

Abstract
Abstract
Intestinal pathogens have a wide variety of strategies for communicating with host epithelial cells. This review highlights a few key examples of those strategies. Enteropathogenic Escherichia coli (EPEC) use a type III secretion system (T3SS) to alter host ion transport through both transcriptional and post-translational mechanisms. Salmonella use a similar T3SS to invade host cells and modify an intracellular vacuole, which also impacts host vesicle trafficking. Helicobacter pylori use host cell integrins to provide a conformational change which drives the type IV secretion system into the host cell for delivery of CagA. The novel type VI section systems are phage-like apparati that deliver VgrG-1, which causes actin cross-linking and fluid accumulation in a suckling mouse model. An entirely different delivery mechanism is the outer membrane vesicle (OMV) which is composed of bacterial outer membrane wrapped around contents of the periplamsic space. Enterotoxigenic E. coli use OMVs to deliver bundles of heat labile enterotoxin to host cells. Finally we discuss the host responses to these varied methods of communication.
Gail Hecht (left) is Professor of Medicine and Microbiology/Immunology and Chief, Digestive Diseases and Nutrition at the University of Illinois at Chicago. She also serves as Editor-in-Chief of a new journal, Gut Microbes. The focus of her research is host–pathogen interactions with specific emphasis on enteropathogenic Escherichia coli and enterohaemorrhagic E. coli. Her goal is to determine how the bacterial effector proteins that are translocated into intestinal epithelial cells by these pathogens alter host cell physiology including tight junction barrier function, active transport processes, and the innate immune response. Her research is funded by both the NIH and the VA. Kim Hodges (right) is a microbiologist who trained at the University of Michigan in Ann Arbor and received her PhD at the University of Minnesota. She studied invasion of Streptococcus pyogenes under Dr Pat Cleary. Her current work focuses on the role of decreased absorption of ions in diarrhea caused by enteropathogenic Escherichia coli (EPEC).
 |
Introduction
The interface of the host intestinal epithelial layer with the lumen provides a unique opportunity for interspecies communication. Bacteria constantly send and receive signals from the host epithelium and the host cells respond and initiate their own messages. For example, the hormones adrenaline and noradrenaline typically target adrenergic receptors and regulate smooth muscle contraction and thus intestinal motility. However, enterohaemorrhagic Escherichia coli are capable of intercepting those messages via the quorum sensing enzyme histidine kinase, QseC, and initiating the production of virulence factors such as the type III secretion system (Hughes et al. 2009)), a molecular syringe that injects bacterial effector proteins into host cells. Intestinal pathogens communicate with the host in a variety of ways but in this review we focus on the delivery mechanisms of various toxins, effector proteins and even viable bacteria into the host cell and the response of the infected epithelium.
Secretion systems
Communication of bacteria with host cells often involves interaction with eukaryotic cell receptors; however, intestinal cells are polarized in such a way that most of the receptors that detect bacteria (i.e. Toll-like receptors) are positioned on the basolateral surface in order to avoid constant stimulation from the bacteria-filled lumen (Abreu et al. 2003)). While there is some evidence that commensal bacteria interact with the host, they are primarily restricted to the mucus layer in part due to surveillance by host Paneth cells, which secrete antimicrobial peptides in a MyD88-dependent manner in response to encroaching bacteria (Ayabe et al. 2000; Vaishnava et al. 2008)). In