferating cells
We employed STED immunofluorescence microscopy to elucidate the arrangement of the Cep164 centriolar proteins at the nanometer scale. Although the centriole and distal appendages have been shown by EM to have 9-fold radial symmetry, it has not been possible to assess the arrangement of Cep164 or any other specific centrosomal protein in the context of this symmetry (Fig. 1 a). A recent report of the application of PALM/STORM to this question shows only a ring-like structure for Cep164, with unresolved detail of clusters (29). We first visualized Cep164 in centrioles of IMCD3 cells, a mouse epithelial cell line. Cells were fixed and labeled with anti-Cep164 primary (1°) antibodies and ATTO647N-secondary (2°) antibodies. The 1° antibody was raised against a fragment of Cep164 encompassing amino acids 1 to 298 (26). With a given PSFSM, the detected fluorescent spot corresponding to molecules of Cep164, which we refer to as a Cep164 cluster, reflects the arrangement of Cep164 and antibody labels on the distal appendage (Fig. 1 a inset, orange).
To quantify the resolution of our microscope, we benchmarked the PSFSM by imaging single molecules of the ATTO647N dye embedded in Mowiol mounting medium. Conventional diffraction-limited confocal imaging (Fig. 1 b) could not distinguish closely spaced single molecules of ATTO647N, whereas in the STED case (Fig. 1 b), the molecules were clearly resolved. The PSFSM width of 58 nm was calculated as the average FWHM of line intensity plots from 25 single molecules. A representative intensity plot is displayed in Fig. 1 c.
In diffraction-limited confocal images of randomly oriented centrioles in asynchronously growing IMCD3 cells, Cep164 appeared as a spot of various sizes, depending on the orientation of the centriole with respect to the imaging plane (Fig. S1, a–c in the Supporting Materials), and in some cases a ring-like appearance was barely present, consistent with previous deconvolved diffraction-limited images (26). However, the far higher resolution of STED microscopy revealed distinct substructure in the same Cep164 rings (Fig. S1, d–f). In the case in which the centriole face was oriented parallel (0°) to the imaging plane, the Cep164 ring appeared to consist of nine clusters of Cep164 signal (Fig. S1 d). When the centriole face was oriented 90° to the imaging plane, the STED image showed a side view of the Cep164 ring, which frequently appeared as a double-lobed structure (Fig. S1 e). Although intermediate angles (Fig. S1 f) resulted in an image with finer detail than corresponding confocal images (Fig. S1 c), the Cep164 clusters were not clearly resolved. Overall, fewer than 10% of IMCD3 cells imaged had clear evidence of a 9-fold symmetric Cep164 ultrastructure (Fig. S1 d), with the remaining majority having Cep164 rings at an angle with the imaging plane similar to Fig. S1, e and f.
Imaging of oriented centrioles in multiciliated epithelial cells reveals the 9-fold symmetry of Cep164 clusters
Although STED imaging was clearly able to resolve previously unappreciated substructure of the Cep164 arrangement in centrioles, the random orientation and small number of the centrioles in IMCD3 cells made quantitative super-resolution measurements difficult. To overcome this problem we imaged Cep164 in centrioles of MTEC, which have hundreds of centrioles at their apical surface, all serving as basal bodies for cilia, oriented parallel to the apical-basolateral axis of the cell. This layer of centrioles is in a plane ∼0.5 μm thick across a typical cell width, providing an ideal sample for measurement of Cep164 protein superstructure. MTECs were cultured as described (27), stimulated to differentiate by culturing the cells at an air-liquid interface for ∼15 days before fixation and imaging. As above, Cep164 was visualized with anti-Cep164 1° antibodies and ATTO647N-2° antibodies. To image centrioles in a MTEC, we first located the approximate plane of the centrioles in an intermediate field of view (∼10 μm2) in confocal mode (Fig. 2 a), and then took STED sections through the Cep164 signal-containing plane (Fig. 2 b). A typical MTEC would have hundreds of Cep164-labeled rings similar to those shown in Fig. 2 b. Diffraction-limited confocal images of a subset (Fig. 2 c) and a representative Cep164 ring (see Fig. 2 e) show a collection of ill-defined spots, whereas the STED images (Fig. 2, d and f), clearly resolve Cep164 clusters in a ring surrounding an unlabeled central core. An intensity profile (Fig. 2 g) of a line across the indicated Cep164 ring (Fig. 2, e and f), shows the substantial increase in resolution of STED compared to conventional fluorescence microscopy. Most importantly, STED microscopy unambiguously reveals that the Cep164 ring around the distal end of the centriole actually consists of nine separate Cep164 clusters (Fig. 2 f).
Figure 2.
