

Abstract
Abstract
The gut epithelium is a barrier between the ‘outside’ and ‘inside’ world. The major function of the epithelium is to absorb nutrients, ions and water, yet it must balance these functions with that of protecting the ‘inside’ world from potentially harmful toxins, irritants, bacteria and other pathogens that also exist in the gut lumen. The health of an individual depends upon the efficient digestion and absorption of all required nutrients from the diet. This requires sensing of meal components by gut enteroendocrine cells, activation of neural and humoral pathways to regulate gastrointestinal motor, secretory and absorptive functions, and also to regulate food intake and plasma levels of glucose. In this way, there is a balance between the delivery of food and the digestive and absorptive capacity of the intestine. Maintenance of the mucosal barrier likewise requires sensory detection of pathogens, toxins and irritants; breakdown of the epithelial barrier is associated with gut inflammation and may ultimately lead to inflammatory bowel disease. However, disruption of the barrier alone is not sufficient to cause frank inflammatory bowel disease. Several recent studies have provided compelling new evidence to suggest that changes in epithelial barrier function and inflammation are associated with and may even lead to altered regulation of body weight and glucose homeostasis. This article provides a brief review of some recent evidence to support the hypothesis that changes in the gut microbiota and alteration of gut epithelial function will perturb the homeostatic humoral and neural pathways controlling food intake and body weight.
Dr Raybould obtained her PhD from the University of Liverpool, UK with Professor Graham Dockray. She completed a postdoctoral fellowship at the UCLA School of Medicine, Los Angeles and continued working there on the gut–brain axis and the regulation of postprandial gastrointestinal function. She moved to School of Veterinary Medicine in 2000, where she is Professor and Chair of the Department of Anatomy, Physiology and Cell Biology. Her research interests are in the role of gut endocrine cells and vagal afferent neurons in the regulation of gastrointestinal function and food intake in health and metabolic disease.
 |
The gut epithelium is a barrier between the ‘outside’ and ‘inside’ world. The major function of the epithelium is to absorb nutrients, ions and water, yet it must balance these functions with that of protecting the ‘inside’ world from potentially harmful toxins, irritants, bacteria and other pathogens that also exist in the gut lumen. The health of an individual depends upon the efficient digestion and absorption of all required nutrients from the diet. This requires sensing of meal components by gut enteroendocrine cells, activation of neural and humoral pathways to regulate gastrointestinal (GI) motor, secretory and absorptive functions, and also to regulate food intake and plasma levels of glucose. In this way, there is a balance between the delivery of food and the digestive and absorptive capacity of the intestine. Maintenance of the mucosal barrier likewise requires sensory detection of pathogens, toxins and irritants; breakdown of the epithelial barrier is associated with gut inflammation and may ultimately lead to inflammatory bowel disease. However, disruption of the barrier alone is not sufficient to cause frank inflammatory bowel disease. Several recent studies have provided compelling new evidence to suggest that changes in epithelial barrier function and inflammation are associated with and may even lead to altered regulation of body weight and glucose homeostasis. This article provides a brief review of some recent evidence to support the hypothesis that changes in the gut microbiota and alteration of gut epithelial function will perturb the homeostatic humoral and neural pathways controlling food intake and body weight.