contrast, pathogens often have direct contact with the cell, and can become internalized or breach the epithelial barrier by passing though M-cells (Jones et al. 1994)). As such, pathogens have become fluent in cellular communication and how to bypass host defences. Flagellum-, pilus- and phage-like structures have evolved into the type III, type IV and type VI secretion systems that deliver effector molecules to the host (Fig. 1)) (Cascales & Christie, 2003)). Type III secretion systems are the most abundant and are used by enteropathogenic E. coli (EPEC), enterohaemorrhagic E. coli (EHEC), Salmonella and Shigella for both extracellular and intracellular communication. EHEC and EPEC are closely related and both utilize the type III secretion system but in slightly different ways to recruit actin beneath bacterial microcolonies in a Tir-dependent process to form what is referred to as an attaching and effacing lesion. For EHEC, Tir is injected into the host cell where it is phosphorylated, which drives the recruitment of actin through an intracellular signalling process involving another effector protein, EspFu, a host linker protein called IRSp53, and finally the actin nucleator N-WASP (Vingadassalom et al. 2009)). In contrast, EPEC tyrosine 474 is phosphorylated and binds directly to the host cell protein Nck, which recruits actin via N-WASP and Arp2/3 (Campellone et al. 2002)). In addition, some 25 putative EPEC effector proteins have been identified with additional phage-localized proteins being expressed by EHEC. Among these effectors are EspF, Map and EspG, which have been shown to have effects on both ion transport and tight junctions (Fig. 2)) (McNamara et al. 2001; Dean & Kenny, 2004; Tomson et al. 2005; Dean et al. 2006; Gill et al. 2007; Hodges et al. 2008)). EspF was first identified for its role in tight junction disruption, which alters paracellular permeability by interfering with the localization of occludin and ZO-1 in intestinal epithelium (McNamara et al. 2001)). More recently it was found that EspF is also responsible for a reduction in sodium uptake through NHE3 (Hodges et al. 2008)). EspF and Map have been implicated in reducing SGLT-1 activity through a brief process involving reduced glucose uptake or a more prolonged process caused by microvillus effacement (Dean et al. 2006)). Finally EspG1 and EspG2 block Cl− uptake via Slc26a3/DRA (solute carrier 26a3/downregulated in adenoma) by breaking down microtubules, thus preventing its surface localization (Gill et al. 2007)). Therefore, EPEC type III secretion effectors block the three major sodium and chloride transport pathways and alter tight junction function resulting in perturbations of the osmotic gradient which aids in salt and water absorption.
Figure 1. Schematic of the type III, IV and VI secretion systems.
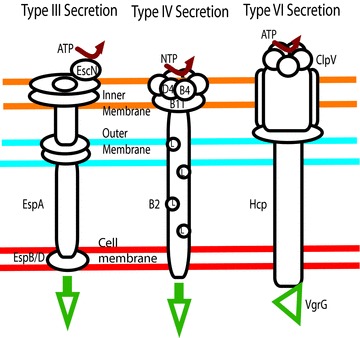
The EPEC type III secretion system uses EscN, an ATPase, to drive secretion through a flagellum-like apparatus. H. pylori uses several NTPases, D4, B4 and B11 to power its pilus-like system. Finally, the phage-like type VI secretion system uses the ATPase ClpV to deliver effectors like VgrG.
Figure 2. Enteropathogenic E. coli (EPEC) use a type III secretion system to deliver effector proteins including Tir, EspF, Map and EspG1 and EspG2.
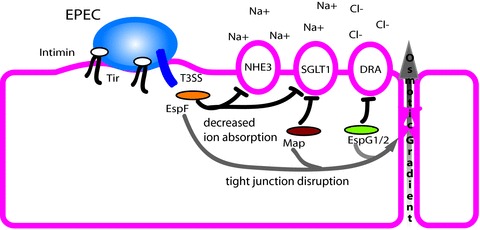
Injected Tir binds to Intimin on the bacterial surface and promotes attaching and effacing lesion formation. EspF decreases both sodium–hydrogen exchanger 3 (NHE3) and sodium–glucose cotransport 1 (SGLT1) activity in addition to disruption of tight junctions. Map acts along with EspF to decrease SGLT1 activity and tight junction function. EspG1 and EspG2 decrease Cl−/OH− exchange by DRA and also impair tight junctions. The combined activities of these proteins on ion transport alter the osmotic gradient leading to subsequent movement of water into the intestinal lumen.
