

Abstract
Liver fibrosis occurs as a wound-healing scar response following chronic liver inflammation including alcoholic liver disease, non-alcoholic steatohepatitis, viral hepatitis, cholestatic liver disease and autoimmune liver diseases. The liver has a unique vascular system within the gastrointestinal tract, as the majority of the liver's blood supply comes from the intestine through the portal vein. When the intestinal barrier function is disrupted, an increase in intestinal permeability leads to the translocation of intestine-derived bacterial products such as lipopolysaccharide (LPS) and unmethylated CpG containing DNA to the liver via the portal vein. These gut-derived bacterial products stimulate innate immune receptors, namely Toll-like receptors (TLRs), in the liver. TLRs are expressed on Kupffer cells, endothelial cells, dendritic cells, biliary epithelial cells, hepatic stellate cells, and hepatocytes. TLRs activate these cells to contribute to acute and chronic liver diseases. This review summarizes recent studies investigating the role of TLRs, intestinal microbiota and bacterial translocation in liver fibrosis, alcoholic liver disease and non-alcoholic steatohepatitis.
Ekihiro Seki (left) is an MD and PhD, and was trained as a surgeon in Hyogo College of Medicine, Japan. His current position is an assistant professor in the Department of Medicine, University of California, San Diego. His current research interests are (i) regulation of innate immune system and inflammation in hepatic fibrosis and cancer, (ii) crosstalk between hepatic stellate cells and immune cells including Kupffer cells in liver diseases, and (iii) modulation of TGFβ signaling by Toll-like receptors in hepatic stellate cells and hepatocytes. Bernd Schnabl (right) is an Assistant Professor in Gastroenterology at UCSD School of Medicine. His main interest as a physician–scientist is the pathogenesis of chronic liver disease resulting in liver fibrosis and cirrhosis. In particular, he is studying the contribution of the intestinal microbiome and microbial products to liver disease.
 |
Introduction
Innate immunity exists widely from insects to mammals and is an evolutionary acquired function as the first line of host defence against pathogenic microorganisms (bacteria, viruses, fungi and parasites). The innate immune system induces the production of inflammatory mediators and anti-microbial peptides, and builds a bridge with the acquired immunity to eradicate invading microorganisms from the host. The innate immune signalling also maintains tissue and organ homeostasis, such as intestinal microflora, proliferation and apoptosis of intestinal epithelial cells, and liver regeneration after the loss of liver mass (Rakoff-Nahoum et al. 2004; Seki et al. 2005; Wen et al. 2008)). Notably, aberrant activation of innate immune signalling may trigger ‘harmful inflammation’ that contributes to sepsis, chronic inflammation, autoimmune diseases, tissue and organ injuries, fibrosis and carcinogenesis (Seki & Brenner, 2008)).
The liver is the first extraintestinal organ that encounters venous blood from the small and large intestines via the portal vein. Due to this unique blood supply system, the liver is vulnerable to exposure of bacterial products translocated from the gut lumen via the portal vein. Disruption of the intestinal epithelial barrier results in a leaky gut, which contributes to bacterial translocation (Seki & Brenner, 2008; Crispe, 2009; Pradere et al. 2010)). Translocation of large amounts of gut-derived products is usually prevented by intact barrier systems provided by intestinal epithelial cells (Crispe, 2009)). Thus, in a healthy organism only minor quantities of translocated bacterial products reach the liver. In general, the hepatic immune system tolerates these bacterial products in order to avoid harmful responses, which is also known as ‘liver tolerance’ (Crispe, 2009)). The liver not only consists of parenchymal hepatocytes, but also contains non-parenchymal cells including immune and non-immune cells. Members of the hepatic immune system are resident liver tissue macrophages (Kupffer cells), natural killer (NK) cells, NKT cells, T cells and B cells; these cell types strictly regulate the liver immune system including liver tolerance (Seki & Brenner, 2008)).
Bacterial translocation is defined as the migration of viable bacteria or bacterial products from the intestinal lumen to mesenteric lymph nodes or other extraintestinal organs and sites (Wiest & Garcia-Tsao, 2005)). Bacterial translocation is caused by increased intestinal permeability as a result of a disrupted intestinal barrier. Intestinal bacterial overgrowth and changes in the composition of bacterial flora in the intestine may also promote bacterial translocation (Berg & Garlington, 1979; Wiest & Garcia-Tsao, 2005; Son et al. 2010)). Increased translocation of bacteria and bacterial products from the intestine may impair liver homeostasis and enhance liver inflammation through activation of the innate immune system. In particular, translocated bacterial products augment the activation of hepatic immune cells through pattern recognition receptors including Toll-like receptors (TLRs). Moreover, hepatic non-immune cells, including endothelial cells, biliary epithelial cells and hepatic stellate cells, respond to bacterial products through TLRs (Seki & Brenner, 2008; Crispe, 2009)). Activated TLR signalling induces innate immune responses including cytokine and type I IFN production in the liver. Alarmins, the products released from damaged cells or tissues, also trigger TLR signalling and cause inflammation without actual infections, referred to sterile inflammation (Chen & Nunez, 2010)). Thus, the activation of TLR signalling through intestine-derived microbial products and alarmins may contribute to the initiation and progression of liver diseases (Seki & Brenner, 2008)). In this review, we highlight the current knowledge about bacterial translocation and its contribution to activation of TLR signalling in fibrogenic liver disease.
Toll-like receptor signalling in the liver
Toll-like receptors (TLRs) were originally identified as mammalian homologues of Drosophila Toll and function as pattern recognition receptors (Takeuchi & Akira, 2010; Yamamoto & Takeda, 2010)). TLRs recognize signature motifs, often referred as pathogen-associated molecular patterns (PAMPs), that activate downstream intracellular signalling pathways of TLRs, resulting in an induction of innate immune response. Currently, more than 10 members of the TLR family have been identified, and all TLRs contain a conserved extracellular domain with leucine rich repeats that is responsible for PAMP recognition (Takeuchi & Akira, 2010; Yamamoto & Takeda, 2010)). The Gram-negative bacterial cell wall component lipopolysaccharide (LPS, also known as endotoxin) binds to TLR4 with co-receptor CD14 and MD-2 (Takeuchi & Akira, 2010; Yamamoto & Takeda, 2010)). TLR2 heterodimerizes with TLR1 or TLR6 to recognize lipoprotein and peptidoglycan derived from Gram-positive bacteria. Bacterial flagellin is recognized by TLR5. Intracellular TLR3 and TLR9 are activated by microbe-derived nucleic acids including double stranded RNA and CpG motif containing unmethylated DNA, respectively (Takeuchi & Akira, 2010; Yamamoto & Takeda, 2010)).