Altered gut microbiota in obesity
Evidence for an alteration in gut microflora in obesity was suggested from studies in genetic models in rodents. Using 16S ribosomal RNA sequencing of the caecal microflora, significant changes in the abundance of two of the major bacterial phyla were observed in leptin-deficient (ob/ob) obese mice compared with lean (ob/+) littermates, with a significant reduction in Bacteriodetes and an increase in Firmicutes (Ley et al. 2005)). Similar findings of an increase in the relative abundance of Firmicutes were also reported in a population of obese humans (Ley et al. 2006)). Data from studies using long-term ingestion of diets high in fat reveal similar changes in the microbiota, with a decrease in overall bacterial abundance and an increase in the ratio of Firmicutes to Bacteriodetes species (Turnbaugh et al. 2008)). Germ-free mice are resistant to high-fat (HF)-diet-induced obesity, and administration of the microbiota from either lean or obese mice to germ-free mice recapitulates the original phenotype (Turnbaugh et al. 2006)). These and other data (see Tilg et al. 2009; Turnbaugh & Gordon, 2009; Ley, 2010)) suggest an association between the gut microbiome and body weight regulation. Data from initial studies suggested that the change in the gut microbiota may influence body weight regulation by increasing nutrient extraction from the diet and by alteration of fat deposition (Turnbaugh et al. 2006, 2008; Backhed et al. 2007)); however, evidence is beginning to emerge that other mechanisms may be involved.
Studies from the group of Cani et al. demonstrated in mice that ingestion of a very HF diet induced changes in the gut microbiota, the expected increase in weight gain, adiposity and other symptoms of the metabolic syndrome. Importantly, these changes were accompanied by an increase in circulating levels of the bacterial product, lipopolysaccharide (LPS; Cani et al. 2007)). In mice fed a low-fat diet, plasma levels of LPS increase during the dark phase, in association with feeding. In contrast, in HF-fed mice, there was no diurnal variation, and LPS remained high throughout the dark and light phases. The levels of LPS were 10–15 times lower than those measured in models of sepsis. This group coined the term ‘metabolic endotoxaemia’ to describe this low, but chronic increase in circulating LPS to distinguish it from high, rapid-onset increases in LPS seen in acute infections and sepsis. Implantation of osmotic minipumps to deliver LPS chronically over 4 weeks induced glucose intolerance and insulinaemia, together with an increase in body weight and adiposity. In mice lacking CD14, the adapter protein for Toll-like receptor 4 (TLR-4; the pattern recognition receptor for LPS), long-term ingestion of a HF diet or chronic administration of LPS had no effect on any parameters, suggesting a role for the TLR-4 receptor in mediating effects of metabolic endotoxaemia on body weight regulation and glucose tolerance (Cani et al. 2007)). Toll-like receptor 4 null mice are resistant to HF-diet-induced obesity (Davis et al. 2008)), lending further support to the concept that products from the gut microbiota play a pivotal role in regulation of body weight. It is well established that obesity is an ‘inflammatory disease’, with marked increases in inflammatory cytokines and other mediators in adipose tissue, liver, muscle and plasma (Hotamisligil, 2006)). Despite the gut being the first place to see changes in the gut microbiota or bacterial breakdown products and toxins, the role of inflammation in the gut epithelium in driving the changes in body weight and adiposity has, until recently, been unexplored.
Gut inflammation and changes in barrier function in obesity
One of the first studies to investigate the possible role of GI inflammation compared the inflammatory markers in the gut induced by HF feeding to that in a model of colitis in mice (Li et al. 2008)). The major change in inflammatory markers in mesenteric fat and liver was an increase in expression of tumour necrosis factor α (TNFα), but in the proximal colon there was a significant increase in interleukin-1β. Other studies have confirmed inflammation in the intestine of HF-fed mice or in genetic models of obesity (Duparc et al. 2011)).
In a convincing demonstration of a possible causative role for gut inflammation in the deleterious systemic effects of a HF diet, Ding et al. (2010) used transgenic mice with the nuclear factor κB (NF-κB) response element–enhanced green fluorescent protein (EGFP) reporter transgene (NF-κBEGFP) to determine the presence and cellular sites of inflammation. In conventionally raised but not in germ-free mice, HF diet increased body weight and adiposity, and this was accompanied by an increase in mRNA for TNFα in the ileum. Importantly, the increase in ileal TNFα occurred early (within 2–4 weeks of initiation of high-fat feeding), preceded weight gain and showed strong correlations with increased body weight, adiposity and increase in plasma glucose and insulin. Nuclear factor κB was activated in epithelial, immune and endothelial cells in the small intestine of conventionally reared HF-fed mice. These data clearly demonstrate that ingestion of a HF diet induces inflammation in the small intestine, and these changes precede obesity. Thus, intestinal inflammation is an early consequence of ingestion of a HF diet and may induce obesity via elevated plasma levels of LPS or some other as yet unidentified mechanism.
