

Abstract
Non-technical summary
The endothelium is a thin layer of cells lining the interior surface of the entire circulatory system. Endothelial progenitor cells (EPCs) have a crucial role in maintaining the integrity of the endothelium, as they are recruited from the bone marrow to sites of endothelial injury where they contribute to blood vessel formation and repair. The factors regulating EPC mobilization and trafficking remain incompletely understood. We evaluated the time-course effects of a single 4 h bout of severe hypoxic breathing (simulating 4100 m altitude) followed by 4 h restoration in room air. We show that hypoxia alone induces a rapid disappearance of EPCs from blood, probably sustained by a prompt cell marginalization followed by a late increase in EPC apoptosis. These observations may broaden our understanding of the mechanisms operated by EPCs to maintain endothelial homeostasis and may help to elucidate the potential role of EPCs in regenerative medicine.
Abstract
There are hints that hypoxia exposure may affect the number of circulating endothelial progenitor cells (EPCs) in humans. To test this hypothesis, the concentration of EPCs was determined by flow cytometry in the peripheral blood of 10 young healthy adults before (0 h), at different times (0.5 h, 1 h, 2 h and 4 h) during a 4 h normobaric hypoxic breathing simulating 4100 m altitude, and in the following recovery breathing room air. Results were interpreted mainly on the basis of the changes in surface expression of CXC chemokine receptor-4 (CXCR-4, a chemokine receptor essential for EPC migration and homing) and the percentage of apoptotic cells, the plasmatic levels of markers of oxidative stress induced by hypoxic breathing. Compared to 0 h, the concentration of EPCs, identified as either CD45dim/CD34+/KDR+ or CD45dim/CD34+/KDR+/CD133+ cells, decreased from 337 ± 83 ml−1 (mean ± SEM) to 223 ± 52 ml−1 (0.5 h; P < 0.005) and 100 ± 37 ml−1 (4 h; P < 0.005), and from 216 ± 91 to 161 ± 50 ml−1 (0.5 h; P < 0.05) and 45 ± 23 ml−1 (4 h; P < 0.005), respectively. Upon return to normoxia, their concentration increased slowly, and after 4 h was still lower than at 0 h (P < 0.05). During hypoxia, CXCR-4 expression and plasmatic stromal derived cell factor-1 (SDF-1) increased abruptly (0.5 h: +126% and +13%, respectively; P < 0.05), suggesting cell marginalization as a possible cause of the rapid hypoxia-induced EPC reduction. Moreover, hypoxia exposure induced an increase in EPC apoptosis and markers of oxidative stress, which was significantly evident only starting from 2 h and 4 h after hypoxia offset, respectively, suggesting that EPC apoptosis may contribute to the later phase of hypoxia-induced EPC reduction. Overall, these observations may provide new insights into the understanding of the mechanisms operated by EPCs to maintain endothelial homeostasis.
Introduction
Circulating endothelial progenitor cells (EPCs) are a rare population of adult mononuclear cells deriving from the bone marrow with properties similar to those of embryonal angioblasts (Asahara et al. 1997; Peichev et al. 2000)). In fact, EPCs are mobilized from the bone marrow and recruited to sites of endothelial injury where they proliferate, differentiate into mature endothelial cells, integrate into the endothelial layer and exert a paracrine function by producing vascular growth factors (Hu et al. 2003; Wassman et al. 2006; Zentilin et al. 2006)). In this way EPCs can contribute to blood vessel formation and repair, thus allowing the maintenance of endothelial integrity (Schatteman et al. 2000; Asahara & Kawamoto 2004)). Accordingly, a reduction in blood EPC concentration might interfere negatively with endothelial function. Indeed, a low concentration of circulating EPCs has been found in patients affected by coronary artery disease and congestive heart failure (Vasa et al. 2001; Valgimigli et al. 2004; Kunz et al. 2006)). On the other hand, conditions characterized by endothelial dysfunction and increased cardiovascular risk such as ageing, smoking, hypertension and diabetes, have been demonstrated to be consistently associated with reduced circulating EPC concentration (reviewed in Sirker et al. 2009)), although the underlying mechanisms have not yet been completely elucidated.
Selective migration and recruitment of EPCs are essential steps during blood vessel formation and repair and the chemokine stromal derived cell factor-1 (SDF-1), via its cognate receptor CXC chemokine receptor-4 (CXCR-4), plays a critical role in this process (Ceradini & Gurtner 2005)). In homeostatic conditions, discrete regions of hypoxia in the bone marrow compartment undergo an increase in the transcriptional activity of hypoxia inducible factor-1 (HIF-1), which is responsible for a robust expression of SDF-1 (Harrison et al. 2002; Ceradini et al. 2005)). The latter binds to CXCR-4 (Ceradini et al. 2004)) expressed on the surface of EPCs which, as a consequence, are kept in the bone marrow. In ischaemic tissues, oxygen tension falls and HIF-1 increases, activating the transcription of SDF-1 by endothelial cells in direct proportion to reduced oxygen tension (Ceradini et al. 2004)). The consequent release of SDF-1 may lead to reversal of the marrow/periphery SDF-1 gradient, allowing EPCs to disengage the bone marrow and to enter the blood stream where their concentration rapidly increases (Aicher et al. 2005; Massa et al. 2005)), before homing to ischaemic tissue.
Besides SDF-1, other angiogenic cytokines such as erythropoietin (Bahlmann et al. 2004)), vascular endothelial growth factor (VEGF) and granulocyte colony-stimulating factor contribute to HIF-1-mediated EPC mobilization (Leone et al. 2009)). Moreover, HIF-1 may be regulated by a variety of other factors besides hypoxia, including oxidative stress (caused by excess production of free radicals), inflammatory cytokines and trophic stimuli (Feldser et al. 1999; Hellwig-Burgel et al. 1999; Sandau et al. 2001; Pialoux et al. 2009)). These factors are also involved in tissue ischaemia, which was consistently present in most animal models used so far to study EPC behaviour, making the role of hypoxia in the mobilization and recruitment of EPCs to the sites of vessel damage still unclear. Moreover, as is well known from cardiovascular studies, systemic hypoxia and tissue ischaemia often elicit different adaptive responses. This might be the case also for the concentration of circulating EPCs. Indeed, this hypothesis seems to be confirmed by two independent observations. The first one is the consistent demonstration that hypoxaemic patients with severe lung disease have reduced numbers of circulating EPCs (Fadini et al. 2006; Palange et al. 2006)). The second one is the direct demonstration in a murine model that ischaemic injury induces the mobilization of bone marrow-derived progenitor cells, whereas chronic systemic hypoxia does not (O’Neill et al. 2005)).
