

 Bromopyridazinedione-mediated bioconjugation to a cysteine containing protein and a disulfide containing peptide is reported. These bioconjugates exhibit excellent hydrolytic stability and cleave in an aqueous, thiol-mediated reducing environment.
Bromopyridazinedione-mediated bioconjugation to a cysteine containing protein and a disulfide containing peptide is reported. These bioconjugates exhibit excellent hydrolytic stability and cleave in an aqueous, thiol-mediated reducing environment.
Abstract
Bromopyridazinedione-mediated bioconjugation to a cysteine containing protein and a disulfide containing peptide is described. The conjugates are cleavable in an excess of thiol, including cytoplasmically-relevant concentrations of glutathione, and show a high level of hydrolytic stability. The constructs have the potential for four points of chemical attachment.
The published sequence of the human genome revealed encoding for only one tenth of the total proteome. The role of protein post-translational modification has therefore become of intense interest in understanding complex, physiological processes. Combining synthetic organic chemistry with molecular biology has enabled the construction of a range of proteins and peptides as probes to aid in this venture. This has allowed the introduction of synthetic post-translational modifications1–3 and a variety of fluorescent,4–6 radiolabelled7 and affinity tags8–10 into a variety of protein and peptide structures. The selective modification of cysteine remains a popular method for achieving protein modification. This is due to its low natural abundance, high nucleophilicity and the ease with which cysteine can be introduced at desired locations in protein sequences using modern molecular biology methods.11–12
In our laboratory, we have recently become interested in the construction of protein bioconjugates that are cleavable in a reducing environment. We believe that such an approach could deliver reversible affinity or fluorescent labels, or constructs cleavable under cytoplasmic conditions with potential as prodrugs. Maleimides are known to react rapidly and selectively, but irreversibly, with thiols. However, we have recently demonstrated an approach to reversible cysteine bioconjugation using bromomaleimides, where retention of the maleimide double bond allows cleavage of the constructs via conjugate addition reaction.13 Using bromomaleimides we have been able to demonstrate the modular construction of complex bioconjugates, without requirement for reagent pre-activation, that have three points of chemical attachment. Following on from this success, we are interested to determine whether the ability to construct reversible constructs is something that is restricted to maleimides or whether other cyclic or acyclic haloeneamide systems will afford alternative opportunities.
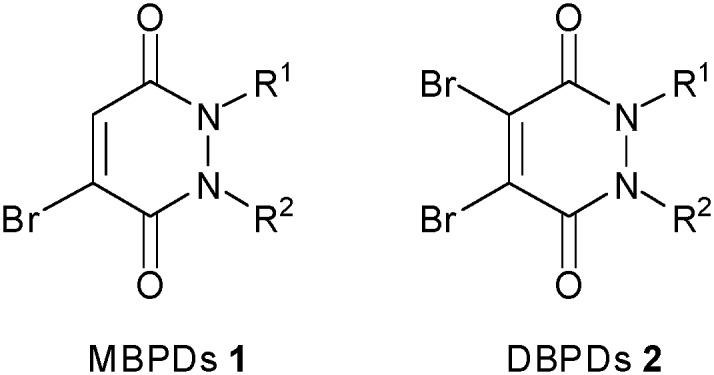
We report herein the use of both monobromo- and dibromo-1,2-dihydro-pyridazine-3,6-diones (MBPDs 1 and DBPDs 2) (Fig. 1) to assemble cleavable bioconjugates. The bioconjugates generated demonstrate exceptional hydrolytic stability with the potential for four points of chemical attachment. We have previously shown that hydrolytic stability is crucial in preserving the thiol-cleavable property of bromomaleimide-linked bioconjugates.13d
Fig. 1. (Di)bromo-1,2-dihydro-pyridazine-3,6-diones.
Our initial studies focused on the functionalisation of a single cysteine mutant (L111C) of the SH2 domain of the Grb2 adapter protein 3,13a a protein that does not otherwise contain any cysteine residues, with pyridazinedione (PD) 4 (100 mol eq., 37 °C, 16 h). No reaction was seen to occur (Scheme 1). The absence of reactivity observed with PD-4 may be rationalised by the acidic nature of its hydrazide protons. At pH 8, PD-4 will predominantly exist in an unreactive, anionic form.14 However, treatment of protein 3 with PD-5 (100 mol eq., 37 °C, 16 h) yielded quantitative conversion to the desired conjugate 6 (Scheme 1). As this protein sequence contains eight lysine residues, the reaction with PD-5 demonstrates remarkable selectivity for cysteine as was evidenced following an absence of reaction with Ellman’s reagent. To our knowledge, this is the first reported example of a pyridazinedione being used to modify cysteine.
Scheme 1. Modification of Grb2-SH2 domain (L111C) 3 with 1,2-dihydro-pyridazine-3,6-diones.