These data fit well with those generated in our laboratory using Sprague–Dawley rats, a non-congenic strain of rats, in which littermates can be either prone or resistant to HF-diet-induced obesity. High-fat feeding was associated with changes in the gut microbiota, but gut inflammation occurred only in obese rats, again suggesting a causative role for gut inflammation in the onset of obesity (de La Serre et al. 2010)). We also found changes in the cellular localization of occludin in epithelial cells in the ileum and an increase in phosphorylation of myosin light chain, both of which are associated with compromised epithelial barrier function. In an interesting model of obesity in rats, in which there is a spontaneous mutation in the gene encoding the cholecystokinin type 1 receptor (CCK1R), there is a separation between the effects of obesity alone (due to hyperphagia because of the lack of the CCK1R) and obesity associated with ingestion of a HF diet (Suzuki & Hara, 2010)). Importantly, there were no differences in intestinal permeability in CCK1R-deficient obese rats versus normal (lean) rats when ingesting laboratory chow; however, intestinal permeability increased in both obese and lean rats when ingesting HF diets. Marked changes in expression of tight junction proteins in the small intestine, but not colon or caecum, were also induced by HF feeding. An increase in intestinal permeability and altered expression of tight junction proteins (ZO-1) has also been reported in response to HF feeding in mice (Cani et al. 2008)) and humans (Osbak et al. 2011)). Taken together, the data from these studies suggest that HF feeding can alter epithelial barrier function and increase intestinal permeability (Fig. 1)). However, caution has to be exercised because similar changes have also been reported in genetic models of obesity. In db/db and ob/ob mice, an decrease in transepithelial resistance, increase flux of horseradish peroxide (HRP) and a decrease in expression of occludin and ZO-1 at tight junctions and distribution of proteins away from apical membrane (Brun et al. 2007)) was observed, suggesting that this can also be secondary to the metabolic effects of an obese state.
Figure 1. A proposed model by which ingestion of a high-fat diet will lead to hyperphagia and obesity.
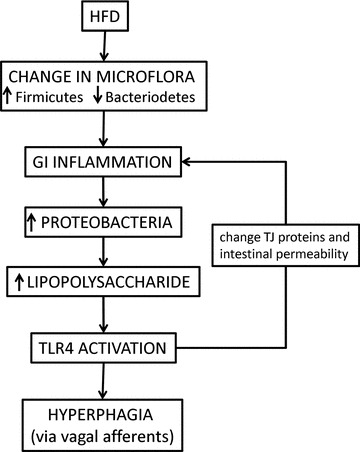
Ingestion of a high-fat diet (HFD) leads to changes in the gut microbiota. In susceptible individuals, the gut may become inflamed, resulting in changes in tight junction (TJ) proteins and an increase in intestinal permeability. The presence of inflammation allows for the increase in Gram-negative Proteobacteria, thus increasing the amount of lipopolysaccaride in the gut lumen. The increased gut permeability allows for an increase in passage of lipopolysaccaride from the gut lumen to the gut interstitium, where it can activate Toll-like receptor 4 (TLR4) on a variety of target tissues, including those localized to vagal afferent nerve terminals. Abbreviation: GI, gastrointestinal.
Sensors within the epithelium
Why would an increase in gut inflammation, leakiness and thus an increase in levels of LPS and possibly other bacterial products and toxins in the lamina propria and plasma induce obesity? Certainly, there is evidence that these bacterial products can produce inflammation in other tissues that are altered in obesity, such as adipose tissue, muscle, pancreas and liver. However, there may be significant effects much closer to home, within the gut epithelium, particularly on cells and pathways involved in the regulation of food intake.