After the binding of corresponding ligands, TLRs activate MyD88-dependent and -independent signalling pathways. The MyD88-dependent pathway is shared by TLR and IL-1 receptor signalling. Except for TLR3, all TLRs activate the MyD88-dependent pathway. An additional adaptor protein, TIRAP, bridges TLR2 and TLR4 to MyD88 (Fig. 1)). Subsequently, MyD88 recruits IL-1R-associated kinase (IRAK)-4, IRAK-1 and IRAK-2, and induces assembly of a multiple protein complex composed of TRAF6, TRAF3, Ubc13, cIAP1/2, TAK1 and NEMO (Takeuchi & Akira, 2010; Tseng et al. 2010; Yamamoto & Takeda, 2010)). Then, TRAF6 and TRAF3 are ubiquitinated and degraded, which leads to the activation of downstream IκB kinase (IKK) complex and MAP kinases. The IKK complex consists of IKKα, IKKβ and NEMO that induces phosphorylation, ubiquitination and degradation of IκBα. After the dissociation from IκBα, NF-κB translocates into the nucleus. MAP kinases including c-Jun N-terminal kinases (JNK) and p38 activate transcription factor AP-1. Activation of these transcription factors induces transcription of proinflammatory cytokines, such as TNF-α, IL-6 and IL-1β. The activation of endosomal TLR7 and TLR9 induces the assembly of the complex consisting of MyD88, IRAK-1, TRAF6, TRAF3 and IKK-α. This complex is required for TLR7- or TLR9-mediated activation of IKKs, MAP kinases and IRF7. IRF7 activation subsequently induces IFN-α production (Takeuchi & Akira, 2010; Yamamoto & Takeda, 2010)).
Figure 1. Overview of TLR signalling.
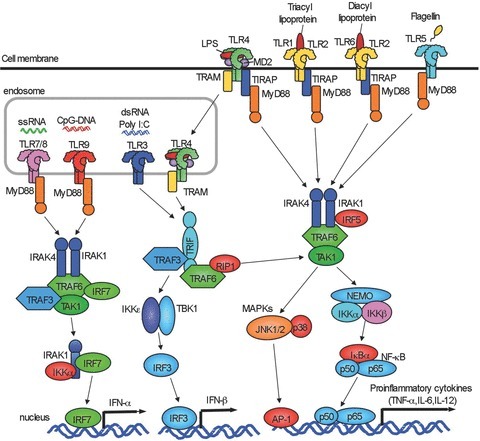
TLR1, TLR2, TLR4, TLR5 and TLR6 are expressed on cell membrane. TLR3, TLR7/8 and TLR9 are expressed in endosome. All TLRs except for TLR3 activate MyD88-dependent pathway to induce NF-κB and p38/JNK activation. TLR2 and TLR4 signalling require TIRAP and MyD88. TLR3 requires TRAF to activate TBK1/IKKɛ. After TLR4 internalization, TLR4 signalling activates TRAM/TRIF-dependent pathway. TLR3/4-dependent TRIF-dependent signalling induces IRF-3 activation and IFN-β production. TLR7/8 and TLR9 induce IFN-α production through IRF7.
TLR3 and TLR4 activate MyD88-independent, TRIF-dependent pathways. When TLR4 binds to another adaptor molecule, TRAM, TLR4 is internalized into cytoplasm from plasma membrane and then interacts with TRIF (Takeuchi & Akira, 2010; Yamamoto & Takeda, 2010)). Subsequently, TRIF associates with TRAF3 and TRAF6 to activate TANK-binding kinase 1 (TBK1) and IKKi, resulting in the activation of transcription factor IRF3 and induction of IFN-β (Takeuchi & Akira, 2010; Tseng et al. 2010; Yamamoto & Takeda, 2010)) (Fig. 1)). The TRIF-dependent pathway requires TRAF3 to induce IL-10 production and TRAF6 to induce late phase activation of NF-κB and MAPK through RIP1 and RIP3 (Takeuchi & Akira, 2010; Tseng et al. 2010; Yamamoto & Takeda, 2010)).
In liver inflammation, Kupffer cells are the primary cells that respond to TLR ligands to produce inflammatory cytokines (e.g. TNF-α, IL-6, IL-1β and IFN-β), chemokines (e.g. KC (CXCL1), MIP-2 (CXCL2), MCP-1 (CCL2), RANTES (CCL5), MIP-1α (CCL3) and MIP-1β (CCL4)) and reactive oxygen species (Seki & Brenner, 2008; Crispe, 2009)). In Kupffer cells, the TLR4–TRIF pathway activates caspase-1, which is required to process the proform of IL-1β and IL-18 into the active form (Seki et al. 2001; Imamura et al. 2009)). Kupffer cells express all TLRs except for TLR5 (Wu et al. 2010)). Hepatic stellate cells (HSCs) reside in the space of Disse and store vitamin A-containing lipid droplets in a healthy normal liver. Upon stimulation with fibrogenic mediators, such as TGF-β and PDGF produced from Kupffer cells, HSCs transdifferentiate into myofibroblasts with loss of vitamin A-containing lipid droplets and an increase in the expression of α-smooth muscle actin (SMA). As a result, hepatic myofibroblasts (activated HSCs) secrete extracellular matrix (ECM) to induce liver fibrosis (Bataller & Brenner, 2005; Friedman, 2008)). HSCs express all TLRs and have the capacity to produce inflammatory cytokines and chemokines, such as KC (CXCL1), MIP-1β (CCL4), MIP-1α (CCL3) MCP-1 (CCL2) and MIP-2 (CXCL2) in response to TLR ligands (Seki et al. 2007; Seki & Brenner, 2008; Wang et al. 2009)).