Encouraged by the successful functionalisation of protein 3 with PD-5, we sought to synthesise and evaluate MBPD-7 and DBPD-8 as reagents for the reversible bioconjugation of proteins. Synthesis of both 7 and 8 was achieved in high yield by the initial condensation of maleic anhydride with diethylhydrazine followed by sequential dibromination/elimination steps (Scheme 2).
Scheme 2. Synthesis of (di)bromo-1,2-dihydro-pyridazine-3,6-diones.

Treatment of protein 3 with MBPD-7 (100 mol eq., 37 °C, 1 h) gave quantitative conversion to the desired adduct 9 most likely via a conjugate addition–elimination mechanism. Retention of the double bond in 9 (cf. adduct 6) created the possibility of reversing this modification in an excess of thiol. Dialysis of the crude product mixture allowed removal of excess MBPD-7. Subsequent treatment of the MBPD-modified protein with 2-mercaptoethanol (BME) (100 mol eq., 20 °C, 1 h) gave quantitative conversion back to protein 3, highlighting the potential of bromopyridazinediones to mediate reversible protein bioconjugation (Scheme 3). Conjugate 9 displayed excellent hydrolytic stability at physiological temperature and was completely stable at 37 °C for 5 h.
Scheme 3. Reversible modification of Grb2-SH2 domain (L111C) 3 with (di)bromo-1,2-dihydro-pyridazine-3,6-diones.

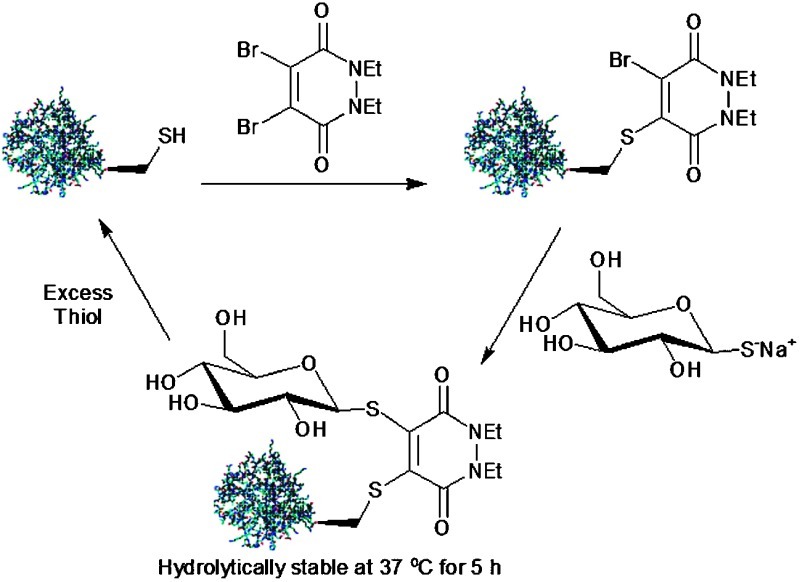
Addition of DBPD-8 (100 mol eq., 37 °C, 1 h) to protein 3 gave adduct 10. Gratifyingly, addition of thioglucose, sodium salt (10 mol eq., 24 °C, 1 h) as a second thiol nucleophile, following dialysis of the crude product mixture, gave bioconjugate 11 in high conversion. Treatment of bioconjugate 11 with an excess of BME (100 mol eq., 24 °C, 1 h) gave disassembly of the conjugate, yielding protein 3. (Scheme 3). Bioconjugate 11 was also seen to demonstrate excellent hydrolytic stability at 37 °C for 5 h.
The observed cleavage of protein/pyridazine constructs in an excess of thiol suggests that this scaffold could potentially form the core of novel prodrug systems. Such prodrugs could be designed and optimised to cleave in the cytoplasm of mammalian cells, which contain a relatively high concentration of the thiol glutathione (GSH) (1–10 mM). As a proof of principle, we subjected bioconjugate 11 to an excess of GSH (1 mM) at physiological pH. Bioconjugate 11 was seen to quantitatively disassemble, yielding protein 3 under these conditions (Scheme 4).
Scheme 4. Cleavage of Grb2-SH2 domain (L111C)/thioglucose conjugate 11 in a cytoplasmically-relevant concentration of GSH.

In addition to the functionalisation of free cysteine in proteins, we are also interested in the labeling of disulfide bonds. We have recently shown the potential of bridging disulfide bonds with dibromomaleimide bridging agents, yielding conjugates that retain biological activity.13a,c We now wish to demonstrate how dibromopyridazinedione 8 can be used to successfully bridge the disulfide bond of peptide hormone somatostatin in a reversible manner. Thus treatment of somatostatin 12 (pH 6.2) with tris-(2-carboxyethyl)-phosphine (TCEP) (1.1 mol eq.) followed by DBPD-8 (5 mol eq., 20 °C, 2 h) gave complete conversion to the desired bridged somatostatin 13. Subsequent treatment of 13 with BME (100 mol eq., 20 °C, 72 h) gave reversion to reduced somatostatin 14 (containing 10–15% of somatostatin/BME adduct) (Scheme 5).
Scheme 5. Reversible bridging of the disulfide bond of somatostatin 12 using 4,5-dibromo-1,2-dihydro-pyridazine-3,6-dione 8.