Approximately 1% of gut epithelial cells are specialized enteroendocrine cells, which act as luminal sensors, capable of detecting ions, nutrients, bacterial products and other luminal contents. These cells are pluripotent, each synthesizing, storing and releasing several different polypeptide hormones and other bioactive molecules. Polypeptide gut hormones are important in many different functions both within the gut, via paracrine actions, and as true hormones acting at distant sites, such as the exocrine and endocrine pancreas and the central nervous system. Moreover, gut hormones can both stimulate and inhibit food intake, and thus play a critical role in matching the entry of nutrients (both in terms of emptying of contents from the stomach and also through alteration of ingestion) to the digestive and absorptive capacity of the small intestine. While the response of gut endocrine cells to nutrient stimuli is well established, evidence is accumulating to suggest that these gut sensors can also respond to other non-nutritive stimuli, such as artificial sweeteners, bitter-tasting compounds, toxins and bacterial products (Sternini et al. 2008; Raybould, 2010)).
Several recent publications have shown that enteroendocrine cells express TLRs, not necessarily a surprising finding given that they share a common lineage with other epithelial cells. In immunohistochemical studies to determine the expression of TLRs in human and murine intestinal tissue, Bogunovic et al. (2007) demonstrated that TLR-1, TLR-2 and TLR-4 were expressed on subpopulations of epithelial cells with enteroendocrine cell morphology located primarily in the crypt in both the colon and the small intestine. Several TLRs are expressed by STC-1 cells, a model of enteroendocrine cells (Bogunovic et al. 2007; Palazzo et al. 2007; Selleri et al. 2008)). Ligands for these receptors cause secretion of cholecystokinin (CCK) from these cells by a mechanism dependent on MyD88 and protein kinase C, intracellular signalling molecules known to be downstream of TLRs. In mice, administration of ligands for each TLR also increased serum levels of CCK; LPS was particularly potent in releasing CCK, an effect absent in TLR-4 null mice. Exposure to LPS was also found to induce both anti- and pro-inflammatory gene expression in STC-1 cells. Thus, it is possible that alteration in the gut microbiota induced by HF feeding produces a different complement of bacterial products that, via activation of TLRs on gut endocrine cells, may influence ingestive and GI function. Considering the important role of gut peptides to regulate gut function and food intake, this could be a key component of induction of hyperphagia in response to HF diets.
However, LPS (or other bacterial products) may have effects beyond the immediate epithelial cell, particularly if the epithelial barrier is leaky and these products accumulate in the lamina propria. It is worth noting that LPS and other inflammatory mediators have been shown to induce signalling events in hypothalamic neurons, rendering them less sensitive to the anorexigenic hormone, leptin (Araujo et al. 2010)). This leptin resistance is a hallmark of HF-diet-induced obesity in rodents and is thought to be a major factor in disordered food intake that leads to obesity. However, vagal afferent neurons that innervate the gut and transmit afferent information from the gut to the brain also express leptin receptors and TLR4 (Burdyga et al. 2002; Araujo et al. 2010)). Vagal afferent terminals lie in the lamina propria and could be exposed to relatively high levels of LPS crossing from the gut lumen.