Therefore, the present study was designed to investigate the effects of acute hypoxia alone on the levels of blood-circulating EPCs in the absence of other confounding factors. To this aim, young healthy adults were exposed to a 4 h hypoxic gas mixture (12.5% O2) breathing in resting conditions. Blood EPC concentration was determined at different times during and after hypoxia exposure. An attempt to cast light upon the mechanisms possibly underlying EPC reduction was also made determining the expression of CXCR-4 on EPCs, the percentage of apoptotic cells and the plasma concentration of metabolic markers of oxidative stress.
Methods
Ethical approval
The study was approved by the ethical committee and research review boards of the involved institutions, and was carried out in accordance with the principles outlined in the 2002 Declaration of Helsinki of the World Medical Association. Written informed consent was obtained by all subjects, after being carefully instructed about aims, methods and experimental procedure.
Subjects
Ten healthy males (25.8 ± 0.9 years of age; 77.4 ± 4.2 kg body mass; mean ± SEM) volunteered for the study. Preliminarily, subjects underwent a clinical screening that included history taking, physical examination and resting ECG. Criteria of exclusion from the study were cigarette smoking, arterial hypertension, hypercholesterolaemia, diabetes and cardiovascular or respiratory diseases, which may affect EPC count. To minimize confounding effects, no subject was above 1000 m altitude in the 4 weeks preceding the study nor was regularly engaged in a training program. In addition, volunteers were required to refrain from any strenuous physical activity and from alcohol- and caffeine-containing beverages in the 2 weeks and 24 h, respectively, prior to the study.
Study design
Subjects reported to the laboratory early in the morning 2 h after a light breakfast, and sat comfortably on a chair. They remained seated over the 8 h procedure and all of them received a light meal for lunch. The room temperature was kept constant at 21°C. A cannula was placed into an antecubital vein. After 15 min a blood sample was withdrawn for EPC and oxidative marker determination (0 h). Subsequently, subjects started breathing a normobaric hypoxic mixture (12.5% O2 in air, simulating about 4100 m altitude) obtained by removing a controlled amount of oxygen from air (MAG-10, Higher Peak LLC, Winchester, MA, USA). The mixture was delivered through a face mask at 30 l min−1. Excess air flow was diverted outside the mask to prevent inspired oxygen pressure from increasing above 90 Torr. Oxygen concentration and pressure in inspired air were carefully checked prior and occasionally during each experiment by a computerized O2–CO2 analyser–flowmeter combination (Vmax 229, SensorMedics, Yorba Linda, CA, USA). Arterialized blood oxygen saturation was relatively constant at 85–90% as frequently monitored by ear lobe pulse oximetry (Biox 3740 Pulse Oximeter, Ohmeda, Denver, CO, USA). An accurate monitoring of cardiorespiratory parameters, including pulmonary ventilation (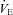 ), respiratory rate (f), heart rate (HR), cardiac index (CI) (which was calculated as cardiac output normalized for body surface area) and gas exchanges, including end-tidal O2 (PET, O 2) and CO2 (PET, CO 2) pressure, arterial oxygen saturation (S aO 2), was performed on a subgroup of subjects. Venous blood samples were withdrawn at different times during the test (0.5 h, 1 h, 2 h and 4 h). At the end of the fourth hour subjects were switched to normoxic breathing conditions and blood samples withdrawn for up to 4 h. Four subjects repeated the same experimental procedure except for breathing through the face mask with the hypoxic apparatus switched to normoxia, at least 4 weeks apart. Blood samples were collected into sodium citrate Vacutainer tubes (Becton Dickinson, San Jose, CA, USA), and processed immediately for EPC analysis. An aliquot of blood was collected in heparinized tubes and immediately centrifuged at 1000 g for 10 min at 4°C. Plasma was stored in multiple aliquots at –80°C for subsequent determination of oxidative stress markers and cytokines. Total blood cells were counted with a standard haematology analyser.
), respiratory rate (f), heart rate (HR), cardiac index (CI) (which was calculated as cardiac output normalized for body surface area) and gas exchanges, including end-tidal O2 (PET, O 2) and CO2 (PET, CO 2) pressure, arterial oxygen saturation (S aO 2), was performed on a subgroup of subjects. Venous blood samples were withdrawn at different times during the test (0.5 h, 1 h, 2 h and 4 h). At the end of the fourth hour subjects were switched to normoxic breathing conditions and blood samples withdrawn for up to 4 h. Four subjects repeated the same experimental procedure except for breathing through the face mask with the hypoxic apparatus switched to normoxia, at least 4 weeks apart. Blood samples were collected into sodium citrate Vacutainer tubes (Becton Dickinson, San Jose, CA, USA), and processed immediately for EPC analysis. An aliquot of blood was collected in heparinized tubes and immediately centrifuged at 1000 g for 10 min at 4°C. Plasma was stored in multiple aliquots at –80°C for subsequent determination of oxidative stress markers and cytokines. Total blood cells were counted with a standard haematology analyser.
Quantification of circulating EPCs
Whole peripheral blood samples were analysed by flow cytometry as previously described (Taddeo et al. 2008; Cobellis et al. 2010)). Briefly, 200 μl of anticoagulated whole blood were incubated for 20 min with biotin-conjugated anti-human kinase insert domain receptor (KDR, a type III receptor tyrosine kinase also known as vascular endothelial growth factor receptor 2 (VEGFR-2); Sigma-Aldrich, St Louis, MO, USA), phycoerythrin (PE)-conjugated anti-human CD133 (Miltenyi-Biotec, GmbH, Bergisch Gladbach, Germany), allophycocyanin (APC)-conjugated anti-human CD34 (Beckman-Coulter Immunotech, Marsille, France) and peridinin–chlorophyll–protein complex (PerCP)–Cy5.5-conjugated anti-human CD45 (e-Bioscience, San Diego, CA, USA) monoclonal antibodies (mAbs). After incubation, erythrocytes were lysed and the remaining cells were further incubated in the dark for 20 min with fluorescein isothiocynate (FITC)-conjugated streptavidin (Sigma-Aldrich) to reveal biotin-conjugated anti-human KDR. Fluorescence minus one (FMO) samples were used as negative controls. Four-colour analysis was performed immediately after staining using a FACSCanto2 flow cytometer and FACSDiva Software (Becton Dickinson). An acquisition gate was established based on forward scatter (FSC) and side scatter (SSC) that included mononuclear cells (MNCs) but excluded most granulocytes and debris. 300,000 events were routinely collected to visualize and gate on this population. The cells were then gated as CD45dim/CD34+ and sequentially characterized based on KDR and CD133 expression (Fig. 1)).
Figure 1. Representative flow cytometry gating strategy for identification of circulating EPCs.