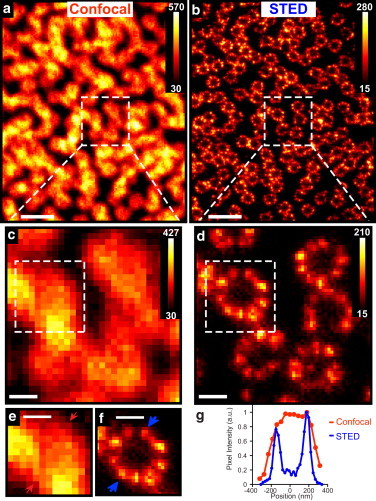
Cep164 ring substructure in multiciliated MTECs. Although (a) confocal microscopy obscures fine detail, (b) STED images of Cep164 labeled with 2000-fold dilution of primary antibody in MTECs resolve the 9-fold symmetric Cep164 localization. (c and d) Closer views of the region indicated by the white dashed boxes in a and b, respectively. (e) Confocal blurs the substructure, but (f) STED exhibits nine Cep164 clusters for a representative centriole. (g) Intensity profile along the lines demarcated by the arrows in (e and f) shows the ability of STED to resolve the Cep164 ring structure. Pixel intensities are represented in the color bars. Scale bars: (a and b) 1 μm and (c–f) 200 nm.
Labeling density effects in STED immunofluorescence images
Although the STED images revealed a 9-fold symmetric ultrastructure of Cep164 in many centrioles (Fig. 2 d) we observed heterogeneity at the single-centriole level, with some rings appearing to consist of 7, 8, 9, or even 10 Cep164 clusters. In addition, there was variation in the observed cluster sizes and cluster brightness even within one ring. To further investigate the distribution of Cep164, we analyzed a large number (>1000) of clusters in MTEC centrioles, under different labeling regimes. Because we were visualizing Cep164 with antibodies, we anticipated that an optimized antibody labeling density was critical for accurate, artifact-free immunofluorescence images (30,31), and mandatory for super-resolution images as the imaging resolution approaches the size of the antibodies used (17,32,33).
To ensure that the antibody labeling density was appropriate to elucidate the Cep164 centriolar structure, we optimized the concentration of the 1° antibody label (Fig. 3). The concentration of the 1° antibody used to label Cep164 was varied over three values covering a factor of 80× while keeping the 2° antibody in excess. In the regime of saturated 2° antibody labeling, we can assume that most of the available epitopes on the 1° antibodies are bound by a 2° antibody. Therefore, the 1° antibody concentration mainly determines the degree of protein labeling, whereas the 2° antibody mainly determines the size and brightness of proteins already labeled with 1° antibody. That is, the 1° antibody has the role of protein labeling, whereas the 2° antibody has the role of signal amplification. As above, we found that in diffraction-limited confocal images (Fig. 3, top row), the Cep164 pattern appears as a blurry image with little detail of fine structure, whereas in the STED images (Fig. 3, bottom row) Cep164 is clearly observed in various patterns depending on antibody concentration. If too low a concentration of 1° antibody was used, a typical centriole showed fewer than nine Cep164 clusters (Fig. 3, underlabeled bottom panel). Increasing the 1° antibody concentration resulted in the 9-fold symmetric structure described previously (Fig. 3, optimized bottom panel). Further increase of the 1° antibody concentration resulted in blurring of the structure (Fig. 3, overlabeled bottom panel); we suspect this is due to denser binding of the relatively large ∼10 nm sized 1° and 2° antibody molecules. We refer to the low, intermediate, and high concentrations of 1° antibody (Fig. 3, bottom row) as underlabeled, optimized-labeled, and overlabeled MTEC, respectively.
Figure 3.
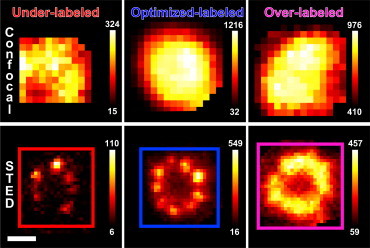
Effect of labeling density in STED images of Cep164 in MTECs. (Top panel) Confocal and (bottom panel) STED images of Cep164 in MTECs immunostained with increasing concentrations of primary antibody and fixed concentration of secondary antibody. With increasing primary antibody concentration, the number of observed clusters increases until saturation at nine clusters. Overlabeling with antibody blurs the Cep164 centriole substructure. Pixel intensities are represented in the color bars. Scale bar: 200 nm.
To determine the influence of labeling density on our ability to quantify Cep164 clusters, we compared STED measurements of the observed cluster number, size, and brightness in underlabeled and optimized-labeled MTEC. First, the observed number of Cep164 clusters per ring was lower in underlabeled MTEC versus the optimized case (Fig. 4 a). Underlabeled MTECs (Fig. 4 a, red series) had 3.3 ± 1.6 Cep164 clusters per ring (N = 38 centrioles), whereas optimized-labeled samples (Fig. 4 a, blue series) had 8.5 ± 0.7 Cep164 clusters per ring (N = 120 centrioles). To evaluate whether the distribution of observed clusters per ring of the optimized-labeled MTEC was consistent with the hypothesis of nine Cep164 clusters per ring, we assumed a simple model in which a centriole had n sites (e.g., distal appendages) for cluster observation and that the resulting distribution of observed clusters was given by binomial statistics; that is, the distribution would be a result of the discrete nature of cluster observation and not due to an underlying heterogeneous population of distal appendages (Fig. S2). We found that a model in which a centriole had n = 9 sites best resembled the STED data distribution (Fig. S2), in agreement with the expected nine distal appendages per centriole. Underlabeled MTEC centrioles had significantly fewer than nine clusters, presumably due to failure to label some Cep164 with 1° and 2° antibodies.