The existence of intestinal feedback mechanisms that can detect the presence of nutrients in the gut lumen predicts that ingestion of food will activate enteroendocrine cells and the vagal afferent pathway to limit further food intake and thus regulate the amount of food ingested. For example, lipid will release CCK, which stimulates the CCK1R on vagal afferent terminals in the lamina propria, resulting in reflex activation of parasympathetic efferent outflow to regulate gut and pancreatic exocrine function and also acts to stop eating (Raybould, 2007)). In response to ingestion of a HF diet, rodents will initially regulate their food intake to compensate for the intake of higher caloric food, the response predicted for the maintenance of homeostatic control of body weight. However, after a period of time (that can vary dependent on the exact experimental protocol), animals will overeat (i.e. become hyperphagic) and no longer regulate the amount of food consumed. In models of HF-diet-induced obesity, hyperphagia leads to an increase in body weight, adiposity and the spectrum of downstream effects characteristic of obesity (Covasa, 2010)). This hyperphagia is associated with a reduction in the ability of nutrients and gut hormones, such as CCK, to activate vagal afferent neurons (Covasa, 2010)). Vagal afferent neurons express TLR4 (Hosoi et al. 2005; de Lartigue et al. 2011)). We have recently demonstrated that LPS induces a change in the phenotype of vagal afferent neurons in short-term culture, rendering them insensitive to leptin (de Lartigue et al. 2011). Moreover, vagal afferent neurons from rats fed HF diets were also leptin resistant. It is well established that leptin and CCK interact at the level of vagal afferent neurons, and thus an inability of these neurons to response to leptin results in a decrease in sensitivity to physiological concentrations of CCK, exactly as described in HF-diet-fed rats (Covasa 2010)). This leptin resistance in vagal afferent neurons occurs prior to that in hypothalamic neurons and may therefore lead to hyperphagia and altered regulation of food intake and body weight (Fig. 2)).
Figure 2. A proposed model by which an increase in lipopolysaccharide (LPS) induces hyperphagia.
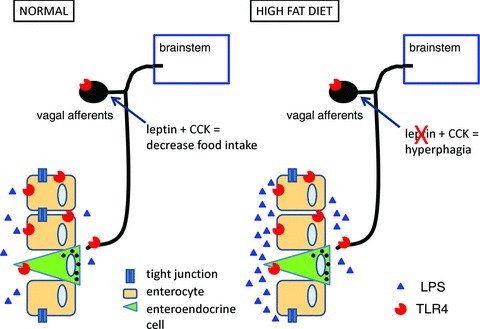
In normal conditions, ingestion of food regulates intake via intestinal feedback mechanisms involving the action of cholecystokinin (CCK) and leptin on vagal afferent neurons. Following chronic ingestion of a high-fat diet, the increase in LPS activates Toll-like receptor 4 (TLR4) on vagal afferent neurons, rendering them leptin resistant and thus unable to respond to CCK, which results in hyperphagia and eventual gain in body weight.
In summary, the presence of intestinal inflammation, resulting in an increase in intestinal permeability to LPS (or other bacterial products), may lead to alteration in the function of nerve terminals in the lamina propria, resulting in disrupted intestinal feedback, hyperphagia and obesity.
Acknowledgments
Work by the author was funded by National Institute for Health, National Institute for Diabetes and digestive and Kidney diseases, National Center for complimentry and alternative medicine and Dairy Research Initiative. The author would like to thank the many graduate students, postdoctoral fellows and faculty colleagues who have contributed to the work, in particular Will de Lartigue and Claire de La Serre.