Salmonella and Shigella also utilize type III secretion systems, initially for invasion and then subsequently to alter the host environment. In fact, invasion has been shown to be required for fluid accumulation in rabbit models of Salmonella infection (Giannella et al. 1973)). Salmonella create specialized intracellular membrane bound compartments called Salmonella containing vacuoles (SCVs) (Fig. 3)). The establishment of SCVs is critical to intracellular survival of Salmonella and also regulates host vesicle trafficking. These compartments are dependent on the host cell microtubule network for both formation and maintenance (Rajashekar & Hensel, 2011)). Microtubule cargo transport is mediated by two opposing motor proteins, dynein and kinesin. The level of kinesin associated with SCVs is very tightly regulated by two proteins, SifA, which interacts with the host cell factor SKIP to exclude kinesin, and PipB2, which binds to it (Boucrot et al. 2005; Henry et al. 2006)). In contrast, dynein, which is oriented toward the negative end of microtubules, is actively recruited to SCVs via the effector SseF where it plays an important role in the formation of bacterial microcolonies near the nucleus (Abrahams et al. 2006)). Bacteria containing vacuoles can traffic on microtubules by utilizing dynein–SseF interaction, thus allowing the bacteria to become clustered into a single SCV. SCVs are often found in association with microtubules and appear to redirect normal exocytic traffic to the SCV. Vesicular stomatitis virus G protein (VSVG) is often used to monitor trafficking and typically VSVG-GFP is randomly distributed; however, in infected cells there is a close association of vesicles with SCVs, suggesting that vesicle traffic is directed to the SCV (Kuhle et al. 2006)). This recruitment was linked to three effector proteins, SifA, SseF and SseG, and was specifically inhibited by the microtubule disrupting agent nocodazole (Kuhle et al. 2006)). Thus not only does Salmonella use type III secretion to create and position the intracellular SCV, it also redirects host vesicle traffic by controlling microtubule motor proteins.
Figure 3. Salmonella containing vacuoles (SCVs) are located at the minus ends of microtubules due to trafficking via dynein, which is recruited to the SCV by the effector SseF.
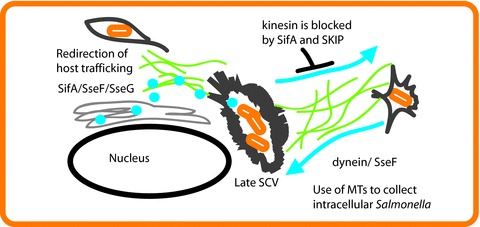
In contrast SifA through its interaction with SKIP inhibits the recruitment of kinesin, the plus end motor, preventing movement toward the cell periphery. Several effector proteins, SifA, SseF and SseG, also alter host cell trafficking toward the SCV.
While type III secretion systems are prevalent, another gastrointestinal pathogen, H. pylori, utilizes a type IV secretion system (T4SS) to deliver its primary effector protein, CagA, into host cells. Type III and type IV secretion systems have similar functions but differ in their origin. Type III systems evolved from flagella while type IV systems more closely resemble conjugative pili (Cascales & Christie, 2003)). It has been shown recently that β1 integrins play a critical role in T4SS effector translocation by H. pylori. Kwok et al. (2007)) proposed that CagL located on the exterior of the T4SS pilus binds to α5β1 integrin via an RGD motif similar to the typical interaction for the endogenous ligand fibronectin. An independent group, however, suggest that the RGD motif is irrelevant and instead propose that multiple T4SS proteins including CagL, CagY, CagI and even CagA bind to the β1 chain of integrins in a novel manner (Jimenez-Soto et al. 2009)). These interactions were identified initially by yeast two-hybrid screening using the extracellular portion of the β1 chain as bait (Jimenez-Soto et al. 2009)). Bead-coupled α1β1 and α5β1 integrins bound to CagA but not CagL or CagY. Subsequent studies suggested that CagL and CagY require an intact T4SS for integrin binding while CagA binds independently of the T4SS. In both studies, interaction of the T4SS with β1 integrin is required for translocation of CagA (Kwok et al. 2007; Jimenez-Soto et al. 2009)) (Fig 4)). CagA translocation did not occur in integrin-deficient cells (GE11) or β1 knock-out cells (GD25); however β1 complementation rescued CagA translocation (Jimenez-Soto et al. 2009)). Clustering of integrins also appears to be a requirement as blocking integrin reorganization in the lipid bilayer using calpeptin prevented CagA translocation. The authors theorize that the conformational changes in integrins after binding help position the T4SS closer to the membrane, thus allowing for translocation (Jimenez-Soto et al. 2009)). Once injected into the cell, CagA binds to upwards of 20 proteins, including host kinases and SH2 domain containing proteins, and precipitates a number of phenotypes including activation of Map kinases, NF-κB, and disruption of the tight junction protein ZO-1 (Amieva et al. 2003; Lamb et al. 2009; Selbach et al. 2009; Backert et al. 2010)). CagA induces a specific alteration in cellular morphology called the hummingbird phenotype, which causes elongation of cells with pointed ‘beak-like’ extensions (Segal et al. 1999)). Perhaps the most interesting aspect of CagA is its ability to mimic the substrate of the host cell kinase Par1b, which is involved in establishing and maintaining host cell polarity (Zeaiter et al. 2008; Nesic et al. 2010)). This is thought to contribute to both the disruption of apical junctions and the establishment of the hummingbird phenotype (Zeaiter et al. 2008; Lu et al. 2009)).