TLR4 signalling in liver fibrosis
Liver fibrosis is a wound healing response following chronic liver inflammation including chronic hepatitis B and C, non-alcoholic steatohepatitis, alcoholic hepatitis and autoimmune hepatitis. Cholestasis caused by biliary obstruction or inflammation also induces liver fibrosis (Bataller & Brenner, 2005; Friedman, 2008)). Liver cirrhosis, the end-stage of liver fibrosis, is histologically characterized by hepatocyte necrosis, inflammatory cell infiltration, bridging fibrosis and the appearance of regenerative nodules, and may result in portal hypertension, liver failure and hepatocellular carcinoma (Bataller & Brenner, 2005; Friedman, 2008)). Clinical evidence demonstrates elevated LPS levels in the systemic and portal circulation in patients with cirrhosis (Lin et al. 1995; Chan et al. 1997)). Additional evidence comes from a large patient cohort demonstrating that the TLR4 single nucleotide polymorphism (SNP) is one of seven SNPs that may predict the risk of liver cirrhosis in patients with chronic hepatitis C infection (Huang et al. 2007)). Therefore, it is postulated that TLR4 and gut microflora-derived LPS contribute to the progression of liver fibrosis. Indeed, systemic plasma levels of LPS are elevated in experimental animal models of liver fibrosis induced by bile duct ligation (BDL) and chronic administration of carbon tetrachloride (CCl4), or thioacetamide (Nolan & Leibowitz, 1978; Grinko et al. 1995; Seki et al. 2007)). Selective decontamination of gut microflora using a cocktail of non-absorbable broad-spectrum antibiotics decreases plasma LPS levels and inhibits experimental liver fibrosis (Rakoff-Nahoum et al. 2004; Seki et al. 2007)). It is suggested that intestine-derived translocated bacterial products such as LPS promote experimental liver fibrosis. Expectedly, experimental liver fibrosis was suppressed in TLR4-mutant C3H/HeJ mice (Seki et al. 2007)). Mice deficient in TLR4 co-receptors, CD14 and LPS-binding protein, and TLR adaptors, MyD88 and TRIF, also have less liver fibrosis mediated by cholestasis (Isayama et al. 2006; Seki et al. 2007)). Importantly, TLR4-mutant mice have similar levels of elevated LPS in the blood compared to wild-type mice (Seki et al. 2007)). This suggests that translocated LPS derived from the gut microflora mediates TLR4 activation in the liver; however, this translocation is independent of intestinal TLR4. The exact mechanism of how experimental liver injury induces a leaky gut barrier is still elusive. The intestinal barrier defect might be caused by disruption of intestinal epithelial tight junctions, imbalance of proliferation and apoptosis of intestinal epithelial cells, intestinal mucosal atrophy and edema associated with portal hypertension or absence of bile acids in intestinal lumen, and systemic increases in inflammatory cytokines and oxidative stress produced from the liver (Assimakopoulos et al. 2007)).
Relative roles of TLR4 signalling between Kupffer cells and hepatic stellate cells
Both Kupffer cells and HSCs express high levels of TLR4 (Seki et al. 2001, 2007; Paik et al. 2003; Seki & Brenner, 2008)). The relative roles of TLR4 between Kupffer cells and HSCs in liver fibrosis were investigated via generation of TLR4-bone marrow (BM) chimeric mice (Seki et al. 2007)). Since the majority of Kupffer cells are radio-resistant, (Kennedy & Abkowitz, 1997; Klein et al. 2007)) a standard style of BM transplantation (BMT) with irradiation is insufficient to engraft donor BM-derived cells as liver macrophages. The modified BMT with depletion of Kupffer cells can replace endogenous Kupffer cells with donor BM derived cells. This protocol can generate two different types of TLR4 BM chimeric mice; one group contains TLR4 mutant Kupffer cells and TLR4 intact HSCs and hepatocytes (Seki et al. 2007)), while the other type contains TLR4 intact Kupffer cells and TLR4 mutant HSCs and hepatocytes. Because Kupffer cells and HSCs, but not hepatocytes, directly respond to TLR4 ligand in vivo (Seki et al. 2001, 2007; Paik et al. 2003; Isogawa et al. 2005)) and because HSCs are radio-resistant and not derived from BM (Kisseleva et al. 2006; Higashiyama et al. 2009)), the relative roles of TLR4 in Kupffer cells and HSCs were characterized by using the modified TLR4 BM-chimeric mice. While mice with TLR4 mutant HSCs and TLR4 intact Kupffer cells lacked induction of significant liver fibrosis, mice with TLR4 intact HSCs and TLR4 mutant Kupffer cells showed a marked induction of fibrosis in the TLR4 chimeric mice (Seki et al. 2007)). Thus, TLR4 signalling in HSCs, but not in Kupffer cells, is crucial for the development of liver fibrosis. Because TLR4-mediated inflammatory and fibrogenic cytokines of Kupffer cells were suggested to be more important than TLR4-mediated HSC activation in liver fibrosis, the results from this study were unexpected and uncovered the new role of TLR4 signalling in HSCs (Seki et al. 2007)).
Currently, at least three mechanisms are identified as roles of TLR4 signalling in HSCs during liver fibrogenesis. The first mechanism is mediated by TLR4-induced chemokines (MCP-1, MIP-1α, MIP-1β and RANTES) and expression of adhesion molecules (ICAM-1, VCAM-1 and E-selectin) (Seki et al. 2007)). These HSC-derived factors lead to chemoattraction of BM-derived monocytes and the accumulation of Kupffer cells in the liver (Seki et al. 2009a,b)). Moreover, HSC-derived MCP-1 and RANTES act in an autocrine manner to activate HSCs (Marra et al. 1999; Marra, 2002; Schwabe et al. 2003; Seki et al. 2009b)). This is also supported by evidence demonstrating that genetic or pharmacological inhibition of chemokines (RANTES, MCP-1) or chemokine receptors (CCR1, CCR2, CCR5) reduces liver fibrosis (Fig. 2)) (Marra et al. 1999; Marra, 2002; Schwabe et al. 2003; Karlmark et al. 2009; Seki et al. 2009a,b; Berres et al. 2010; Baeck et al. 2011)). The second mechanism is mediated by the crosstalk between TLR4 and TGF-β signalling. TGF-β is a potent fibrogenic cytokine that activates HSCs and induces liver fibrosis. In quiescent HSCs, Bambi, an endogenous decoy receptor of TGF-β receptor, is highly expressed and inhibits TGF-β receptor signalling (Seki et al. 2007)). Upon TLR4 stimulation, Bambi is immediately downregulated, which allows full activation of TGF-β receptor signalling in HSCs, leading to HSC activation (Fig. 2)). TLR4-mediated Bambi regulation is dependent on MyD88 and NF-κB, but not TRIF (Seki et al. 2007)). In addition, Bambi not only functions as decoy, but also directly interacts with Smad7 to interfere with the association between TGF-β receptors (type I and type II) and Smad3, resulting in inhibition of TGF-β signalling (Yan et al. 2009)). A recent paper demonstrated that in renal fibrosis, Bambi regulates TLR4-mediated fibrogenesis, indicating the universal role of Bambi in fibrosis (Pulskens et al. 2011)). The third mechanism is mediated by TLR4-regulated microRNA (miR) expression in liver fibrosis. LPS stimulation reduces the expression of miR-29 expression in HSCs. Moreover, miR-29 expression is suppressed in the liver of humans and animals with liver fibrosis (Roderburg et al. 2011)). miR-29 negatively regulates transcription of collagen α1 (I) mRNA, suggesting that TLR4 signalling downregulates miR-29 expression, which in turn promotes collagen production in liver fibrosis (Roderburg et al. 2011)).
Figure 2. TLR4 signalling in hepatic stellate cells during liver fibrosis.