Figure 4.
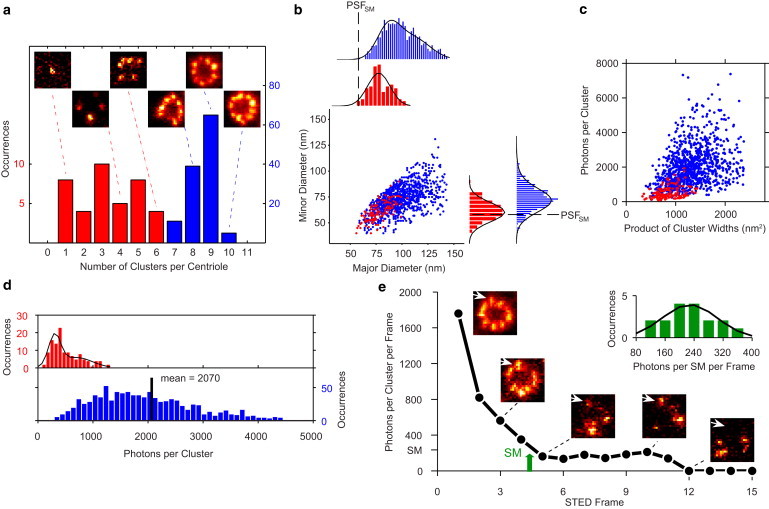
Quantification of Cep164 superstructure in MTECs using STED imaging. (a) Cep164 centriole superstructure in underlabeled (red) and optimized-labeled (blue) MTECs. The histogram shows the distribution of the number of clusters observed per centriole with mean of 3.3 ± 1.6 clusters for N = 38 centrioles in the underlabeled case and 8.5 ± 0.7 clusters for N = 120 centrioles in the optimized case. Representative STED images illustrate the various Cep164 arrangements observed. (b) Scatterplot of cluster major versus minor diameters for (red) underlabeled and (blue) optimized samples with axes histograms (outliers beyond two standard deviations from the mean are not shown). For the underlabeled MTEC, the black line is a single Gaussian fit; in the optimized case, the black line is a double Gaussian fit. The PSFSM of 58 nm FWHM is shown for reference. (c) Scatterplot of cluster area (product of Gaussian widths) versus cluster brightness for (red) underlabeled and (blue) optimized samples. (d) Histogram showing the brightness of each cluster in the underlabeled (red) and optimized-labeled (blue) MTEC sample. Black line is a double Gaussian fit for the underlabeled series. (e) Brightness as a function of STED frame for the representative Cep164 cluster indicated in the centriole showing bleaching. The single dye brightness (SM, green arrow) is determined as the size of the last step before photobleaching to the background level. (Inset) Histogram showing the single dye brightness determined from 17 cluster measurements. Black line is a single Gaussian fit with mean 233 ± 66 photons.
We used STED imaging to assess the apparent size of Cep164 clusters. Each Cep164 cluster was fit to a two-dimensional Gaussian with different widths along two orthogonal axes to extract major and minor cluster diameters (defined as the FWHM of each Gaussian); the results are shown in Fig. 4 b as a scatterplot and as distributions of major and minor diameter. The distributions (Fig. 4 b) were fit with both a single and double Gaussian model to probe for two populations of cluster sizes. The model that better fit the data was selected via the Bayesian information criterion (Materials and Methods in the Supporting Material). The Cep164 cluster minor diameter distribution of the optimized-labeled MTEC exhibited an 80% subpopulation of 71 nm ± 11 nm (one standard deviation) and 20% subpopulation of 80 nm ± 17 nm, whereas the major diameter distribution was fit by a 52% subpopulation of 110 ± 17 nm and 48% subpopulation of 87 ± 11 nm (Fig. 4 b, blue series). In contrast, the Cep164 clusters of the underlabeled MTEC showed a minor diameter with mean of 61 ± 9 nm (limited by the size of the PSFSM) and major diameter with mean of 78 ± 11 nm (Fig. 4 b, red series). STED not only discerns the smaller sizes of the underlabeled MTEC but also suggests a mere ∼10 nm difference in diameter between the underlabeled case and the subpopulation of smaller clusters in the optimized case. This difference of ∼10 nm in the cluster sizes could be due to more binding of the antibody labels, which would be consistent with the ∼10 nm size of the IgG antibody as predicted by crystal structure studies (34). In addition, the subpopulation of the larger clusters in optimized-labeled MTEC could represent two nearby clusters that are unresolvable given a PSFSM of ∼60 nm. In both samples, the clusters are asymmetric, with a mean ratio of diameters of 0.7–0.8. The cluster asymmetry could be a result of the numerous possibilities for antibody binding of the multiple protein epitopes in the indirect polyclonal immunolabeling scheme. The cluster asymmetry also could reflect the underlying cylindrical shape of the distal appendage, which we address in the following sections. In addition, in both samples, the brighter clusters were also larger, with a 2.5-fold difference in brightness between the largest and smallest clusters in the underlabeled samples and 3.8-fold difference in the optimized ones (Fig. 4 c). Both of these observations are consistent with the binding of >1 primary antibody to Cep164 molecules in the distal appendage structures.