Left, mononuclear cells in whole blood samples were gated in a forward-scatter (FSC)–side-scatter (SSC) plot to exclude granulocytes, dead cells and debris. Right, cells were then sequentially gated based on CD45, CD34, KDR and CD133 expression. Circulating EPCs were defined as either CD45dim/CD34+/KDR+ (third panel from left) or CD45dim/CD34+/KDR+/CD133+ cells.
Estimates of the absolute numbers of peripheral blood progenitor cells were calculated from the proportion of cells recorded by flow cytometry in the mononuclear gate multiplied by absolute mononuclear cell count measured using a standard haematology analyser.
Surface expression of CXCR-4 on EPCs
PE-conjugated anti-human CXCR-4 (e-Bioscience) was used in combination with anti-CD45, anti-CD34 and anti-KDR mAbs. The same procedure described for EPC count was applied, and the expression of CXCR-4 was analysed gated on CD45dim/CD34+/KDR+ EPCs.
Spontaneous apoptosis of circulating EPCs
EPC apoptosis was assessed by 7-aminoactinomycin D (7AAD, Sigma-Aldrich) staining, as previously described (Mancuso et al. 2006)). Briefly, 200 μl of anticoagu-lated whole blood were incubated for 20 min with biotin-conjugated anti-human KDR, PE-conjugated anti-human CD45 (e-Bioscience), APC-conjugated anti-human CD34 mAbs and 7AAD. The samples were then processed as described for EPC counting, and appropriate analysis gates were used to enumerate apoptotic EPCs.
SDF-1 and VEGF measurement in plasma samples
The amounts of plasmatic SDF-1 and VEGF were determined by DuoSet ELISA and Quantikine ELISA kits, respectively (R&D Systems, Minneapolis, MN, USA). All individual steps were performed according to the manufacturer's instructions.
Plasma oxidative stress markers
The measurement of thiobarbituric acid-reactive substances (TBARS) is a well-established method to detect lipid peroxidation. We used a TBARS assay kit (Cayman Chemical, Ann Arbur, MI, USA) that allows a rapid photometric detection at 535 nm of the thiobarbituric acid malondialdehyde (TBAMDA) adduct. Samples were read by an Infinite M200 microplate reader spectrophotometer (TECAN, Männedorf, Switzerland). A linear calibration curve was computed from pure MDA-containing reactions.
The protein carbonyl (PC) content, an index of protein oxidation, was determined by means of a commercial kit (Cayman Chemical), through the reaction of 2,4-dinitrophenylhydrazine (DNPH) and carbonyls. This reaction forms a Schiff base producing the correspondent hydrazone. The latter was analysed by spectrophotometry, reading the absorbance signal in the 360–385 nm range. Values were normalized to the total protein concentration in the final pellet (absorbance reading at 280 nm) in order to consider protein loss during the washing steps.
Data analysis and statistics
Data were shown as mean ± standard error of the mean (SEM). Intra-individual and inter-individual variability was expressed as standard deviation to mean value ratio (coefficient of variation: CV %). The intra-individual variability of EPC determination was assessed in a resting subject breathing room air for 8 h. Eight blood samples were withdrawn at the same time points as hypoxia-exposed subjects. The coefficient of variation of CD45dim/CD34+/KDR+ and CD45dim/CD34+/KDR+/CD133+ cell concentration was very low (6.1% and 5.9%, respectively).
To determine the statistical significance of changes with time of hypoxia exposure the Wilcoxon matched-pairs signed-ranks test was performed. All statistical analyses assumed a 2-sided significance level of 0.05. Data analyses were performed with Openstat3 software.
Results
Cardiorespiratory parameters and gas exchanges
The values of pulmonary ventilation ( ), respiratory rate (f), end-tidal O2 (
), respiratory rate (f), end-tidal O2 (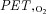 ) and CO2 (
) and CO2 ( ) pressure, heart rate (HR), cardiac index (CI) and arterial oxygen saturation (S aO 2) measured in normoxia and at different time points after hypoxia exposure are reported in Table 1. As expected, a significant marked decrease of
) pressure, heart rate (HR), cardiac index (CI) and arterial oxygen saturation (S aO 2) measured in normoxia and at different time points after hypoxia exposure are reported in Table 1. As expected, a significant marked decrease of  was evident as soon as 1 min after onset of hypoxic breathing (–37%, P < 0.05; after 30 min (0.5 h) it was –50% of the 0 h value (P < 0.05) and remained similarly low throughout the entire hypoxia exposure period (–47% after 4 h, P < 0.05).
was evident as soon as 1 min after onset of hypoxic breathing (–37%, P < 0.05; after 30 min (0.5 h) it was –50% of the 0 h value (P < 0.05) and remained similarly low throughout the entire hypoxia exposure period (–47% after 4 h, P < 0.05).  remained almost constant during the first hour of hypoxic breathing, it decreased slightly but significantly after 2 h (–12% of the 0 h value, P < 0.05) and a mild hypocapnia persisted until the end of hypoxia exposure. A modest increase in CI was observed immediately after the onset of hypoxic breathing (+5% at 1 min, P < 0.05), remained slightly increased for 1 h (+11% at 1 h, P < 0.05); thereafter CI stabilized at initial pre-hypoxia values.
remained almost constant during the first hour of hypoxic breathing, it decreased slightly but significantly after 2 h (–12% of the 0 h value, P < 0.05) and a mild hypocapnia persisted until the end of hypoxia exposure. A modest increase in CI was observed immediately after the onset of hypoxic breathing (+5% at 1 min, P < 0.05), remained slightly increased for 1 h (+11% at 1 h, P < 0.05); thereafter CI stabilized at initial pre-hypoxia values.
Table 1.