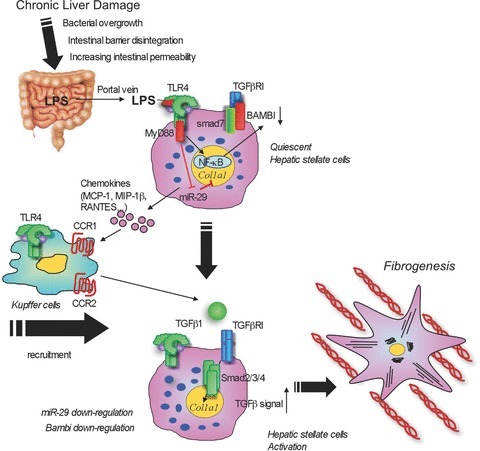
In chronic liver damage, intestinal permeability is increased due to systemic inflammation, portal hypertension, intestinal dysbiosis or tight junction disintegrity, which allows translocation of gut microflora-derived LPS into the liver through the portal vein. Translocated LPS stimulates TLR4 on hepatic stellate cells (HSCs). High expression of Bambi prevents TGF-β signalling in quiescent HSCs. Upon activation of TLR4, HSCs produce chemokines (MCP-1, MIP-1β and RANTES) that recruit Kupffer cells through CCR1 and CCR2. TLR4-activated HSCs downregulate Bambi and increase its sensitivity to TGF-β released from Kupffer cells. The fully activated TGF-β signalling then induces HSC activation. TLR4 signalling-mediated Bambi downregulation requires MyD88 and NF-κB.
Bacterial translocation and liver cirrhosis
Bacterial translocation results from increased intestinal permeability, which might be facilitated by a disruption of intestinal tight junctions and by intestinal bacterial overgrowth. Biliary obstruction or cirrhosis-mediated liver dysfunction may decrease the secretion of bile acids that causes bacterial overgrowth and may change bacterial composition in intestine (Miettinen, 1972; Sung et al. 1993)). Moreover, liver cirrhosis and portal hypertension may influence intestinal motility that might also contribute to intestinal bacterial overgrowth (Gunnarsdottir et al. 2003)).
Currently, it is not entirely clear how changes in the microbiome contribute to chronic liver disease. The analysis of fecal microbiome in patients with hepatitis B and alcoholic liver cirrhosis demonstrated an increase in pathogenic Enterobacteriaceae and Streptococcaceae, while beneficial Bifidobacteria and Lachnospiraceae were decreased (Chen et al. 2011; Lu et al. 2011)). In addition, liver cirrhosis induced by carbon tetrachloride in rats was also associated with high levels of Enterobacteraceae (Zhang et al. 2010)). Another study reported a decrease in Gram-positive anaerobic Clostridium groups and an increase in the aerobic/anaerobic bacterial ratio in mice with fibrosis (Gomez-Hurtado et al. 2011)). For animals with cirrhosis, treatment with antibiotics (e.g. norfloxacin) or probiotics (e.g. Bifidobacterium) reduces Enterobacter, while it increases Bifidobacterium and Lactobacillus, resulting in decreased systemic endotoxin levels and improved liver function (Zhang et al. 2010)). Thus, it is suggested that bacterial translocation and intestinal flora dysfunction are associated with the development of liver fibrosis.
TLR4 signalling, bacterial translocation and alcoholic liver disease
Chronic alcohol abuse causes hepatic steatosis, alcoholic hepatitis and cirrhosis. Not only alcohol itself, but also its metabolite, acetaldehyde, is a potent hepatotoxin (Petrasek et al. 2010; Szabo & Bala, 2010)). LPS derived from gut microflora is also considered to be a crucial factor inducing alcohol-mediated pathological changes in the liver (Rao, 2009; Szabo & Bala, 2010)). Indeed, LPS levels in systemic and portal blood are significantly increased in patients and animals with chronic alcohol intake (Bode et al. 1987; Fukui et al. 1991; Uesugi et al. 2001; Yan et al. 2011)). Mice deficient in TLR4, LBP and CD14 are shown to be resistant to alcohol-induced liver diseases (Uesugi et al. 2001, 2002; Yin et al. 2001)). Moreover, gut sterilization with antibiotics decreased plasma LPS levels and liver steatosis, inflammation and injury after chronic ethanol treatment (Adachi et al. 1995)). Most importantly, plasma LPS levels are similarly increased in wild-type and TLR4 mutant mice (Uesugi et al. 2001)). Together, it is evident that translocation of LPS in alcoholic liver disease activates TLR4 in the liver, whereas bacterial translocation does not require TLR4 in the intestine. Despite the failure of LPS alone to mimic the condition of alcoholic steatohepatitis, ethanol administration increases the sensitivity to LPS-induced hepatocyte injury and cytokine production in the animal model (Pennington et al. 1997; von Montfort et al. 2008)). As noted above, HSCs, but not Kupffer cells, are responsible for TLR4-mediated liver fibrosis. Previous studies emphasized the pathophysiological importance of TLR4 on Kupffer cells in alcoholic liver disease, but they did not investigate the role of TLR4 on HSCs (Adachi et al. 1994; Uesugi et al. 2001)). A recent study investigating the relative contribution of TLR4 expressed on Kupffer cells and HSCs in alcoholic liver disease has been published (Inokuchi et al. 2011)). In addition to the previous concept, we demonstrate that TLR4 signalling is important in both BM-derived cells including Kupffer cells, and endogenous liver cells including HSCs for alcohol-induced hepatocyte injury, steatosis, inflammation and fibrogenesis (Inokuchi et al. 2011)) (Fig. 3)). Unexpectedly, the TLR4-TRIF-IRF3 pathway was found to be more important than the TLR4-MyD88 dependent pathway in the development of alcoholic steatohepatitis (Hritz et al. 2008a; Petrasek et al. 2011)).
Figure 3. Bacterial translocation and hepatic TLR4 signalling in alcoholic liver disease.
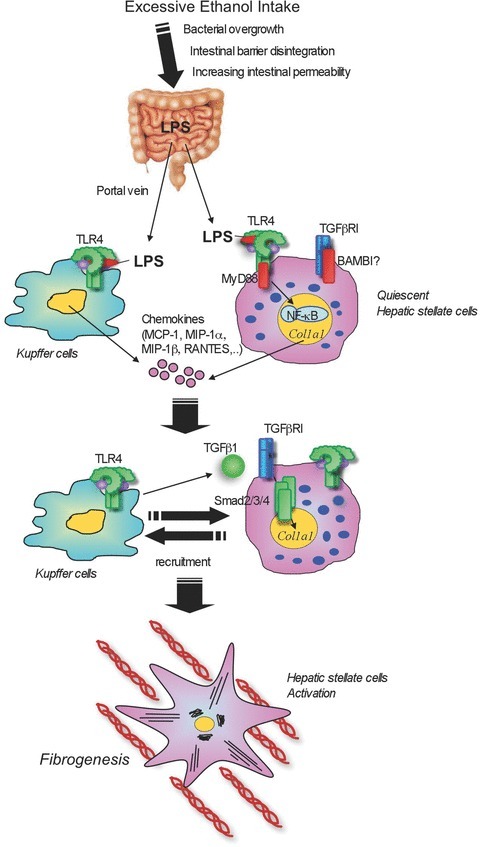
Excessive intake of alcohol induces changes in composition of intestinal microflora and bacterial overgrowth. In addition, tight junction disruption causes an increase in intestinal permeability, leading to translocation of gut microflora-derived LPS into the liver through the portal vein. Translocated LPS stimulates TLR4 on both Kupffer cells and hepatic stellate cells (HSCs). Upon activation of TLR4, Kupffer cells and HSCs produce chemokines (MCP-1, MIP-1α, MIP-1β and RANTES) that recruit Kupffer cells as well as HSCs. This activation of cells participates in liver inflammation, hepatocyte steatosis and fibrosis.