Figure 4. H. pylori type IV secreted effector proteins Cag Y, L, I and A form an association with the type IV secretion system (T4SS) and bind to the β1 chain of integrins.
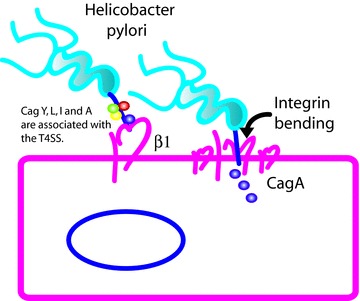
Injection of Cag A via the T4SS requires a conformational change in the β1 chain in addition to integrin clustering. An antibody that locks the conformation of β1 integrin was able to prevent CagA delivery. It is concluded therefore that the physical change of the integrin upon binding is needed for insertion of the T4SS tip and subsequent injection of effector proteins.
Type VI secretions systems were only recently identified and appear to be limited to only a few pathogens. Interestingly, they target both eukaryotic and prokaryotic hosts (Russell et al. 2011)). The type VI secretion system is derived from bacteriophage tail proteins and delivers effector proteins that are fused to the spike protein VgrG (Bonemann et al. 2010)). The type VI system exists in Vibrio cholerae, Salmonella enterica, and a variety of non-intestinal pathogens including Pseudomonas aeruginosa and Legionella pneumophila. In V. cholerae, the cap protein VgrG breaks away from the tip of the type VI apparatus after insertion into the host cell resulting in an alternative form of effector delivery (Pukatzki et al. 2006; Pukatzki et al. 2007)). There are several variants of VgrG which are designated by a number, such as VgrG-1, which has an actin cross-linking domain. V. cholerae strains with a deletion in VgrG-1, VgrG-2 or VasK, part of the type VI secretion apparatus, induced less fluid accumulation in a suckling mouse model suggesting less diarrhea (Ma & Mekalanos, 2010)). Type VI secretion systems are in the early stages of discovery and only a limited number of host effects have been described to date.
Outer membrane vesicles
Some bacterial toxins, including heat labile enterotoxin (LT) of enterotoxigenic E. coli, have been demonstrated to associate with outer membrane vesicles (OMVs) (Fig. 5)). OMVs are composed of lipid and protein components of the outer membrane of bacteria such as lipopolysaccharide (LPS) and outer membrane proteins (OMPs) that surround a residual periplasmic space containing characteristic proteins such as alkaline phosphatase (Horstman & Kuehn, 2000)). LT has been shown to reside both in the periplasm of these OMVs and in a LPS-associated fraction. Antibodies against the LT-receptor GM1 blocked cell rounding caused by LT-containing OMVs, suggesting that vesicles and their contents are directly internalized in a process that depends on GM1 (Kesty et al. 2004)). A number of Gram negative bacteria produce OMVs which enhance toxin delivery, including V. cholerae and H. pylori (Parker et al. 2010; Chatterjee & Chaudhuri, 2011)).
Figure 5. Enterotoxigenic E. coli (ETEC) form outer membrane vesicles (OMVs) from lipid and proteins constituents of the bacterial outer membrane.