In conclusion, we have shown how both mono- and di-bromopyridazinediones can be used to mediate reversible protein and peptide bioconjugation. More specifically, we have demonstrated the selective modification of a free cysteine residue in a protein and also that dibromopyridazinedione 8 can be used to successfully bridge the disulfide bond of the peptide hormone somatostatin. These modifications have been shown to be reversible in an excess of thiol such as BME, and the protein modification was shown to cleave in a physiologically relevant concentration of GSH.
We envisage numerous potential applications for this methodology that will exploit the thiol-cleavable and hydrolytically stable nature of this modification. These include the synthetic PTM of proteins; cleavable affinity tags for protein purification, enrichment and immobilisation; cleavable fluorophores, radiolabels and quantum dots for imaging; cleavable PEGylation for protein stabilisation; prodrugs designed to cleave in the cytoplasmic environment of cells. In addition, the potential for four points of attachment creates the possibility of the multi-labelling of protein conjugates via a single cysteine or disulfide modification. Application and further evaluation of this methodology will be reported in due course.
The authors are grateful to RCUK, EPSRC, BBSRC, UCL and the Wellcome Trust and UCLB for support of our programme.
Footnotes
References
- Kasteren S. I. Van, Kramer H. B., Jensen H. H., Campbell S. J., Kirkpatrick J., Oldham N. J., Anthony D. C., Davis B. G. Nature. 2007;446:1105. doi: 10.1038/nature05757. [DOI] [PubMed] [Google Scholar]
- Davis B. G. Science. 2004;303:480. doi: 10.1126/science.1093449. [DOI] [PubMed] [Google Scholar]
- Kiick K. L., Saxon E., Tirrell D. A., Bertozzi C. R. Proc. Natl. Acad. Sci. U. S. A. 2002;99:19. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K. Chem. Soc. Rev. 2010;39:2048. doi: 10.1039/b819316a. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos K., Kiel A., Seefeld A., Stohr K., Herten D.-K. ChemPhysChem. 2010;11:43. doi: 10.1002/cphc.200900359. [DOI] [PubMed] [Google Scholar]
- Johnsson N., Johnsson K. ACS Chem. Biol. 2007;2:31. doi: 10.1021/cb6003977. [DOI] [PubMed] [Google Scholar]
- Tolmachev V., Stone-Elander S. Biochim. Biophys. Acta, Gen. Subj. 2010:487. doi: 10.1016/j.bbagen.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Dirksen A., Yegneswaren S., Dawson P. E. Angew. Chem. Int. Ed. 2010;49:2023. doi: 10.1002/anie.200906756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers A. E., Cravatt B. F. J. Am. Chem. Soc. 2005;127:10018. doi: 10.1021/ja0532842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhelst S. H. L, Fonovic M., Bogyo M. Angew. Chem., Int. Ed. 2007;46:1284. doi: 10.1002/anie.200603811. [DOI] [PubMed] [Google Scholar]
- Lundblad R. L., Chemical Reagents for Protein Modification, CRC Press, Boca Raton, FL, 3rd edn, 2005 [Google Scholar]
- Chalker J. M., Bernardes G. J. L., Lin Y. A., Davis B. G. Chem.–Asian J. 2009;4:630. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- (a) Smith M. E. B., Schumacher F. F., Ryan C. P., Tedaldi L. M., Papaioannou D., Waksman G., Caddick S., Baker J. R. J. Am. Chem. Soc. 2010;132:1960. doi: 10.1021/ja908610s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tedaldi L. M., Smith M. E. B., Nathani R., Baker J. R. Chem. Commun. 2009:6583. doi: 10.1039/b915136b. [DOI] [PubMed] [Google Scholar]; (c) Schumacher F. F., Nobles M., Ryan C. P., Smith M. E. B., Tinker A., Caddick S., Baker J. R. Bioconjugate Chem. 2011;22:132. doi: 10.1021/bc1004685. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ryan C. P., Smith M. E. B., Schumacher F. F., Grohmann D., Papaioannou D., Waksman G., Werner F., Baker J. R., Caddick S. Chem. Commun. 2011;47:5452. doi: 10.1039/c1cc11114k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Smith M. E. B., Caddick S. and Baker J. R., Patent applications WO 2011/018611 A1 (reversible covalent linkage of functional moieties), WO 2011/018612 A2 (thiol protecting group), WO 2011/018613 A1 (functionalization of solid substrates).
- Albert A., Philips J. N. J. Chem. Soc. 1956:1294. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.