Pulmonary ventilation ( ), respiratory rate (f), end tidal O2 (PET, O 2) and CO2 (PET, CO 2) pressure, heart rate (HR), cardiac index (CI) and arterial oxygen saturation (S aO 2) of subjects breathing air and at different times (from 1 min to 4 h) in normobaric hypoxia (12.5% O2)
), respiratory rate (f), end tidal O2 (PET, O 2) and CO2 (PET, CO 2) pressure, heart rate (HR), cardiac index (CI) and arterial oxygen saturation (S aO 2) of subjects breathing air and at different times (from 1 min to 4 h) in normobaric hypoxia (12.5% O2)
| Air | 1 min | 5 min | 10 min | 15 min | 0.5 h | 1 h | 2 h | 3 h | 4 h | |
|---|---|---|---|---|---|---|---|---|---|---|
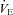 (l min−1) (l min−1) |
11.3 ± 2.0 | 16.2 ± 3.7* | 14.3 ± 2.5* | 12.8 ± 3.2 | 13.5 ± 2.7 | 13.4 ± 2.7* | 13.4 ± 2.1 | 13.4 ± 3.0 | 14.1 ± 4.1 | 14.1 ± 3.3* |
| f (breaths min−1) | 14.3 ± 2.4 | 14.9 ± 2.8 | 15.4 ± 1.9 | 13.5 ± 2.7 | 13.9 ± 2.8 | 14.9 ± 1.9 | 14.8 ± 2.4 | 14.4 ± 2.2 | 17.1 ± 2.0 | 17.5 ± 3.7 |
| PET, O 2 (mmHg) | 103.1 ± 9.9 | 64.7 ± 6.9* | 54.9 ± 3.3* | 53.2 ± 6.6* | 52.3 ± 5.0* | 51.8 ± 5.9* | 52.4 ± 4.2* | 54.6 ± 5.5* | 57.2 ± 7.7* | 54.8 ± 5.5* |
| PET, CO 2 (mmHg) | 39.2 ± 5.2 | 37.0 ± 3.0 | 36.2 ± 2.9 | 36.6 ± 4.4 | 36.6 ± 3.3 | 36.5 ± 4.4 | 36.0 ± 4.4 | 34.4 ± 5.6* | 32.4 ± 5.7* | 33.6 ± 3.7* |
| HR (beats min−1) | 69.8 ± 6.6 | 73.0 ± 5.7* | 77.3 ± 7.3* | 74.7 ± 9.6 | 73.5 ± 9.1 | 73.7 ± 9.9 | 72.8 ± 11.1 | 68.8 ± 9.7 | 68.2 ± 11.6 | 66.8 ± 11.0 |
| CI (l min−1m−2) | 3.75 ± 0.64 | 3.95 ± 0.66* | 4.27 ± 0.82* | 4.11 ± 0.88 | 4.06 ± 0.85 | 4.12 ± 0.67 | 4.17 ± 0.60* | 3.17 ± 0.68 | 3.58 ± 0.66 | 3.60 ± 0.96 |
| S aO 2 (%) | 99.4 ± 0.7 | 96.1 ± 2.5* | 91.9 ± 2.3* | 90.1 ± 2.0* | 90.7 ± 1.2* | 87.7 ± 2.1* | 88.9 ± 1.8* | 87.7 ± 3.8* | 90.0 ± 1.1* | 90.3 ± 3.2* |
Values are mean ± SD obtained from 6 subjects. *P < 0.05 compared with pre-hypoxia, as assessed by Wilcoxon matched-pairs signed-ranks test.
Circulating EPCs
In normoxia prior to hypoxia exposure (0 h), the concentration of circulating EPCs, identified as either CD45dim/CD34+/KDR+ or CD45dim/CD34+/KDR+/CD133+ cells, was 337 ± 83 and 216 ± 91 cells ml−1 (mean ± SEM), respectively, showing a high inter-individual variability (CV 65% and 94%), as expected (Xiao et al. 2008)). At the onset of hypoxic breathing, the concentration of both types of cells underwent a fast reduction in all the subjects (Fig. 2)). After 30 min (0.5 h) it was already significantly reduced compared to the 0 h value (–32% and –20%, P < 0.005 and P < 0.05, respectively). As shown in Fig. 3A, the concentration of circulating CD45dim/CD34+/KDR+ cells continued to decrease with time, attaining the lowest value after 4 h hypoxia (100 ± 37 cells ml−1, i.e. 24% of 0 h value; P < 0.005). During the following 4 h in normoxia, the blood concentration of CD45dim/CD34+/KDR+ cells progressively increased. However, it is important to note that at the end of the fourth hour in normoxia it was still significantly lower than that observed at 0 h (–26%; P < 0.05). In three examined subjects pre-hypoxia EPC concentration values were restored after 20 h in normoxia. As shown in Fig. 3B, similar results were obtained when circulating EPCs were identified as CD45dim/CD34+/KDR+/CD133+ cells. No concomitant reduction of other blood cells, including neutrophils, lymphocytes and monocytes, was observed. Importantly, to exclude the possibility that the observed variations in EPC count may depend on experimental procedures other than hypoxia, four subjects repeated the same protocol in normoxic control conditions. As shown in Fig. 3A and B, the number of EPCs did not decrease during normoxia.
Figure 2. Impact of hypoxic breathing on EPC count: a fast reduction of EPCs was observed in all subjects.

The concentration of circulating EPCs, identified as either CD45dim/CD34+/KDR+ (A) or CD45dim/CD34+/KDR+/CD133+ (B) cells, rapidly declined upon hypoxia exposure. EPC reduction was already evident in most subjects after 0.5 h and progressed, becoming evident in all subjects, after 1 h of hypoxic breathing. *P < 0.05 and **P < 0.005 compared with pre-hypoxia, as assessed by Wilcoxon matched-pairs signed-ranks test.
Figure 3. Time-course of circulating EPCs in 10 healthy subjects before, during and after 4.

h continuous exposure to hypoxia The concentration of circulating EPCs, identified as either CD45dim/CD34+/KDR+ (A) or CD45dim/CD34+/KDR+/CD133+ (B) cells, decreased in a time-dependent manner during hypoxic breathing and increased progressively during recovery in room air. At the end of the fourth hour in normoxia it still remained lower than that observed at 0 h. The concentration of EPCs did not change in 4 subjects who repeated the same procedure with the hypoxic apparatus switched to normoxia. Data shown as means ± SEM, *P < 0.05 and **P < 0.005 compared with pre-hypoxia; §P < 0.05 compared with 1 h in hypoxia; ‡P < 0.05 compared with 2 h in hypoxia; †P < 0.05 compared with the end of hypoxia, as assessed by Wilcoxon matched-pairs signed-ranks test.
CXCR-4 expression on EPCs
To investigate whether EPC reduction may be related to cell marginalization, EPC expression of CXCR-4, which is critical in EPC mobilization and homing, was determined in six subjects. Upon hypoxia exposure, the expression of CXCR-4 on EPCs underwent a rapid upregulation that was as prompt as the marked initial decrease in EPC count (Fig. 4A)). It significantly increased by +126% 30 min after hypoxia onset, with mean fluorescence intensity (MFI) increasing from 106 ± 8 before hypoxia to 240 ± 14 at 0.5 h (P < 0.05). CXCR-4 expression remained high for the whole period of hypoxia exposure and the following recovery in room air.
Figure 4. Impact of hypoxic breathing on EPC phenotype.