To further study the nature of the antibody binding, we first investigated whether there was a relationship between the asymmetry of the cluster and its brightness, which could indicate the manner in which the 2° and 1° antibodies were binding to Cep164. However, we found no significant correlation of the cluster asymmetry to brightness for the two labeling series (Fig. S3). The lack of correlation is not unexpected and could be due to the distribution throughout the distal appendage of the epitopes for the 1° and 2° antibodies. We also examined in more detail the orientation of the asymmetric cluster shape and its possible correlation with the local orientation of the distal appendage. The model of centrioles derived from averaged electron tomography (Fig. 1 a) shows the distal appendages as cylinders of ∼100 nm × 20 nm, tilted ∼45° clockwise with respect to the plane of the centriole ring, as viewed from the distal end. The arrangement of Cep164 molecules on the distal appendage is unknown, but if antibody labels were bound along the whole length of the distal appendage, the Cep164 clusters might show a similar orientation. However, we found no such correlation; the orientation of the major axis of each cluster with respect to the local tangent of the centriole under optimized labeling conditions revealed a random cluster angle spanning −90° to 90° (Fig. S4). The most likely explanation for lack of observed orientation is the aforementioned indeterminacy in the spatial organization of the polyclonal antibodies.
Estimation of the number of antibody labels per Cep164 cluster
The brightness of Cep164 clusters in the optimized MTEC are higher than in the underlabeled counterpart, as expected, where cluster brightness is defined as photons detected above the background in one STED scan comprising a total integration time at the cluster of ∼10 ms with 15 μW pump and 80 mW STED powers. The Cep164 brightness distribution in the optimized sample exhibited a mean brightness of 2070 photons (Fig. 4 d, blue series), whereas in the underlabeled case, the distribution featured a 54% subpopulation with Gaussian mean of 300 photons ± 110 photons per frame (one standard deviation) and a 46% subpopulation with mean of 700 ± 290 photons per frame (Fig. 4 d, red series). It is intriguing that the underlabeled cluster brightness distribution showed two peaks with means differing by a factor of two. This suggests that one and two 2° antibodies were present, to be discussed more carefully below. In the optimized case, the broad brightness in distribution could be due to stochasticity in binding and to heterogeneity in the local cellular environment of the ATTO647N-2° antibody because the fluorescence of the dye ATTO647N has been shown to be sensitive to the hydrophilicity of its microenvironment (34–36).
The number of ATTO647N-2° antibody labels bound per Cep164 cluster, N2°, is an important parameter, determined by the following equation:
where the brackets denote an average over the sample ensemble, and N647N is the number of Atto647N dyes per 2° antibody. The average brightness of a single ATTO647N in the local environment of the immunostained Cep164 MTEC sample, measured directly on the dye-antibody-Cep164 cellular complex embedded in the Mowiol medium, was obtained via sequential STED imaging of the MTEC sample until single-step photobleaching was observed (Fig. 4 e), resulting in 〈photons/647N〉 = 233 ± 66 photons per frame. Separate bulk absorption and single-molecule ATTO647N-2° antibody fluorescence imaging measurements (Materials and Methods in the Supporting Material) yielded 〈N647N〉= 1.5.
For the underlabeled sample (Fig. 4 d, red), the subpopulations of 300 photons and 700 photons corresponded to an average of 〈N2°〉 = 0.9 ATTO647N-2° antibodies and 〈N2°〉 = 2.0 ATTO647N-2° antibodies, respectively, suggesting that we are observing the binding of one and two 2° antibody labels in the Cep164 cluster. However, due to the poorly controlled nature of the binding of the polyclonal antibodies used, it is difficult to pinpoint whether subpopulations present in the raw data correspond to specific numbers of Cep164 or 1° antibody. For example, the subpopulation at 300 photons in the underlabeled case might result from, on average, one 2° antibody that bound one 1° antibody, which bound one Cep164 on the distal appendage. However, the brighter subpopulation at 700 photons could represent, on average, two 2° antibodies, which bound different epitopes on the same 1° antibody bound to one Cep164, two 2° antibodies, which bound two different 1° antibodies bound to different epitopes of the same Cep164, or two independent binding events of one 2° antibody binding one 1° antibody on one Cep164 (all average binding ratios). Furthermore, for the optimized labeling case, which involves the binding of more antibody-epitope combinations (Fig. 4 d, blue), the resulting brightness distribution is a convolution of many overlapping probability density functions for the possible number of antibodies bound (37,38), and no discrete peaks in the brightness distribution are observed. Therefore, we characterize the optimized labeling brightness distribution by its ensemble average of 〈N2°〉 = 5.9. We note that the average numbers of ATTO647N-2° antibody labels determined here by STED are reasonable given the indirect polyclonal antibody binding scheme, size of IgG antibodies, and electron tomography distal appendage ultrastructure. Our data suggest that the number of epitopes on a single 1° antibody available for the 2° antibody to bind to is quite small, and the degree of labeling of Cep164 clusters is determined by 1° antibody concentration. A similar concentration of 2° antibody was used in both the underlabeled and optimized-labeled case. If 2° antibody concentrations were not in excess, in the underlabeled case we would expect fewer clusters that were closer in brightness to optimized-labeled clusters. Instead, both cluster number and cluster brightness were considerably lower (Fig. 4 d) in the underlabeled case. This confirms that 2° antibody concentration was indeed in excess and had negligible effect on Cep164 cluster brightness. One possible effect of multiple bindings of 2° antibody is the bimodal distribution of optimized labeled cluster sizes (Fig. 4 b).