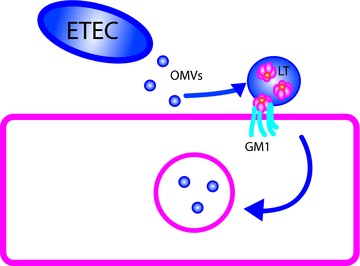
These vesicles contain a periplasmic space which carries heat labile enterotoxin (LT) in addition to other proteins such as alkaline phosphatase. LT in OMVs can interact with its receptor GM1 and mediate delivery of OMVs into the host cell, thus enhancing toxin delivery.
Understanding the host: mouse models
While a number of bacterial communication strategies with the host have been discussed, the host has its own repertoire of responses. To fully explore this response, animal models are needed. Citrobacter rodentium is a natural murine pathogen often used to mimic the human pathogen EPEC and EHEC infection as they all belong to the attaching and effacing (A/E) family of pathogens. However, the response of most mouse breeds to C. rodentium infection is intestinal crypt hyperplasia. A mouse model for diarrhoea has long been lacking. A specific inbred strain of mouse, FVB, derived from Swiss–Webster mice, is uniquely susceptible to C. rodentium infection. Specifically, mice appear ill at 6 days post-infection and begin to die at 9 days with only a 23% survival rate (Borenshtein et al. 2007)). There is a correlation between death and weight loss, but inflammatory markers such as serum TNF-α are not increased and systemic infection, characterized by colonization of the spleen, does not occur. Instead, mice exhibit signs of dehydration including ruffled coat, decreased skin turgor and sunken eyes. Importantly, most of the infected animals exhibit severe diarrhoea, and fluid rehydration by subcutaneous injection prevented mortality. Rehydration did not, however, alter bacterial counts suggesting that death was due to fluid loss.
To gain further insight into the host response, microarray analysis was performed. Interestingly, a number of host intestinal transporter levels were modified to a greater extent than even inflammatory response genes. Aquaporin 8, which contributes to water absorption, was decreased 268-fold. In another C. rodentium model system, aquaporin 2 and 3 proteins were mislocalized (Guttman et al. 2007)). In addition to changes in water absorption, there were major changes in the expression of ion transporters, thus altering the osmotic gradient and increasing luminal water. Both sodium and chloride absorptive pathways, but not secretion, were impaired by C. rodentium infection. Levels of epithelial sodium channel (ENaC/Scnn Ia), and sodium–hydrogen exchanger NHE2 (Slc9a2) and NHE3 (Slc9a3) transcripts were decreased between 2- and 13-fold leading to a decrease in serum sodium from 144 to 139 mEq l-1 in FVB mice. The chloride/bicarbonate transporter down-regulated in adenoma (DRA) was impacted even more dramatically with a 1100-fold decrease in transcript after 9 days of infection reducing serum chloride levels from 103 to 92 mEq l-1. DRA is also controlled at the post-translational level. DRA is known to recycle and was found to exist predominantly in subcellular pools as opposed to the apical surface in EPEC infected mouse colon (Gill et al. 2007)). Of some interest the transcriptional changes in DRA can be counteracted by using microflora of C57/BL/6 mice which are resistant to the effects of C. rodentium (Ghosh et al. 2011)). Interestingly, CFTR levels remained unchanged; however A2B adenosine receptor increased. A2B interacts with free adenosine, generated during inflammation, thus increasing cAMP and activating CFTR. Therefore, infection by C. rodentium decreases the transcription of a number of intestinal absorptive transporters in the host that are directly involved in salt and water movement. A number of other transporters that indirectly alter salt transport by affecting H+, HCO3− and Na+ levels in the cell were also decreased. These include carbonic anhydrase I and IV, peptide transporters Pept1 and Petp2 and the Na+/K+-ATPase (Borenshtein et al. 2008)). Thus the role of absorption, long overlooked, is key to diarrhoea caused by A/E, and probably other pathogens. In addition to these changes in ion transport, mice have been shown to respond to EPEC infection with decreased resistance, initially due to the bacterial effector EspF but with a subsequent EspF-independent effect at 5 days post-infection, which is thought to depend on TNF-α (Shifflett et al. 2005)). Importantly, host inflammation differs between EPEC and C. rodentium in this case.