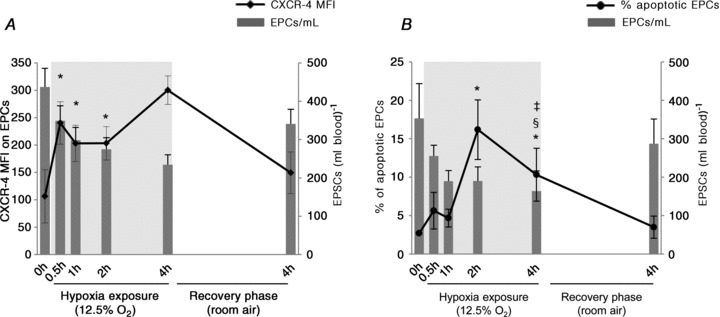
A, upon hypoxia exposure, surface expression of CXCR-4 on EPCs underwent a rapid upregulation that was as prompt as the marked initial decrease in EPC count. B, changes in the frequency of apoptotic circulating EPCs were observed later, with significant increases observed only 2 h and 4 h after starting hypoxic breathing, followed by reversal to initial control values within 4 h of normoxic breathing. Data shown as means ± SEM of 4 independent experiments. *P < 0.05 compared with pre-hypoxia; §P < 0.05 compared with 1 h in hypoxia; ‡P < 0.05 compared with 2 h in hypoxia, as assessed by Wilcoxon matched-pairs signed-ranks test.
Hypoxia-induced EPC apoptosis
In normoxia, apoptotic circulating EPCs, assessed by 7AAD staining, were 2.68 ± 0.10%. As shown in Fig. 4B, compared to pre-exposure, the percentage of apoptotic CD45dim/CD34+/KDR+ cells remained almost constant during the first hour of hypoxic breathing, whereas it significantly increased by 6- and 3-fold, after 2 h and 4 h in hypoxia, respectively (P < 0.05 in both cases). After 4 h normoxic breathing the concentration of apoptotic EPCs was back to initial control values. Similar results were obtained when the count of apoptotic CD45dim/CD34+/KDR+ cells in the blood was considered, with apoptotic EPCs increasing from 10 ± 3 cells ml−1 before hypoxia to 31 ± 12 cells ml−1 after 2 h.
SDF-1 and VEGF plasmatic levels
Soluble circulating SDF-1 increased promptly, although slightly, after hypoxia, with levels increasing from 134 ± 7 pg ml−1 before hypoxia to 152 ± 9 pg ml−1 at 0.5 h (P < 0.05). SDF-1 plasma levels continued to increase with time, attaining the highest value after 4 h hypoxia (182 ± 9 pg ml−1; P < 0.05 compared with 0.5 h). During the following normoxia exposure, the blood concentration of SDF-1 progressively decreased, returning to pre-hypoxia levels after 4 h in normoxia (130 ± 13 pg ml−1). The plasma levels of VEGF did not significantly change in response to hypoxia (57 ± 17 pg ml−1 before hypoxia; 47 ± 11 pg ml−1 after 4 h in hypoxia; 46 ± 10 pg ml−1 after 4 h in normoxia).
Oxidative stress markers
Figure 5 shows the plasma concentration of TBARS and PC during hypoxia and in the following normoxia. Before hypoxia exposure, the plasma concentration of TBARS (Fig. 5A)) was 6.9 ± 0.3 mm. During hypoxia TBARS underwent only a 10% increase at 4 h. By contrast, 1 h after hypoxia offset the plasma concentration of TBARS was significantly (P < 0.005) increased to 9.1 ± 0.6 mm, returning back to pre-hypoxia control value in the subsequent 2 h.
Figure 5. Impact of hypoxic breathing on plasma oxidative stress markers.

Plasmatic concentrations of TBARS, assessed as marker of lipid peroxidation (A) and protein carbonyls (PC), assessed as marker of protein oxidation (B), increased after 4 h of hypoxia exposure and 1 h after its discontinuation, respectively, and returned back to pre-hypoxia control values within 4 h of recovery breathing in room air. Data shown as means ± SEM of 10 independent experiments. *P < 0.05 compared with pre-hypoxia, as assessed by Wilcoxon matched-pairs signed-ranks test.
At 0 h, the blood concentration of protein carbonyl (PC, Fig. 5B)) was 0.7 ± 0.1 nmol (mg protein)−1. During hypoxia, PC concentration significantly increased, attaining the highest value (1.1 ± 0.2 nmol (mg protein)−1, i.e. +75% 0 h value, P < 0.005) at 4 h. At hypoxia offset, PC concentration decreased slowly, being still about 20% higher after 4 h than at 0 h.
Discussion
To our knowledge, this is the first study specifically designed to evaluate the effects of a single 4 h bout of severe hypoxic breathing on circulating EPCs of healthy humans, under controlled resting conditions and in the absence of other confounding factors. The major novel finding of this study is the great reduction in EPC concentration, occurring within 30 min of hypoxic breathing onset and persisting throughout the subsequent 4 h of hypoxia. Moreover, although the mechanism/s underlying the hypoxia-induced reduction in EPCs is/are still unknown, evidence provided in this study suggests that cell marginalization and, at least in part, increased apoptosis of EPCs are likely to play a role in hypoxia-induced EPC disappearance from blood.
EPCs and hypoxia exposure in healthy humans
The rapid and pronounced reduction of EPCs we observed upon hypoxia exposure was clearly evident when EPCs were identified as CD45dim/CD34+/KDR+ cells. This is important because, although a unique consensus on the optimal markers to be used to define EPCs is still lacking, current literature supports the idea that CD45dim/CD34+/KDR+ represents the best antigenic combination to define EPCs in terms of detection accuracy, biological meaning and clinical usefulness (Sandri et al. 2005; Fadini et al. 2007)). In fact, only the level of CD45dim/CD34+/KDR+ cells has been repeatedly and convincingly shown to be an independent predictor of cardiovascular events (Schmidt-Lucke et al. 2005; Werner et al. 2005)). However, in our study we obtained similar results when CD133 was used as an additional marker to restrict EPCs, thus further confirming with a more stringent and rigorous flow cytometric criterion (Urbich & Dimmeler 2004)) that exposure of healthy subjects to systemic hypoxia induces a marked decrease of circulating endothelial progenitors.
To our knowledge, so far few studies have addressed the effects of acute or subacute exposure to hypoxia on EPC homeostasis, yet reporting controversial results. In fact either a reduction (Mancuso et al. 2008)) or an increase (Theiss et al. 2008)) in EPCs was found after a 7–12 day sojourn at altitudes ranging from 1700 to 5000 m. By contrast, a 3 h intermittent hypobaric hypoxia exposure equivalent to 5000 m altitude for three consecutive days failed to change EPC concentration in four subjects (Viscor et al. 2009)), whereas a 1 h bout of normobaric hypoxia equivalent to 4850 m increased EPCs by approximately 70% (Ciulla et al. 2007)). These conflicting results may depend on a variety of factors, including degree and mode of hypoxia exposure, number and homogeneity of investigated subjects, concomitant physical activity, timing of blood sampling, time-span before analysis, analytical procedures and inter-individual variability of the measure.