Comparison of Cep164 STED and EM measurements
The enhanced resolution of STED Cep164 measurements invites comparison to electron tomography studies of centrioles (21) and immuno-EM of Cep164 (26). The binding sites and locations of Cep164 on the distal appendage are unknown, but if we assume that the Cep164 protein binds along the length of the distal appendage, and take the average of the range of centriole diameters reported by Ibrahim et al. (21) plus a contribution from the size of antibody molecules (34), we would predict a range of immunolabeled Cep164 ring diameters of 230–430 nm from EM (Table 1). Our STED measurements of Cep164 ring diameters for both MTEC and proliferating cell samples agree with this prediction. The Cep164 ring diameters of the underlabeled, optimized-labeled, and overlabeled MTEC were consistent (290 ± 18 nm, 310 ± 16 nm, and 310 ± 25 nm, respectively), whereas those of the proliferating cell samples were ∼20% larger (360 ± 16 nm). In conjunction with our observation of 9-fold symmetry of both optimized-labeled MTEC and proliferating cells, our results suggest that the distal appendages of both proliferating cells and differentiated multiciliated cells are similar. Interestingly, overlaying a representative STED image of a proliferating cell with the model from the electron tomographic reconstruction on the same scale shows the Cep164 clusters positioned near the mid-point of the appendages (Fig. 5), as opposed to near either of the ends of the distal appendage. Given our PSFSM width of ∼60 nm, our results suggest that the Cep164 epitope observed in our experiments is present near the mid-point of the distal appendage.
Table 1.
Comparison of Cep164 structural measurements by STED and EM
| Cep164 clusters/centriole | Ring diameter∗ | Cluster diameter† | 2° Antibodies/cluster | |
|---|---|---|---|---|
| STED | ||||
| Underlabeled MTECs | 3.3 ± 1.6 | 290 ± 18 nm | 78 ± 11 nm x | 0.9, 2.0 |
| 61 ± 9 nm | ||||
| Optimized-labeled MTECs | 8.5 ± 0.7 | 310 ± 16 nm | 87 ± 11 nm, 110 ± 17 nm x | 5.9‡ |
| 71 ± 11 nm, 80 ± 17 nm | ||||
| Overlabeled MTECs | N/A | 310 ± 25 nm | N/A | N/A |
| Proliferating IMCD3 | 8–9§ | 360 ± 16 nm | N/A | N/A |
| Electron microscopy | ||||
| Immuno-EM | N/A | 260 nm¶ | N/A | N/A |
| Electron tomography | N/A | 230–430 nm‖ | N/A | N/A |
Figure 5.

Representative STED image of Cep164 in an IMCD3 cell overlaid on the electron tomographic model of the mother centriole on the same scale (reprinted with permission from (21)), scale bar, 200 nm.
Discussion
In this work, we have directly observed the 9-fold symmetric arrangement of a centrosomal protein in two distinct cell types, reflecting the underlying 9-fold symmetry of the centriole. We have also quantified labeling density effects in super-resolution immunofluorescence imaging, and demonstrated the importance of appropriate labeling to achieving validity of the observed structure.
Both electron and far-field optical microscopy have been widely employed to probe the anatomy of the centrosome and centriole, with each technique having strengths and limitations. EM is unrivaled in its ability to spatially resolve details of subcellular architecture to fractions of a nanometer. For example, the morphological features of centriole components, including their 9-fold symmetry, have been shown in remarkable detail (21,27,39–41). However, EM reconstructions often make an assumption of symmetry that is imposed on the fitting model, and many images are averaged together, rendering undetectable any heterogeneity in the observed structures. The use of antibody-conjugated gold particles in immuno-EM permits the localization of specific proteins (26), but the sample preparation conditions are often incompatible with preserving antigen reactivity, and may introduce other artifacts such as metallic particle clustering (42). Thus, quantitative characterization of protein localization using immuno-EM is challenging.