Bacterial translocation is a crucial factor for development of alcohol liver disease (Rao, 2009)). Ethanol and its metabolite, acetaldehyde, directly influence dissociation of tight junction protein ZO-1 in the intestinal epithelium, suggesting a mechanism of intestinal barrier dysfunction and subsequent bacterial translocation in alcoholic liver disease (Ma et al. 1999)). Moreover, ethanol ingestion suppresses intestinal motility, which contributes to bacterial overgrowth (Bode & Bode, 2003)). However, as only 15–20% of the enteric microflora can be cultured using conventional bacterial culture systems, a comprehensive analysis of characteristic changes in the gut flora could not be performed. As noted above, with the invention of culture-independent systems, such as massively parallel pyrosequencing, the composition of fecal microbiome was demonstrated to be different in patients with alcoholic liver cirrhosis (Chen et al. 2011)). A recent study has characterized bacterial translocation and changes in intestinal microbiome in an animal model of continuous intragastric ethanol infusion. One week after ethanol infusion, translocated bacteria were detected in the systemic blood, and systemic LPS levels were elevated (Yan et al. 2011)). While intestinal bacterial overgrowth was not detected 1 week after enteral ethanol feeding in contrast to the induction of bacterial translocation, the number of aerobic and anaerobic bacteria was increased 3 weeks after continuous intragastric ethanol feeding as assessed by conventional bacterial culture methods (Yan et al. 2011)). Thus, intestinal bacterial overgrowth occurs after early bacterial translocation. Subsequent massively parallel pyrosequencing using caecal contents demonstrated that ethanol treatment decreased operational taxonic units (OTUs) of several Firmicutes, such as Lactococcuss, Pediococcuss and Lactobacillus, and increased OTUs of unknown bacteria, Verrucomicrobia and Bacteroidetes, such as Bacteroidales, Bacteroides and Porphyromonadaceae (Yan et al. 2011)). Interestingly, most of the suppressed bacteria are considered ‘good’ bacteria or probiotic bacteria. The principle component analysis (PCA) indicated distinct enteric microbiomes in control and ethanol treated groups (Yan et al. 2011)).
The study also found significantly suppressed intestinal expression of antimicrobial proteins Reg3b and Reg3g in animals and patients with chronic ethanol consumption, which suggests that gut dysbiosis following ethanol treatment is induced by the deregulation of antimicrobial molecule expression (Yan et al. 2011)). Because antimicrobial Reg3g levels were increased in B. thetaiotaomicron mono-associated mice, and because administration of probiotic Lactobacillus had a beneficial effect in alcoholic liver disease, it is postulated that pre- or probiotic treatment regulates intestinal expression of endogenous antimicrobial molecules that control homeostasis of the intestinal microflora (Nanji et al. 1994; Sonnenburg et al. 2006; Mutlu et al. 2009)). Upon administration of prebiotic fructo-oligosaccharides (FOS), indigestive short-chain saccharides that stimulate probiotic bacteria Lactobacilli and Bifdobacteria, the intestinal expression of Reg3g was partially restored even after enteral ethanol treatment (Yan et al. 2011)). Partially restored Reg3g expression suppressed enteric bacterial overgrowth and alcohol-induced steatohepatitis (Yan et al. 2011)). Thus, pre- or probiotics are promising therapeutic approaches to ameliorate intestinal dysbiosis, bacterial overgrowth and subsequent reversal of alcoholic steatohepatitis. Besides LPS, bacterial DNA, a natural ligand for TLR9, is also recognized as a part of microbial products that are able to translocate from the intestine. Therefore, we highly expect TLR9 signalling to influence disease progression in alcohol liver disease. However, this hypothesis has not yet been tested (Gao, 2011)).
TLR9 signalling in non-alcoholic steatohepatitis
Non-alcoholic steatohepatitis (NASH) is a hepatic feature of the metabolic syndrome. Obesity and insulin resistance are often demonstrated as promoting factors for NASH (Miura et al. 2010b; Sorrentino et al. 2010)). NASH is histologically characterized by hepatocyte steatosis, ballooning, inflammatory cell infiltration and fibrosis (Miura et al. 2010b)), and might progress to cirrhosis (Bataller & Brenner, 2005)). There is an interesting animal study investigating the relationship between gut microbiota and obesity. When wild-type germfree mice fed a standard chow diet are colonized with a microbiota harvested from ob/ob or lean donors, adiposity in recipients of the obese microbiota increased more than in recipients of a lean microbiota, indicating a crucial role of the microbiota in obesity and hence fatty liver disease (Backhed et al. 2004; Ley et al. 2006; Turnbaugh et al. 2006)). There are at least two mechanisms to be suggested here. First, microflora itself alters nutrition and metabolism in the intestine contributing to obese and fatty livers in animals (Kim & Sears, 2010; Son et al. 2010)). Second, microbiota from obese mice is associated with bacterial translocation that promotes obese and fatty liver. Small intestinal bacterial overgrowth (SIBO), tight junction disruption and increased intestinal permeability were seen in NAFLD patients. These factors are associated with the severity of steatosis (Miele et al. 2009; Vanni & Bugianesi, 2009)). This clinical observation corroborates previous reports demonstrating the importance of TLR4 and intestine-derived LPS in the animal model of NASH (Rivera et al. 2007; Hritz et al. 2008b; Spruss et al. 2009; Miura et al. 2010b; Csak et al. 2011)). Besides LPS, bacterial products such as bacterial DNA, a ligand for TLR9, was detected by PCR for bacterial 16S rRNA in the blood of the murine NASH model developed by 22 weeks of choline deficient amino acid defined (CDAA) diet feeding (Miura et al. 2010a)). This evidence suggested that activation of TLR9 signalling plays an important role in NASH. CDAA diet induced severe steatosis, hepatocyte damage, inflammatory cell infiltration and fibrosis in wild-type mice, but not TLR9-deficient mice (Miura et al. 2010a)). The decreased steatohepatitis in TLR9-deficient mice was associated with a decrease in hepatic levels of IL-1β, but lacked association with other cytokines (Miura et al. 2010a)). IL-1β was preferentially expressed and produced in response to the TLR9 ligand CpG-DNA in Kupffer cells, but not in other hepatic cells (Miura et al. 2010a)). Subsequently, mature IL-1β secreted from Kupffer cells stimulates hepatocytes and HSCs in NASH. IL-1β directly increases lipid accumulation through the expression of lipogenesis gene DGAT2 in hepatocytes (Miura et al. 2010a)). Surprisingly, IL-1β also induces cell death in lipid-accumulated hepatocytes with an upregulation of the proapoptotic gene Bax and a downregulation of the antiapoptotic gene Bcl-2 (Miura et al. 2010a)). Moreover, IL-1β induces fibrogenic responses with upregulated fibrogenic genes and downregulated Bambi in HSCs (Miura et al. 2010a). Finally, mice deficient in IL-1 receptor and MyD88 showed a significant reduction of diet-induced steatohepatitis (Miura et al. 2010a)). In NASH, translocated bacterial DNA binds to TLR9 on Kupffer cells to produce IL-1β, which in turn stimulates hepatocytes for lipid accumulation and cell death. In parallel, IL-1β activates HSCs to induce liver fibrosis.