In summary, pathogens have devised a number of ways to manipulate intestinal epithelial cell function. Some pathogens initiate the infectious process by intercepting host signals to induce their own virulence via quorum sensing. The type III secretion system of EPEC alters surface levels of a number of intestinal transporters including NHE3, SGLT1 and DRA/Slc26a3 while the related pathogen C. rodentium dramatically decreases the transcriptional regulation of NHE2, NHE3 and DRA/Slc26a3. In Salmonella and Shigella, diarrhoea is dependent on invasion for which type III secretion is required. H. pylori uses a type IV secretion system in a manner that depends on host cell integrin clustering and conformational change to inject the primary virulence determinant CagA. The type VI secretion system injects a modified VgrG, which causes actin cross-linking and fluid accumulation. Finally, enterotoxigenic E. coli uses small vesicles made from their own outer membrane to carry LT to host cells. These examples illustrate that pathogens have acquired a wide variety of mechanisms for communicating with cells. Our exploration and understanding of these mechanisms stand to provide insight into the development of new therapeutic strategies for the treatment of infectious diarrhoea and other pathogen-mediated gastrointestinal diseases.
Glossary
Abbreviations
- CFTR
cystic fibrosis transmembrane conductance regulator
- DRA
downregulated in adenoma
- EHEC
enterohaemorrhagic E. coli
- EPEC
enteropathogenic E. coli
- ENAC
epithelial sodium channel
- IRSp53
insulin receptor substrate p53
- LT
heat labile enterotoxin
- LPS
lipopolysaccharide
- NHE
sodium–hydrogen exchanger
- OMP
outer membrane protein
- OMV
outer membrane vesicle
- QseC
quorum sensing enzyme C
- SCV
Salmonella containing vacuole
- SGLT-1
sodium glucose cotransport
- SKIP
SifA and kinesin interacting protein
- T3SS
type III secretion system
- T4SS
type IV secretion system
- VSVG
vesicular stomatitis virus G protein
- ZO-1
zona occludens 1
References
- Abrahams GL, Muller P, Hensel M. Functional dissection of SseF, a type III effector protein involved in positioning the Salmonella-containing vacuole. Traffic. 2006;7:950–965. doi: 10.1111/j.1600-0854.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- Abreu MT, Thomas LS, Arnold ET, Lukasek K, Michelsen KS, Arditi M. TLRsignalling at the intestinal epithelial interface. J Endotoxin Res. 2003;9:322–330. doi: 10.1179/096805103225002593. [DOI] [PubMed] [Google Scholar]
- Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal α-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- Backert S, Tegtmeyer N, Selbach M. The versatility of Helicobacter pylori CagA effector protein functions: The master key hypothesis. Helicobacter. 2010;15:163–176. doi: 10.1111/j.1523-5378.2010.00759.x. [DOI] [PubMed] [Google Scholar]
- Bonemann G, Pietrosiuk A, Mogk A. Tubules and donuts: a type VI secretion story. Mol Microbiol. 2010;76:815–821. doi: 10.1111/j.1365-2958.2010.07171.x. [DOI] [PubMed] [Google Scholar]
- Borenshtein D, Fry RC, Groff EB, Nambiar PR, Carey VJ, Fox JG, Schauer DB. Diarrhea as a cause of mortality in a mouse model of infectious colitis. Genome Biol. 2008;9:R122. doi: 10.1186/gb-2008-9-8-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borenshtein D, Nambiar PR, Groff EB, Fox JG, Schauer DB. Development of fatal colitis in FVB mice infected with Citrobacter rodentium. Infect Immun. 2007;75:3271–3281. doi: 10.1128/IAI.01810-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucrot E, Henry T, Borg JP, Gorvel JP, Meresse S. The intracellular fate of Salmonella depends on the recruitment of kinesin. Science. 2005;308:1174–1178. doi: 10.1126/science.1110225. [DOI] [PubMed] [Google Scholar]
- Campellone KG, Giese A, Tipper DJ, Leong JM. A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol Microbiol. 2002;43:1227–1241. doi: 10.1046/j.1365-2958.2002.02817.x. [DOI] [PubMed] [Google Scholar]