In contrast, conventional far-field optical microscopy can be easily used to image specific proteins in fixed cells using immunofluorescence labeling, and in live cells by tagging proteins of interest with fluorescent markers, but it suffers from the optical diffraction limit of spatial resolution (∼250 nm) (1). The arrangement of most centriole proteins cannot be accurately determined by diffraction-limited optical methods because the centriole itself is typically 250 nm wide and 500 nm long, roughly the resolution limit of such methods. With the development of super-resolution microscopy methods such as STED, SIM, PALM, F-PALM, and STORM (4,7–9,43), the resolution of far-field optical microscopy has been extended down to the 10's of nm (3,44,45). Characterizing the organization of centriole proteins should be greatly facilitated by super-resolution microscopy, and indeed, several super-resolution microscopy studies of centriole proteins were recently reported. These include our preliminary STED images of Cep164 (28), SIM images of the centriole proteins Sas-4 (46), CEP63 and CEP152 (47), and PALM and STORM images of distal appendage proteins CEP164 and CEP123 (29).
The studies using PALM, STORM, and SIM revealed the centriolar ring-like structures of the studied proteins, but with little resolved substructure within the rings. In contrast, we were able, using STED, to further resolve the localization of Cep164 into clusters with 9-fold symmetry about the centriole, consistent with the known structure of the distal appendages of the centriole. We harnessed several strengths of STED super-resolution microscopy in this study. First, the optical sectioning ability allowed rejection of out-of-focus background and imaging of the centrioles in a cell type, MTEC, that has many centrioles in a plane, but is relatively thick compared to typical cultured cells (28,48). Combining STED with the MTEC cultures made it possible to assess the distribution of Cep164 molecules in hundreds of centrioles, allowing us to quantify distributions of cluster number, size and brightness. In contrast, immuno-EM would have been limited to no more than a few centrioles. Finally, because the resolution enhancement in STED is essentially all-optical and independent of the active concentration of dye emitters, it does not rely on postacquisition image reconstruction or stochastic switching, which may lead to image artifacts. Immuno-EM localization of Cep164 to distal appendages hinted that Cep164 might localize with 9-fold radial symmetry, but the measurements were confounded by the aggregation of the metal nanoparticles and potential underlabeling; it was conceivable based solely on immuno-EM that Cep164 might localize to a subset of distal appendages. Using only raw STED images and optimization of antibody labeling with no deconvolution, we were able to show in both proliferating and multiciliated cells that Cep164 localizes in a 9-fold radially symmetric ring of ∼350 nm diameter.
The resolving power of super-resolution immunofluorescence microscopy is influenced by the size of the antibody labels used, whereas in diffraction-limited immunofluorescence microscopy, the resolution limit is an order of magnitude coarser than the label size. As mentioned previously, optimized label density is necessary for obtaining accurate super-resolution immunofluorescence images. We have demonstrated that labeling density significantly affects the observed distribution of Cep164 protein, and have quantified these labeling effects on observed protein cluster number, size, and brightness. Given the measured mean cluster size of ∼80 nm in the optimized-labeled samples and our PSFSM width of ∼60 nm, we estimate the true cluster size to be ∼60 nm. A 60 nm object (Cep164 plus bound antibodies) is reasonable given that there are many possible patterns for the binding of ∼5–10 antibodies of ∼10 nm size to the N-terminal domain of Cep164 on each distal appendage. Thus, under optimized labeling conditions, our measurements of Cep164 cluster size and brightness are consistent with expectation. In contrast, underlabeling results in underestimation of observed protein cluster number, size, and brightness, and over-labeling results in blurring of fine features due to the large size of the antibody labels. This shows that antibody labeling artifacts can cause inaccurate measurements of protein localization at the nanometer scale and should be avoided in high-resolution immunofluorescence microscopy.
The optimal antibody concentration will have to be determined empirically for each protein of interest and immunolabeling scheme. Here, the optimized concentration can be defined as the highest 1° antibody concentration used before the observed structure becomes blurred due to crowding of antibody labels. The observed structure using this optimized labeling density represents the most accurate structure obtainable for a given resolution. Whether the measured structure is an accurate depiction of the true structure is a general question that the microscopist should carefully evaluate. In this study, we determined a 9-fold symmetric Cep164 structure independent of previous knowledge of the 9-fold symmetry of centrioles. The agreement with results from EM, which is a different method with different assumptions and sample preparation conditions, further strengthens our findings. Indeed, it is conceivable and would have been interesting had Cep164 been found to localize to fewer or more than nine clusters per centriole. Had we observed fewer than nine clusters per centriole, for example due to blurring of the observed structure from crowding of antibody labels, one possible improvement would be to directly conjugate the fluorophore to an anti-Cep164 monoclonal antibody (or Fab fragments thereof). Because only one antibody molecule can bind to each molecule of Cep164, the contribution of primary and secondary antibodies to observed cluster size would be greatly reduced. Alternatively, tagging with a fluorescent protein such as TagRFP657 (49) would eliminate the antibody component and approach one label per protein, further reducing observed cluster size, and might also allow super-resolution imaging of the centrosome in live cells. However, fluorescent protein tags can disrupt the function or stability of the protein of interest (50), or photodamage to cells from live imaging might also affect localization of tagged proteins of interest (51).