Conclusions and future perspective
While clinical symptoms may not be overt in patients with mild to moderate liver fibrosis/cirrhosis, the complications of liver cirrhosis, including hepatic failure, portal hypertension and hepatocellular carcinoma, are major causes of morbidity and mortality. Unfortunately, effective antifibrotic therapies are not yet established. Because TLR signalling and intestinal microbiota are important in the development of liver fibrosis, targeting innate immune signalling, such as TLRs and intestinal microbiota may become an effective therapy for chronic liver disease including liver fibrosis, alcoholic liver disease and obesity associated NASH. A number of issues concerning TLRs, bacterial translocation and intestinal microbiota in liver fibrosis are yet to be addressed. First, since the role of dysbiosis in bacterial translocation during liver fibrosis has not been studied in detail, the changes in gut microbiome in chronic liver disease should be comprehensively analysed using 454 massively parallel pyrosequencing. Second, the regulation of intestinal expression of antimicrobial molecules and its effects on dysbiosis and liver fibrosis need to be examined. Third, the influences of specific bacterial strains in translocation and liver fibrosis should be investigated. Fourth, the therapeutic effects of pre-, pro- and antibiotics on intestinal microbiota in liver fibrosis remain to be assessed. Moreover, endogenous TLR ligands associated with bacterial translocation and chronic liver disease should be studied. A recent report has identified HMGB-1 and fatty acids to be endogenous TLR4 ligands in NASH (Li et al. 2011)). Finally, while it is known that hepatocellular carcinoma is strongly associated with liver fibrosis and cirrhosis, it is still unclear whether TLRs, intestinal microbiota and bacterial translocation are involved in fibrosis-mediated hepatocellular carcinoma. One study has clearly shown the critical role of TLR4 and intestine-derived LPS in chemical-induced experimental liver cancer (Yu et al. 2010)). Future studies will uncover additional clinical relevance of TLRs and intestinal microbiota in liver fibrosis, cirrhosis and cancer.
Acknowledgments
This study is supported by the pilot grants from the UCSD Digestive Diseases Research Development Centre (DK080506) (E.S. and B.S.), the research grants from ABMRF (E.S. and B.S.), NIH grant R01AA02172 (E.S.) and R01DK085252 (E.S.), and NIH grant K08DK081830 (B.S.) and R01AA020703 (B.S.). We thank Ms Jingyi Isabelle Song and Ms Karin Diggle (Department of Medicine at UC San Diego) for editing the manuscript. There is no conflict of interest to disclose for either author.
Glossary
Abbreviations
- BDL
bile duct ligation
- BM
bone marrow
- BMT
BM transplantation
- CCl4
carbon tetrachloride
- CDAA
choline deficient amino acid defined
- ECM
extracellular matrix
- FOS
fructo-oligosaccharides
- HSC
hepatic stellate cell
- LPS
lipopolysaccharide
- NASH
non-alcoholic steatohepatitis
- NK
natural killer
- OTU
operational taxonic unit
- PCA
principle component analysis
- SIBO
small intestinal bacterial overgrowth
- SMA
smooth muscle actin
- SNP
single nucleotide polymorphism
- TLR
Toll-like receptor
References
- Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. 1994;20:453–460. [PubMed] [Google Scholar]
- Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology. 1995;108:218–224. doi: 10.1016/0016-5085(95)90027-6. [DOI] [PubMed] [Google Scholar]
- Assimakopoulos SF, Scopa CD, Vagianos CE. Pathophysiology of increased intestinal permeability in obstructive jaundice. World J Gastroenterol. 2007;13:6458–6464. doi: 10.3748/wjg.v13.i48.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D, Luedde T, Trautwein C, Tacke F. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2011 doi: 10.1136/gutjnl-2011-300304. (in press) [DOI] [PubMed] [Google Scholar]
- Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg RD, Garlington AW. Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model. Infect Immun. 1979;23:403–411. doi: 10.1128/iai.23.2.403-411.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berres ML, Koenen RR, Rueland A, Zaldivar MM, Heinrichs D, Sahin H, Schmitz P, Streetz KL, Berg T, Gassler N, Weiskirchen R, Proudfoot A, Weber C, Trautwein C, Wasmuth HE. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J Clin Invest. 2010;120:4129–4140. doi: 10.1172/JCI41732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode C, Bode JC. Effect of alcohol consumption on the gut. Best Pract Res Clin Gastroenterol. 2003;17:575–592. doi: 10.1016/s1521-6918(03)00034-9. [DOI] [PubMed] [Google Scholar]
- Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol. 1987;4:8–14. doi: 10.1016/s0168-8278(87)80003-x. [DOI] [PubMed] [Google Scholar]
- Chan CC, Hwang SJ, Lee FY, Wang SS, Chang FY, Li CP, Chu CJ, Lu RH, Lee SD. Prognostic value of plasma endotoxin levels in patients with cirrhosis. Scand J Gastroenterol. 1997;32:942–946. doi: 10.3109/00365529709011206. [DOI] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang F, Lu H, Wang B, Lei D, Wang Y, Zhu B, Li L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54:562–572. doi: 10.1002/hep.24423. [DOI] [PubMed] [Google Scholar]
- Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–163. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, Catalano D, Mandrekar P, Dolganiuc A, Kurt-Jones E, Szabo G. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G433–441. doi: 10.1152/ajpgi.00163.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol. 1991;12:162–169. doi: 10.1016/0168-8278(91)90933-3. [DOI] [PubMed] [Google Scholar]
- Gao B. Innate immunity and steatohepatitis: a critical role of another toll (TLR-9) Gastroenterology. 2011;139:27–30. doi: 10.1053/j.gastro.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Hurtado I, Santacruz A, Peiro G, Zapater P, Gutierrez A, Perez-Mateo M, Sanz Y, Frances R. Gut microbiota dysbiosis is associated with inflammation and bacterial translocation in mice with CCl4-induced fibrosis. PLoS One. 2011;6:e23037. doi: 10.1371/journal.pone.0023037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinko I, Geerts A, Wisse E. Experimental biliary fibrosis correlates with increased numbers of fat-storing and Kupffer cells, and portal endotoxemia. J Hepatol. 1995;23:449–458. doi: 10.1016/0168-8278(95)80204-5. [DOI] [PubMed] [Google Scholar]