References
- Araujo EP, Torsoni MA, Velloso LA. Hypothalamic inflammation and obesity. Vitam Horm. 2010;82:129–143. doi: 10.1016/S0083-6729(10)82007-2. [DOI] [PubMed] [Google Scholar]
- Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104:979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic M, Dave SH, Tilstra JS, Chang DT, Harpaz N, Xiong H, Mayer LF, Plevy SE. Enteroendocrine cells express functional Toll-like receptors. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1770–G1783. doi: 10.1152/ajpgi.00249.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palu G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G518–G525. doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Spiller D, Morris R, Lal S, Thompson DG, Saeed S, Dimaline R, Varro A, Dockray GJ. Expression of the leptin receptor in rat and human nodose ganglion neurones. Neuroscience. 2002;109:339–347. doi: 10.1016/s0306-4522(01)00474-2. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- Covasa M. Deficits in gastrointestinal responses controlling food intake and body weight. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1423–R1439. doi: 10.1152/ajpregu.00126.2010. [DOI] [PubMed] [Google Scholar]
- Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring) 2008;16:1248–1255. doi: 10.1038/oby.2008.210. [DOI] [PubMed] [Google Scholar]
- de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Diet-induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab. 2011;301:E187–E195. doi: 10.1152/ajpendo.00056.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol. 2010;299:G440–G448. doi: 10.1152/ajpgi.00098.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, Jobin C, Lund PK. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One. 2010;5:e12191. doi: 10.1371/journal.pone.0012191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duparc T, Naslain D, Colom A, Muccioli GG, Massaly N, Delzenne NM, Valet P, Cani PD, Knauf C. Jejunum inflammation in obese and diabetic mice impairs enteric glucose detection and modifies nitric oxide release in the hypothalamus. Antioxid Redox Signal. 2011;14:415–423. doi: 10.1089/ars.2010.3330. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Okuma Y, Matsuda T, Nomura Y. Novel pathway for LPS-induced afferent vagus nerve activation: possible role of nodose ganglion. Auton Neurosci. 2005;120:104–107. doi: 10.1016/j.autneu.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Ley RE. Obesity and the human microbiome. Curr Opin Gastroenterol. 2010;26:5–11. doi: 10.1097/MOG.0b013e328333d751. [DOI] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Li H, Lelliott C, Hakansson P, Ploj K, Tuneld A, Verolin-Johansson M, Benthem L, Carlsson B, Storlien L, Michaelsson E. Intestinal, adipose, and liver inflammation in diet-induced obese mice. Metabolism. 2008;57:1704–1710. doi: 10.1016/j.metabol.2008.07.029. [DOI] [PubMed] [Google Scholar]
- Osbak PS, Bindslev N, Hansen MB. Relationships between body mass index and short-circuit current in human duodenal and colonic mucosal biopsies. Acta Physiol (Oxf) 2011;201:47–53. doi: 10.1111/j.1748-1716.2010.02202.x. [DOI] [PubMed] [Google Scholar]
- Palazzo M, Balsari A, Rossini A, Selleri S, Calcaterra C, Gariboldi S, Zanobbio L, Arnaboldi F, Shirai YF, Serrao G, Rumio C. Activation of enteroendocrine cells via TLRs induces hormone, chemokine, and defensin secretion. J Immunol. 2007;178:4296–4303. doi: 10.4049/jimmunol.178.7.4296. [DOI] [PubMed] [Google Scholar]
- Raybould HE. Mechanisms of CCK signaling from gut to brain. Curr Opin Pharmacol. 2007;7:570–574. doi: 10.1016/j.coph.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould HE. Gut chemosensing: interactions between gut endocrine cells and visceral afferents. Auton Neurosci. 2010;153:41–46. doi: 10.1016/j.autneu.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selleri S, Palazzo M, Deola S, Wang E, Balsari A, Marincola FM, Rumio C. Induction of pro-inflammatory programs in enteroendocrine cells by the Toll-like receptor agonists flagellin and bacterial LPS. Int Immunol. 2008;20:961–970. doi: 10.1093/intimm/dxn055. [DOI] [PubMed] [Google Scholar]
- Sternini C, Anselmi L, Rozengurt E. Enteroendocrine cells: a site of ‘taste’ in gastrointestinal chemosensing. Curr Opin Endocrinol Diabetes Obes. 2008;15:73–78. doi: 10.1097/MED.0b013e3282f43a73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Hara H. Dietary fat and bile juice, but not obesity, are responsible for the increase in small intestinal permeability induced through the suppression of tight junction protein expression in LETO and OLETF rats. Nutr Metab (Lond) 2010;7:19. doi: 10.1186/1743-7075-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H, Moschen AR, Kaser A. Obesity and the microbiota. Gastroenterology. 2009;136:1476–1483. doi: 10.1053/j.gastro.2009.03.030. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]