- Cascales E, Christie PJ. The versatile bacterial type IV secretion systems. Nat Rev Microbiol. 2003;1:137–149. doi: 10.1038/nrmicro753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee D, Chaudhuri K. Association of cholera toxin with Vibrio cholerae outer membrane vesicles which are internalized by human intestinal epithelial cells. FEBS Lett. 2011;585:1357–1362. doi: 10.1016/j.febslet.2011.04.017. [DOI] [PubMed] [Google Scholar]
- Dean P, Kenny B. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol Microbiol. 2004;54:665–675. doi: 10.1111/j.1365-2958.2004.04308.x. [DOI] [PubMed] [Google Scholar]
- Dean P, Maresca M, Schuller S, Phillips AD, Kenny B. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proc Natl Acad Sci U S A. 2006;103:1876–1881. doi: 10.1073/pnas.0509451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Dai C, Brown K, Rajendiran E, Makarenko S, Baker J, Ma C, Halder S, Montero M, Ionescu VA, Klegeris A, Vallance BA, Gibson DL. Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am J Physiol Gastrointest Liver Physiol. 2011;301:G39–49. doi: 10.1152/ajpgi.00509.2010. [DOI] [PubMed] [Google Scholar]
- Giannella RA, Formal SB, Dammin GJ, Collins H. Pathogenesis of salmonellosis. Studies of fluid secretion, mucosal invasion, and morphologic reaction in the rabbit ileum. J Clin Invest. 1973;52:441–453. doi: 10.1172/JCI107201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill RK, Borthakur A, Hodges K, Turner JR, Clayburgh DR, Saksena S, Zaheer A, Ramaswamy K, Hecht G, Dudeja PK. Mechanism underlying inhibition of intestinal apical Cl/OH exchange following infection with enteropathogenic E. coli. J Clin Invest. 2007;117:428–437. doi: 10.1172/JCI29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman JA, Samji FN, Li Y, Deng W, Lin A, Finlay BB. Aquaporins contribute to diarrhoea caused by attaching and effacing bacterial pathogens. Cell Microbiol. 2007;9:131–141. doi: 10.1111/j.1462-5822.2006.00773.x. [DOI] [PubMed] [Google Scholar]
- Henry T, Couillault C, Rockenfeller P, Boucrot E, Dumont A, Schroeder N, Hermant A, Knodler LA, Lecine P, Steele-Mortimer O, Borg JP, Gorvel JP, Meresse S. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc Natl Acad Sci U S A. 2006;103:13497–13502. doi: 10.1073/pnas.0605443103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges K, Alto NM, Ramaswamy K, Dudeja PK, Hecht G. The enteropathogenic Escherichia coli effector protein EspF decreases sodium hydrogen exchanger 3 activity. Cell Microbiol. 2008;10:1735–1745. doi: 10.1111/j.1462-5822.2008.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstman AL, Kuehn MJ. Enterotoxigenic Escherichia coli secretes active heat-labile enterotoxin via outer membrane vesicles. J Biol Chem. 2000;275:12489–12496. doi: 10.1074/jbc.275.17.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DT, Clarke MB, Yamamoto K, Rasko DA, Sperandio V. The QseC adrenergicsignalling cascade in enterohemorrhagic E. coli (EHEC) PLoS Pathog. 2009;5:e1000553. doi: 10.1371/journal.ppat.1000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, Rohde M, Pirch T, Jung K, Retta SF, Terradot L, Fischer W, Haas R. Helicobacter pylori type IV secretion apparatus exploits β1 integrin in a novel RGD-independent manner. PLoS Pathog. 2009;5:e1000684. doi: 10.1371/journal.ppat.1000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BD, Ghori N, Falkow S. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer's patches. J Exp Med. 1994;180:15–23. doi: 10.1084/jem.180.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesty NC, Mason KM, Reedy M, Miller SE, Kuehn MJ. Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J. 2004;23:4538–4549. doi: 10.1038/sj.emboj.7600471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhle V, Abrahams GL, Hensel M. Intracellular Salmonella enterica redirect exocytic transport processes in a Salmonella pathogenicity island 2-dependent manner. Traffic. 2006;7:716–730. doi: 10.1111/j.1600-0854.2006.00422.x. [DOI] [PubMed] [Google Scholar]
- Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, Konig W, Backert S. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449:862–866. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- Lamb A, Yang XD, Tsang YH, Li JD, Higashi H, Hatakeyama M, Peek RM, Blanke SR, Chen LF. Helicobacter pylori CagA activates NF-κB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009;10:1242–1249. doi: 10.1038/embor.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Murata-Kamiya N, Saito Y, Hatakeyama M. Role of partitioning-defective 1/microtubule affinity-regulating kinases in the morphogenetic activity of Helicobacter pylori CagA. J Biol Chem. 2009;284:23024–23036. doi: 10.1074/jbc.M109.001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma AT, Mekalanos JJ. In vivo actin cross-linking induced by Vibrio cholerae type VI secretion system is associated with intestinal inflammation. Proc Natl Acad Sci U S A. 2010;107:4365–4370. doi: 10.1073/pnas.0915156107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara BP, Koutsouris A, O’Connell CB, Nougayrede JP, Donnenberg MS, Hecht G. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J Clin Invest. 2001;107:621–629. doi: 10.1172/JCI11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesic D, Miller MC, Quinkert ZT, Stein M, Chait BT, Stebbins CE. Helicobacter pylori CagA inhibits PAR1-MARK family kinases by mimicking host substrates. Nat Struct Mol Biol. 2010;17:130–132. doi: 10.1038/nsmb.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker H, Chitcholtan K, Hampton MB, Keenan JI. Uptake of Helicobacter pylori outer membrane vesicles by gastric epithelial cells. Infect Immun. 2010;78:5054–5061. doi: 10.1128/IAI.00299-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci U S A. 2007;104:15508–15513. doi: 10.1073/pnas.0706532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A. 2006;103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajashekar R, Hensel M. Dynamic modification of microtubule-dependent transport by effector proteins of intracellular Salmonella enterica. Eur J Cell Biol. 2011 doi: 10.1016/j.ejcb.2011.05.008. (in press) [DOI] [PubMed] [Google Scholar]
- Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD. Type VI secretion delivers bacteriolytic effectors to target cells. Nature. 2011;475:343–347. doi: 10.1038/nature10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci U S A. 1999;96:14559–14564. doi: 10.1073/pnas.96.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Paul FE, Brandt S, Guye P, Daumke O, Backert S, Dehio C, Mann M. Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe. 2009;5:397–403. doi: 10.1016/j.chom.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Shifflett DE, Clayburgh DR, Koutsouris A, Turner JR, Hecht GA. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab Invest. 2005;85:1308–1324. doi: 10.1038/labinvest.3700330. [DOI] [PubMed] [Google Scholar]
- Tomson FL, Viswanathan VK, Kanack KJ, Kanteti RP, Straub KV, Menet M, Kaper JB, Hecht G. Enteropathogenic Escherichia coli EspG disrupts microtubules and in conjunction with Orf3 enhances perturbation of the tight junction barrier. Mol Microbiol. 2005;56:447–464. doi: 10.1111/j.1365-2958.2005.04571.x. [DOI] [PubMed] [Google Scholar]
- Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingadassalom D, Kazlauskas A, Skehan B, Cheng HC, Magoun L, Robbins D, Rosen MK, Saksela K, Leong JM. Insulin receptor tyrosine kinase substrate links the E. coli O157:H7 actin assembly effectors Tir and EspF(U) during pedestal formation. Proc Natl Acad Sci U S A. 2009;106:6754–6759. doi: 10.1073/pnas.0809131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeaiter Z, Cohen D, Musch A, Bagnoli F, Covacci A, Stein M. Analysis of detergent-resistant membranes of Helicobacter pylori infected gastric adenocarcinoma cells reveals a role for MARK2/Par1b in CagA-mediated disruption of cellular polarity. Cell Microbiol. 2008;10:781–794. doi: 10.1111/j.1462-5822.2007.01084.x. [DOI] [PubMed] [Google Scholar]