- Gunnarsdottir SA, Sadik R, Shev S, Simren M, Sjovall H, Stotzer PO, Abrahamsson H, Olsson R, Bjornsson ES. Small intestinal motility disturbances and bacterial overgrowth in patients with liver cirrhosis and portal hypertension. Am J Gastroenterol. 2003;98:1362–1370. doi: 10.1111/j.1572-0241.2003.07475.x. [DOI] [PubMed] [Google Scholar]
- Higashiyama R, Moro T, Nakao S, Mikami K, Fukumitsu H, Ueda Y, Ikeda K, Adachi E, Bou-Gharios G, Okazaki I, Inagaki Y. Negligible contribution of bone marrow-derived cells to collagen production during hepatic fibrogenesis in mice. Gastroenterology. 2009;137:1459–1466. doi: 10.1053/j.gastro.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, Szabo G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008a;48:1224–1231. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hritz I, Velayudham A, Dolganiuc A, Kodys K, Mandrekar P, Kurt-Jones E, Szabo G. Bone marrow-derived immune cells mediate sensitization to liver injury in a myeloid differentiation factor 88-dependent fashion. Hepatology. 2008b;48:1342–1347. doi: 10.1002/hep.22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ, Layden TJ, Wright TL, White T, Cheung RC. A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology. 2007;46:297–306. doi: 10.1002/hep.21695. [DOI] [PubMed] [Google Scholar]
- Imamura M, Tsutsui H, Yasuda K, Uchiyama R, Yumikura-Futatsugi S, Mitani K, Hayashi S, Akira S, Taniguchi S, Van Rooijen N, Tschopp J, Yamamoto T, Fujimoto J, Nakanishi K. Contribution of TIR domain-containing adapter inducing IFN-β-mediated IL-18 release to LPS-induced liver injury in mice. J Hepatol. 2009;51:333–341. doi: 10.1016/j.jhep.2009.03.027. [DOI] [PubMed] [Google Scholar]
- Inokuchi S, Tsukamoto H, Park EJ, Liu Z-X, Brenner DA, Seki E. Toll-like receptor 4 mediates alcohol-induced steatohepatitis through bone marrow-derived and endogenous liver cells in mice. Alcohol Clin Exp Res. 2011;35:1509–1518. doi: 10.1111/j.1530-0277.2011.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isayama F, Hines IN, Kremer M, Milton RJ, Byrd CL, Perry AW, McKim SE, Parsons C, Rippe RA, Wheeler MD. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1318–1328. doi: 10.1152/ajpgi.00405.2005. [DOI] [PubMed] [Google Scholar]
- Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol. 2005;79:7269–7272. doi: 10.1128/JVI.79.11.7269-7272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, Merad M, Luedde T, Trautwein C, Tacke F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261–274. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- Kennedy DW, Abkowitz JL. Kinetics of central nervous system microglial and macrophage engraftment: analysis using a transgenic bone marrow transplantation model. Blood. 1997;90:986–993. [PubMed] [Google Scholar]
- Kim JJ, Sears DD. TLR4 and insulin resistance. Gastroenterol Res Pract. 2010;2010:212563. doi: 10.1155/2010/212563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, Brenner DA. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–438. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, Pierce RH, Crispe IN. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110:4077–4085. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, Hou YJ, Chang YX, Tu QQ, Feng GS, Shen F, Wu MC, Wang HY. Nuclear factor high-mobility group box1 mediating the activation of toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54:1620–1630. doi: 10.1002/hep.24552. [DOI] [PubMed] [Google Scholar]
- Lin RS, Lee FY, Lee SD, Tsai YT, Lin HC, Lu RH, Hsu WC, Huang CC, Wang SS, Lo KJ. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol. 1995;22:165–172. doi: 10.1016/0168-8278(95)80424-2. [DOI] [PubMed] [Google Scholar]
- Lu H, Wu Z, Xu W, Yang J, Chen Y, Li L. Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection. Intestinal microbiota of HBV cirrhotic patients. Microb Ecol. 2011;61:693–703. doi: 10.1007/s00248-010-9801-8. [DOI] [PubMed] [Google Scholar]
- Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 1999;276:G965–974. doi: 10.1152/ajpgi.1999.276.4.G965. [DOI] [PubMed] [Google Scholar]
- Marra F. Chemokines in liver inflammation and fibrosis. Front Biosci. 2002;7:d1899–1914. doi: 10.2741/A887. [DOI] [PubMed] [Google Scholar]
- Marra F, Romanelli RG, Giannini C, Failli P, Pastacaldi S, Arrighi MC, Pinzani M, Laffi G, Montalto P, Gentilini P. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology. 1999;29:140–148. doi: 10.1002/hep.510290107. [DOI] [PubMed] [Google Scholar]
- Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Masciana R, Forgione A, Gabrieli ML, Perotti G, Vecchio FM, Rapaccini G, Gasbarrini G, Day CP, Grieco A. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- Miettinen TA. Lipid absorption, bile acids, and cholesterol metabolism in patients with chronic liver disease. Gut. 1972;13:682–689. doi: 10.1136/gut.13.9.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology. 2010a;139:323–334. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K, Seki E, Ohnishi H, Brenner DA. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic fatty liver disease. Gastroenterol Res Pract. 2010b;2010:362847. doi: 10.1155/2010/362847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res. 2009;33:1836–1846. doi: 10.1111/j.1530-0277.2009.01022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanji AA, Khettry U, Sadrzadeh SM. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease) Proc Soc Exp Biol Med. 1994;205:243–247. doi: 10.3181/00379727-205-43703. [DOI] [PubMed] [Google Scholar]
- Nolan JP, Leibowitz AI. Endotoxin and the liver. III. Modification of acute carbon tetrachloride injury by polymyxin b – an antiendotoxin. Gastroenterology. 1978;75:445–449. [PubMed] [Google Scholar]
- Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. [DOI] [PubMed] [Google Scholar]
- Pennington HL, Hall PM, Wilce PA, Worrall S. Ethanol feeding enhances inflammatory cytokine expression in lipopolysaccharide-induced hepatitis. J Gastroenterol Hepatol. 1997;12:305–313. doi: 10.1111/j.1440-1746.1997.tb00426.x. [DOI] [PubMed] [Google Scholar]