Our study has the key advantage of examining the impact of systemic hypoxia alone, avoiding the confounding effects of physical exercise or other variables that may affect EPC homeostasis (Mobius-Winkler et al. 2009; Sirker et al. 2009)). Moreover, it clearly indicates that timing of sampling is crucial. In fact, as is shown in Fig. 3 (A and B) within 1 h of hypoxia onset blood concentration of EPCs is more than halved and keeps on going down throughout the ongoing duration of exposure. As soon as subjects’ breathing is switched to normoxia, EPC concentration increases rapidly. Nevertheless, after 4 h EPC concentration is still slightly lower than the pre-hypoxia value.
It was theoretically possible that, due to somewhat elevated blood flow during systemic hypoxia, the total number of circulating EPCs per unit of time was not reduced in our subjects to the same extent as the concentration measures of EPCs indicated. However, the small increase in cardiac output (CI: +11% at 1 h) in comparison to the observed reductions in EPCs (–76% at 4 h) suggests that hypoxia induces indeed a marked reduction in the total number of circulating EPCs.
Overall, these findings raise questions concerning the mechanism/s probably underlying EPC disappearance from blood and its physiological meaning.
Putative mechanism/s involved in hypoxia-induced EPC reduction
Mobilization and homing of EPCs
The present study has shown that in healthy humans acute exposure to hypoxia induced a rapid increase of CXCR-4 expression on circulating EPCs. This finding is in accordance with preclinical observations demonstrating the ability of hypoxia to induce CXCR-4 upregulation on other cell types (Chacko et al. 2010)). Moreover, the rapidity of CXCR-4 upregulation observed in our experimental setting is not surprising. In fact, the surface expression of CXCR-4 is strictly regulated by processes that involve constitutive internalization of CXCR-4 and equilibration with its intracellular pool (Zhang et al. 2004)). A rapid upregulation of CXCR-4, required to allow a rapid response to SDF-1, may then be achieved through CXCR-4 protein stabilization, as suggested elsewhere (Takagi et al. 2009)).
Our study has also shown that acute exposure to hypoxia induced a rapid although mild increase in the plasmatic levels of SDF-1. SDF-1 is transcribed by endothelial cells under hypoxic conditions, so that SDF-1 is supposed to act as a luminal signal that facilitates EPC adhesion to ischaemic endothelium and egress from circulation into ischaemic tissue (Peled et al. 1999; Ceradini et al. 2005)). SDF-1 can be either rapidly expressed upon induction or released from intracellular stores (Jin et al. 2006)). This last mechanism may possibly explain the rapid increase in the plasma levels of SDF-1 that was already evident after 30 min in hypoxia in our study and that probably reflected a prompt increase of SDF-1 in hypoxia-exposed peripheral tissues.
Starting from these observations, several different scenarios can be hypothesized to explain our finding of a prompt EPC decrease in response to hypoxia. One possibility is that the mild hypoxia-induced increase in circulating SDF-1 may be enough to sustain a recruitment of EPCs, although at low rate, from the bone marrow to the blood stream, similar to what has been observed in models of tissue ischaemia. In this respect, it should be noted that the levels of SDF-1 measured upon hypoxia exposure in our subjects were tenfold lower than the levels reported in patients 1 day after acute stroke (Bogoslovsky et al. 2011)), usually associated with massive recruitment of EPCs from the bone marrow into the bloodstream. In line with these observations, we did not observe consensual changes in plasma VEGF. As we observed a rapid increase of CXCR-4 expression on EPCs, it may be possible that while some subtypes of circulating EPCs decreased upon hypoxia exposure, a highly positive CXCR-4 subpopulation was retained in the blood stream. Another possibility is that hypoxia exposure may induce a prompt upregulation of CXCR-4 expression on EPCs present in the blood stream. EPCs that have upregulated the expression of CXCR-4 molecules on their surface may then be recruited from the blood towards tissues expressing high amounts of SDF-1, such as the bone marrow or the tissues most sensitive to low oxygen tension. Because of hypoxia exposure persistence, CXCR-4 upregulation may involve a progressively increasing number of EPCs, which may then be recruited into highly positive SDF-1 tissues, thus possibly sustaining the progressive reduction of EPCs in the blood stream.
Increased apoptosis
Our results indicated that exposure to hypoxia induced a transient increase in apoptotic EPCs, peaking after 2 h and then decreasing slowly in the ongoing hypoxia and subsequent normoxia (shown in Fig. 4B)). This is the first demonstration, to our knowledge, that circulating EPCs are susceptible to hypoxia-induced apoptosis. Indeed, this result is not surprising, as a major proapoptotic effect of hypoxia is commonly observed in most cell types, including endothelial cells (Jin et al. 2009)). EPC apoptosis has been described in metabolic and cardiovascular diseases (Verma et al. 2004; Fadini et al. 2006, 2007). In such conditions, apoptosis probably results from an impaired balance between nitric oxide bioavailability and oxidative stress (Fleissner & Thum, 2010)). The latter may be defined as ‘an imbalance between oxidants and antioxidants in favour of oxidants, leading to disruption of redox signalling and control and/or molecular damage’ (Jones et al. 2006)). Such imbalance may occur also in healthy lowlanders exposed to chronic hypoxia, where indirect evidence of oxidative stress was found at skeletal muscle level (Martinelli et al. 1990; Gelfi et al. 2004)).
Many tissues produce reactive oxygen species (ROS) during reoxygenation after hypoxia. However, recently it has been shown in isolated rat diaphragm strips that a burst of intracellular ROS is formed in the transition to low intracellular O2 tension (Zuo & Clanton, 2005)). The present study provides indirect evidence that a single 4 h hypoxia bout may cause an increase in ROS production also in humans, extending previous observations (Magalhaes et al. 2004)). In fact, compared to control conditions at 0 h, a statistically significant rise in protein carbonyl (PC) content in the blood was observed during hypoxia exposure. By contrast, the increase in TBARS content followed a slower kinetics attaining a peak value only 1 h after return to normoxia. As hypoxia-induced increase in ROS, which are known inducers of apoptosis in many different cell types (Simon et al. 2000)), precedes the appearance of oxidative stress-induced damage (measured as increase in TBARS and PC), the overall findings of our study may suggest a link between oxidative stress and transient EPC apoptosis, although they cannot provide any cause–effect relationship. Since EPC apoptosis, that peaked 2 h after hypoxia onset, was followed by a further mild but significant reduction in EPC concentration (from 2 h to 4 h of hypoxic exposure), it may be hypothesized that EPC apoptosis may contribute to the later phase of hypoxia-induced EPC reduction.