The similarity of Cep164 organization in both proliferating and multiciliated cells suggests that Cep164 has a similar function in both cell types, probably in cilia formation as previously characterized in human RPE-1 cells (26). Because Cep164 was, until recently, the only known molecular component of distal appendages, our result further suggests that distal appendages are identical in structure and function in proliferating and multiciliated cells. Because the localization of proteins to structures within the centrosome can provide clues to their function, STED and other super-resolution methods are likely to become critical tools for the investigation of centrosome organization. With its ability for high-throughput quantitative measurements using simple labeling protocols, STED super-resolution microscopy is a complimentary technique to EM for probing centrosome structure.
Acknowledgments
We gratefully acknowledge support from M. Koenig (PicoQuant, Berlin, Germany), M. Hsu (National Measurement Institute), and the Hell lab (Max Planck Institute for Biophysical Chemistry, Göttingen, Germany), for initial help on the STED setup. We thank Q. Wang, M. Lew, R. Goldsmith, Y. Jiang, H. Lee, and W. Duim for helpful discussions and E. Nigg (University of Basel, Basel, Switzerland) for the Cep164 antibody. The project described was supported in part by award No. R01GM085437 from the National Institute of General Medical Sciences (W.E.M.) and by award No. R01GM52022 from the National Institute of General Medical Sciences (T.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. L.L. acknowledges support from a Stanford Lieberman Fellowship and a Stanford Diversifying Academia, Recruiting Excellence Fellowship. Y.L.L. acknowledges fellowship support from the Agency for Science, Technology and Research, Singapore.
Supporting Material
References
- 1.Abbe E. Contributions to the theory of the microscope and microscopic detection (translated from German) Arch. Mikrosk. Anat. 1873;9:413–468. [Google Scholar]
- 2.Moerner W.E. New directions in single-molecule imaging and analysis. Proc. Natl. Acad. Sci. USA. 2007;104:12596–12602. doi: 10.1073/pnas.0610081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hell S.W. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 4.Hell S.W., Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 5.Gustafsson M.G.L. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hell S.W. Microscopy and its focal switch. Nat. Methods. 2009;6:24–32. doi: 10.1038/nmeth.1291. [DOI] [PubMed] [Google Scholar]
- 7.Hess S.T., Girirajan T.P.K., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Betzig E., Patterson G.H., Hess H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 9.Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson R.E., Larson D.R., Webb W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang B., Babcock H., Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biteen J.S., Thompson M.A., Moerner W.E. Super-resolution imaging in live Caulobacter crescentus cells using photoswitchable enhanced yellow fluorescent protein. Proc. SPIE. 2009;7185:71850I. [Google Scholar]
- 13.Willig K.I., Harke B., Hell S.W. STED microscopy with continuous wave beams. Nat. Methods. 2007;4:915–918. doi: 10.1038/nmeth1108. [DOI] [PubMed] [Google Scholar]
- 14.Punge A., Rizzoli S.O., Hell S.W. 3D reconstruction of high-resolution STED microscope images. Microsc. Res. Tech. 2008;71:644–650. doi: 10.1002/jemt.20602. [DOI] [PubMed] [Google Scholar]
- 15.Hein B., Willig K.I., Hell S.W. Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proc. Natl. Acad. Sci. USA. 2008;105:14271–14276. doi: 10.1073/pnas.0807705105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westphal V., Rizzoli S.O., Hell S.W. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science. 2008;320:246–249. doi: 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- 17.Shroff H., Galbraith C.G., Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat. Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singla V., Reiter J.F. The primary cilium as the cell's antenna: signaling at a sensory organelle. Science. 2006;313:629–633. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- 19.Basto R., Lau J., Raff J.W. Flies without centrioles. Cell. 2006;125:1375–1386. doi: 10.1016/j.cell.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 20.Nigg E.A., Raff J.W. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 21.Ibrahim R., Messaoudi C., Marco S. Electron tomography study of isolated human centrioles. Microsc. Res. Tech. 2009;72:42–48. doi: 10.1002/jemt.20637. [DOI] [PubMed] [Google Scholar]
- 22.Kitagawa D., Vakonakis I., Steinmetz M.O. Structural basis of the 9-fold symmetry of centrioles. Cell. 2011;144:364–375. doi: 10.1016/j.cell.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Breugel M., Hirono M., Zuber B. Structures of SAS-6 suggest its organization in centrioles. Science. 2011;331:1196–1199. doi: 10.1126/science.1199325. [DOI] [PubMed] [Google Scholar]
- 24.Nigg E.A., Stearns T. The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 2011;13:1154–1160. doi: 10.1038/ncb2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawe H.R., Farr H., Gull K. Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J. Cell Sci. 2007;120:7–15. doi: 10.1242/jcs.03305. [DOI] [PubMed] [Google Scholar]