- Petrasek J, Dolganiuc A, Csak T, Nath B, Hritz I, Kodys K, Catalano D, Kurt-Jones E, Mandrekar P, Szabo G. Interferon regulatory factor 3 and type I interferons are protective in alcoholic liver injury in mice by way of crosstalk of parenchymal and myeloid cells. Hepatology. 2011;53:649–660. doi: 10.1002/hep.24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrasek J, Mandrekar P, Szabo G. Toll-like receptors in the pathogenesis of alcoholic liver disease. Gastroenterol Res Pract. 2010;2010:710381. doi: 10.1155/2010/710381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradere JP, Troeger JS, Dapito DH, Mencin AA, Schwabe RF. Toll-like receptor 4 and hepatic fibrogenesis. Semin Liver Dis. 2010;30:232–244. doi: 10.1055/s-0030-1255353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulskens WP, Rampanelli E, Teske GJ, Butter LM, Claessen N, Luirink IK, van der Poll T, Florquin S, Leemans JC. TLR4 promotes fibrosis but attenuates tubular damage in progressive renal injury. J Am Soc Nephrol. 2011;21:1299–1308. doi: 10.1681/ASN.2009070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–644. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571–579. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi M, Tacke F, Trautwein C, Luedde T. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- Schwabe RF, Bataller R, Brenner DA. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am J Physiol Gastrointest Liver Physiol. 2003;285:G949–958. doi: 10.1152/ajpgi.00215.2003. [DOI] [PubMed] [Google Scholar]
- Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, Llovet JM, Brenner DA, Schwabe RF. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest. 2009a;119:1858–1870. doi: 10.1172/JCI37444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, Schwabe RF, Brenner DA. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009b;50:185–197. doi: 10.1002/hep.22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- Seki E, Tsutsui H, Iimuro Y, Naka T, Son G, Akira S, Kishimoto T, Nakanishi K, Fujimoto J. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology. 2005;41:443–450. doi: 10.1002/hep.20603. [DOI] [PubMed] [Google Scholar]
- Seki E, Tsutsui H, Nakano H, Tsuji N, Hoshino K, Adachi O, Adachi K, Futatsugi S, Kuida K, Takeuchi O, Okamura H, Fujimoto J, Akira S, Nakanishi K. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1β. J Immunol. 2001;166:2651–2657. doi: 10.4049/jimmunol.166.4.2651. [DOI] [PubMed] [Google Scholar]
- Son G, Kremer M, Hines IN. Contribution of gut bacteria to liver pathobiology. Gastroenterol Res Pract. 2010;2010:453563. doi: 10.1155/2010/453563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg JL, Chen CT, Gordon JI. Genomic and metabolic studies of the impact of probiotics on a model gut symbiont and host. PLoS Biol. 2006;4:e413. doi: 10.1371/journal.pbio.0040413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino P, Terracciano L, D’Angelo S, Ferbo U, Bracigliano A, Vecchione R. Predicting fibrosis worsening in obese patients with NASH through parenchymal fibronectin, HOMA-IR, and hypertension. Am J Gastroenterol. 2010;105:336–344. doi: 10.1038/ajg.2009.587. [DOI] [PubMed] [Google Scholar]
- Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology. 2009;50:1094–1104. doi: 10.1002/hep.23122. [DOI] [PubMed] [Google Scholar]
- Sung JY, Shaffer EA, Costerton JW. Antibacterial activity of bile salts against common biliary pathogens. Effects of hydrophobicity of the molecule and in the presence of phospholipids. Dig Dis Sci. 1993;38:2104–2112. doi: 10.1007/BF01297092. [DOI] [PubMed] [Google Scholar]
- Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol. 2010;16:1321–1329. doi: 10.3748/wjg.v16.i11.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol. 2010;11:70–75. doi: 10.1038/ni.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- Uesugi T, Froh M, Arteel GE, Bradford BU, Wheeler MD, Gabele E, Isayama F, Thurman RG. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol. 2002;168:2963–2969. doi: 10.4049/jimmunol.168.6.2963. [DOI] [PubMed] [Google Scholar]
- Vanni E, Bugianesi E. The gut-liver axis in nonalcoholic fatty liver disease: Another pathway to insulin resistance? Hepatology. 2009;49:1790–1792. doi: 10.1002/hep.23036. [DOI] [PubMed] [Google Scholar]
- von Montfort C, Beier JI, Guo L, Kaiser JP, Arteel GE. Contribution of the sympathetic hormone epinephrine to the sensitizing effect of ethanol on LPS-induced liver damage in mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1227–1234. doi: 10.1152/ajpgi.00050.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Trippler M, Pei R, Lu M, Broering R, Gerken G, Schlaak JF. Toll-like receptor activated human and murine hepatic stellate cells are potent regulators of hepatitis C virus replication. J Hepatol. 2009;51:1037–1045. doi: 10.1016/j.jhep.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005;41:422–433. doi: 10.1002/hep.20632. [DOI] [PubMed] [Google Scholar]
- Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, Bucchi A, Krux F, Dittmer U, Yang D, Roggendorf M, Gerken G, Lu M, Schlaak JF. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology. 2010;129:363–374. doi: 10.1111/j.1365-2567.2009.03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Takeda K. Current views of toll-like receptor signaling pathways. Gastroenterol Res Pract. 2010;2010:240365. doi: 10.1155/2010/240365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan AW, Fouts DE, Brandl J, Starkel P, Torralba M, Schott E, Tsukamoto H, Nelson KE, Brenner DA, Schnabl B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53:96–105. doi: 10.1002/hep.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Lin Z, Chen F, Zhao X, Chen H, Ning Y, Chen YG. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-β signaling. J Biol Chem. 2009;284:30097–30104. doi: 10.1074/jbc.M109.049304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, Bradford BU, Wheeler MD, Uesugi T, Froh M, Goyert SM, Thurman RG. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol. 2001;166:4737–4742. doi: 10.4049/jimmunol.166.7.4737. [DOI] [PubMed] [Google Scholar]
- Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, Tang L, Lin Y, He YQ, Zou SS, Wang C, Zhang HL, Cao GW, Wu MC, Wang HY. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology. 2010;52:1322–1333. doi: 10.1002/hep.23845. [DOI] [PubMed] [Google Scholar]
- Zhang W, Gu Y, Chen Y, Deng H, Chen L, Chen S, Zhang G, Gao Z. Intestinal flora imbalance results in altered bacterial translocation and liver function in rats with experimental cirrhosis. Eur J Gastroenterol Hepatol. 2010;22:1481–1486. doi: 10.1097/MEG.0b013e32833eb8b0. [DOI] [PubMed] [Google Scholar]