- 26.Graser S., Stierhof Y.D., Nigg E.A. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007;179:321–330. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vladar E.K., Stearns T. Molecular characterization of centriole assembly in ciliated epithelial cells. J. Cell Biol. 2007;178:31–42. doi: 10.1083/jcb.200703064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lau L., Lee Y.L., Moerner W.E. STED super-resolution microscopy in Drosophila tissue and in mammalian cells. Proc. SPIE. 2011;7910:79101N. doi: 10.1117/12.881221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sillibourne J.E., Specht C.G., Bornens M. Assessing the localization of centrosomal proteins by PALM/STORM nanoscopy. Cytoskeleton (Hoboken) 2011;68:619–627. doi: 10.1002/cm.20536. [DOI] [PubMed] [Google Scholar]
- 30.Beesley J.E. IRL Press; Oxford: 1993. Immunocytochemistry: A Practical Approach. [Google Scholar]
- 31.Harlow E., Lane D. Cold Spring Harbor Press; New York: 2008. Antibodies: A Laboratory Manual. [Google Scholar]
- 32.Huang B., Wang W., Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aquino D., Schönle A., Egner A. Two-color nanoscopy of three-dimensional volumes by 4Pi detection of stochastically switched fluorophores. Nat. Methods. 2011;8:353–359. doi: 10.1038/nmeth.1583. [DOI] [PubMed] [Google Scholar]
- 34.Harris L.J., Larson S.B., McPherson A. The three-dimensional structure of an intact monoclonal antibody for canine lymphoma. Nature. 1992;360:369–372. doi: 10.1038/360369a0. [DOI] [PubMed] [Google Scholar]
- 35.Clifford J.N., Bell T.D., Yeow E.K. Fluorescence of single molecules in polymer films: sensitivity of blinking to local environment. J. Phys. Chem. B. 2007;111:6987–6991. doi: 10.1021/jp072864d. [DOI] [PubMed] [Google Scholar]
- 36.Kasper R., Harke B., Sauer M. Single-molecule STED microscopy with photostable organic fluorophores. Small. 2010;6:1379–1384. doi: 10.1002/smll.201000203. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt T., Schütz G.J., Schindler H. Local stoichiometries determined by counting individual molecules. Anal. Chem. 1996;68:4397–4401. [Google Scholar]
- 38.Mutch S.A., Fujimoto B.S., Chiu D.T. Deconvolving single-molecule intensity distributions for quantitative microscopy measurements. Biophys. J. 2007;92:2926–2943. doi: 10.1529/biophysj.106.101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen E., Binet S., Meininger V. Ciliogenesis and centriole formation in the mouse embryonic nervous system. An ultrastructural analysis. Biol. Cell. 1988;62:165–169. [PubMed] [Google Scholar]
- 40.Fisch C., Dupuis-Williams P. Ultrastructure of cilia and flagella - back to the future! Biol. Cell. 2011;103:249–270. doi: 10.1042/BC20100139. [DOI] [PubMed] [Google Scholar]
- 41.Bornens M., Paintrand M., Karsenti E. Structural and chemical characterization of isolated centrosomes. Cell Motil. Cytoskeleton. 1987;8:238–249. doi: 10.1002/cm.970080305. [DOI] [PubMed] [Google Scholar]
- 42.Rash J.E., Yasumura T. Direct immunogold labeling of connexins and aquaporin-4 in freeze-fracture replicas of liver, brain, and spinal cord: factors limiting quantitative analysis. Cell Tissue Res. 1999;296:307–321. doi: 10.1007/s004410051291. [DOI] [PubMed] [Google Scholar]
- 43.Gustafsson M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- 44.Lippincott-Schwartz J., Patterson G.H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009;19:555–565. doi: 10.1016/j.tcb.2009.09.003. (Abstr.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moerner W.E. Single-molecule mountains yield nanoscale cell images. Nat. Methods. 2006;3:781–782. doi: 10.1038/nmeth1006-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gopalakrishnan J., Mennella V., Avidor-Reiss T. Sas-4 provides a scaffold for cytoplasmic complexes and tethers them in a centrosome. Nat. Commun. 2011;2:359. doi: 10.1038/ncomms1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sir J.H., Barr A.R., Gergely F. A primary microcephaly protein complex forms a ring around parental centrioles. Nat. Genet. 2011;43:1147–1153. doi: 10.1038/ng.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urban N.T., Willig K.I., Nägerl U.V. STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys. J. 2011;101:1277–1284. doi: 10.1016/j.bpj.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morozova K.S., Piatkevich K.D., Verkhusha V.V. Far-red fluorescent protein excitable with red lasers for flow cytometry and super-resolution STED nanoscopy. Biophys. J. 2010;99:L13–L15. doi: 10.1016/j.bpj.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carminati J.L., Stearns T. Microtubules orient the mitotic spindle in yeast through dynein-dependent interactions with the cell cortex. J. Cell Biol. 1997;138:629–641. doi: 10.1083/jcb.138.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van de Linde S., Heilemann M., Sauer M. Live-cell super-resolution imaging with synthetic fluorophores. Annu. Rev. Phys. Chem. 2012;63:519–540. doi: 10.1146/annurev-physchem-032811-112012. [DOI] [PubMed] [Google Scholar]
- 52.You Y., Richer E.J., Brody S.L. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;283:L1315–L1321. doi: 10.1152/ajplung.00169.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.