Limitations
We recognize two main limitations of our study. First, because of the experimental procedure used in our study we could not rule out possible effects of hyperventilation-mediated hypocapnia on the observed reduction in EPCs. However, the slow and mild reduction in  (a significant reduction was observed 2 h after hypoxia onset: –12% of the 0 h value), in comparison to the prompt and marked reduction in
(a significant reduction was observed 2 h after hypoxia onset: –12% of the 0 h value), in comparison to the prompt and marked reduction in 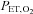 (a significant reduction was observed 1 min after hypoxia onset, and after 30 min it was –50% of the 0 h value) probably suggests that, compared with hypoxia, hypocapnia may play a rather marginal role, if any, in the EPC reduction observed in our study. Future studies enabling normocapnic hypoxia exposure will be needed to definitely clarify this aspect and to further address the complexity of the response to hypoxia. Second, some weakly positive results that we observed in response to hypoxia exposure could be secondary to the mild hypoxaemia reached during the experiments. For instance, it is possible that the oxidative stress markers were only mildly elevated because hypoxic stimulation was insufficient to evoke a larger response. Future dose–response experiments performed in more severe hypoxic conditions may be helpful to better elucidate this point.
(a significant reduction was observed 1 min after hypoxia onset, and after 30 min it was –50% of the 0 h value) probably suggests that, compared with hypoxia, hypocapnia may play a rather marginal role, if any, in the EPC reduction observed in our study. Future studies enabling normocapnic hypoxia exposure will be needed to definitely clarify this aspect and to further address the complexity of the response to hypoxia. Second, some weakly positive results that we observed in response to hypoxia exposure could be secondary to the mild hypoxaemia reached during the experiments. For instance, it is possible that the oxidative stress markers were only mildly elevated because hypoxic stimulation was insufficient to evoke a larger response. Future dose–response experiments performed in more severe hypoxic conditions may be helpful to better elucidate this point.
Physiological meaning and clinical implication
Taken together, the results shown in the present study indicate that a single bout of hypoxia alone induces a rapid disappearance of EPCs from blood, probably sustained, at least in part, by a prompt cell marginalization followed by a late increase in EPC apoptosis. These observations may broaden our understanding of the mechanisms operated by EPCs to maintain endothelial homeostasis in physiological and pathological conditions. Our results may also suggest a possible role of EPCs in hypoxic preconditioning, a pro-adaptive response to mild hypoxic exposure conferring cell protection from subsequent hypoxic or ischaemic insult. In fact, it may be hypothesized that, similarly to what has been demonstrated in a mouse model of myocardial ischaemia (Ii et al. 2005)), during hypoxia exposure EPCs may be rapidly recruited to hypoxic tissues and mediate protective effects. It may be reasoned that the experimental procedure we have adopted in this study may serve as a ‘human in vivo hypoxic preconditioning model’, whose effects in the long run are unknown, therefore deserving further investigations that may help to elucidate the potential role of EPCs in regenerative medicine.
Acknowledgments
This study was supported by grant 2008A3E9WM-003 from Ministero dell’Istruzione, dell’Università e della Ricerca (S.D.B.), and by grant 2008-12 from Ministero lavoro, salute e politiche sociali – Commissione di vigilanza, controllo sul doping e tutela della salute nelle attività sportive (C.M.). E.C., A.T. and M.C. were supported by a fellowship of the Doctorate School of Molecular Medicine, University of Milan.
Glossary
Abbreviations
- 7AAD
7-aminoactinomycin D
- APC
allophycocyanin
- CXCR-4
CXC chemokine receptor-4
- EPC
endothelial progenitor cell
- HIF-1
hypoxia inducible factor-1
- KDR
kinase insert domain receptor
- MFI
mean fluorescence intensity
- SDF-1
stromal derived cell factor-1
- VEGF
vascular endothelial growth factor
Author contributions
Experiments in the manuscript were conducted in the Laboratory of Human Exercise Physiology of the Italian National Research Council and in the Laboratory of Immunology of the School of Medicine of the University of Milan, at Segrate (Milan). All authors approved the final version of the manuscript to be published and all authors contributed to drafting the article and revising it critically for important intellectual content. All authors contributed to the conception and design, or analysis and interpretation of the data in the manuscript.
References
- Aicher A, Zeiher AM, Dimmeler S. Mobilizing endothelial progenitor cells. Hypertension. 2005;45:321–325. doi: 10.1161/01.HYP.0000154789.28695.ea. [DOI] [PubMed] [Google Scholar]
- Asahara T, Kawamoto A. Endothelial progenitor cells for postnatal vasculogenesis. Am J Physiol Cell Physiol. 2004;287:C572–C579. doi: 10.1152/ajpcell.00330.2003. [DOI] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Bahlmann FH, De Groot K, Spandau JM, Landry AL, Hertel B, Duckert T, et al. Erythropoietin regulates endothelial progenitor cells. Blood. 2004;103:921–926. doi: 10.1182/blood-2003-04-1284. [DOI] [PubMed] [Google Scholar]
- Bogoslovsky T, Spatz M, Chaudhry A, Maric D, Luby M, Frank J, Warach S. Stromal-derived factor-1 correlates with circulating endothelial progenitor cells and with acute lesion volume in stroke patients. Stroke. 2011;42:618–625. doi: 10.1161/STROKEAHA.110.596007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceradini DJ, Gurtner GC. Homing to hypoxia: HIF-1 as a mediator of progenitor cell recruitment to injured tissue. Trends Cardiovasc Med. 2005;15:57–63. doi: 10.1016/j.tcm.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- Chacko SM, Ahmed S, Selvendiran K, Kuppusamy ML, Khan M, Kuppusamy P. Hypoxic preconditioning induces the expression of prosurvival and proangiogenic markers in mesenchymal stem cells. Am J Physiol Cell Physiol. 2010;299:C1562–C1570. doi: 10.1152/ajpcell.00221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciulla M, Cortiana M, Silvestris I, Matteucci E, Ridolfi E, Giofrè F, et al. Effects of simulated altitude (normobaric hypoxia) on cardiorespiratory parameters and circulating endothelial precursors in healthy subjects. Respir Res. 2007;8:58. doi: 10.1186/1465-9921-8-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobellis G, Botti C, Taddeo A, Silvestroni A, Lillo S, Da Ponte A, et al. Successful bone marrow transplantation reveals the lack of endothelial progenitor cells mobilization in a patient with critical limb ischemia: a case report. Transplant Proc. 2010;42:2816–2820. doi: 10.1016/j.transproceed.2010.04.047. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Sartore S, Agostini C, Avogaro A. Significance of endothelial progenitor cells in subjects with diabetes. Diabetes Care. 2007;30:1305–1313. doi: 10.2337/dc06-2305. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Schiavon M, Cantini M, Baesso I, Facco M, Miorin M, et al. Circulating progenitor cells are reduced in patients with severe lung disease. Stem Cells. 2006;24:1806–1813. doi: 10.1634/stemcells.2005-0440. [DOI] [PubMed] [Google Scholar]
- Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1α and insulin-like growth factor 2. Cancer Res. 1999;59:3915–3918. [PubMed] [Google Scholar]
- Fleissner F, Thum T. Critical role of the nitric oxide/reactive oxygen species balance in endothelial progenitor dysfunction. Antioxid Redox Signal. 2010;15:933–948. doi: 10.1089/ars.2010.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfi C, De Palma S, Ripamonti M, Eberini I, Wait R, Bajracharya A, et al. New aspects of altitude adaptation in Tibetans: a proteomic approach. FASEB J. 2004;18:612–614. doi: 10.1096/fj.03-1077fje. [DOI] [PubMed] [Google Scholar]
- Harrison JS, Rameshwar P, Chang V, Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood. 2002;99:394. doi: 10.1182/blood.v99.1.394. [DOI] [PubMed] [Google Scholar]
- Hellwig-Burgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin-1β and tumor necrosis factor-α stimulate DNA binding of Hypoxia-inducible factor-1. Blood. 1999;94:1561–1567. [PubMed] [Google Scholar]
- Hu Y, Davison F, Zhang Z, Xu Q. Endothelial replacement and angiogenesis in arteriosclerotic lesions of allografts are contributed by circulating progenitor cells. Circulation. 2003;108:3122–3127. doi: 10.1161/01.CIR.0000105722.96112.67. [DOI] [PubMed] [Google Scholar]
- Ii M, Nishimura H, Iwakura A, Wecker A, Eaton E, Asahara T, Losordo DW. Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via ‘imported’ nitric oxide synthase activity. Circulation. 2005;111:1114–1120. doi: 10.1161/01.CIR.0000157144.24888.7E. [DOI] [PubMed] [Google Scholar]
- Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12:557–567. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, An X, Ye Z, Cully B, Wu J, Li J. RGS5, a hypoxia-inducible apoptotic stimulator in endothelial cells. J Biol Chem. 2009;284:24436–24443. doi: 10.1074/jbc.M109.032664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP. Redifining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- Kunz GA, Liang G, Cuculi F, Gregg D, Vata KC, Shaw LK, et al. Circulating endothelial progenitor cells predict coronary artery disease severity. Am Heart J. 2006;152:190–195. doi: 10.1016/j.ahj.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Leone AM, Valgimigli M, Giannico MB, Zaccone V, Perfetti M, D’Amario D, et al. From bone marrow to the arterial wall: the ongoing tale of endothelial progenitor cells. Eur Heart J. 2009;30:890–899. doi: 10.1093/eurheartj/ehp078. [DOI] [PubMed] [Google Scholar]
- Magalhães J, Ascensão A, Viscor G, Soares J, Oliveira J, Marques F, Duarte J. Oxidative stress in humans during and after 4 h of hypoxia at a simulated altitude of 5500 m. Aviat Space Environ Med. 2004;75:16–22. [PubMed] [Google Scholar]
- Mancuso P, Colleoni M, Calleri A, Orlando L, Maisonneuve P, Pruneri G, et al. Circulating endothelial-cell kinetics and viability predict survival in breast cancer patients receiving metronomic chemotherapy. Blood. 2006;108:452–459. doi: 10.1182/blood-2005-11-4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso P, Peccatori F, Rocca A, Calleri A, Antoniotti P, Rabascio C, et al. Circulating endothelial cell number and viability are reduced by exposure to high altitude. Endothelium. 2008;15:53–58. doi: 10.1080/10623320802092344. [DOI] [PubMed] [Google Scholar]
- Martinelli M, Winterhalder R, Cerretelli P, Howald H, Hoppeler H. Muscle lipofuscin content and satellite cell volume is increased after high altitude exposure in humans. Experientia. 1990;46:672–676. doi: 10.1007/BF01939930. [DOI] [PubMed] [Google Scholar]
- Massa M, Rosti V, Ferrario M, Campanelli R, Ramajoli I, Rosso R, et al. Increased circulating hematopoietic and endothelial progenitor cells in the early phase of acute myocardial infarction. Blood. 2005;105:199–206. doi: 10.1182/blood-2004-05-1831. [DOI] [PubMed] [Google Scholar]
- Mobius-Winkler S, Hilberg T, Menzel K, Golla E, Burman A, Schuler G, Adams V. Time-dependent mobilization of circulating progenitor cells during strenuous exercise in healthy individuals. J Appl Physiol. 2009;107:1943–1950. doi: 10.1152/japplphysiol.00532.2009. [DOI] [PubMed] [Google Scholar]
- O’Neil TJ, Wamhoff BR, Owens GK, Skalak TC. Mobilization of bone marrow-derived cells enhances the angiogenic response to hypoxia without transdifferentiation into endothelial cells. Circ Res. 2005;97:1027–1035. doi: 10.1161/01.RES.0000189259.69645.25. [DOI] [PubMed] [Google Scholar]
- Palange P, Testa U, Huertas A, Calabrò L, Antonucci R, Petrucci E, et al. Circulating haemopoietic and endothelial progenitor cells are decreased in COPD. Eur Resp J. 2006;27:529–541. doi: 10.1183/09031936.06.00120604. [DOI] [PubMed] [Google Scholar]
- Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, et al. Expression of VEGFR-2 and AC133 by circulating human CD34+ cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–958. [PubMed] [Google Scholar]
- Peled A, Grabovsky V, Habler L, Sandbank J, Arenzana-Seisdedos F, Petit I, et al. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34+ cells on vascular endothelium under shear flow. J Clin Invest. 1999;104:1199–1211. doi: 10.1172/JCI7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pialoux V, Mounier R, Brown AD, Steinback CD, Rawling JM, Poulin MJ. Relationship between oxidative stress and HIF-1α mRNA during sustained hypoxia in humans. Free Radic Biol Med. 2009;46:321–326. doi: 10.1016/j.freeradbiomed.2008.10.047. [DOI] [PubMed] [Google Scholar]
- Sandau KB, Fandrey J, Brune B. Accumulation of HIF-1α under the influence of nitric oxide. Blood. 2001;97:1009–1015. doi: 10.1182/blood.v97.4.1009. [DOI] [PubMed] [Google Scholar]
- Sandri M, Adams V, Gielen S, Linke A, Lenk K, Kränkel N, et al. Effects of exercise and ischemia on mobilization and functional activation of blood-derived progenitor cells in patients with ischemic syndromes: results of 3 randomized studies. Circulation. 2005;111:3391–3399. doi: 10.1161/CIRCULATIONAHA.104.527135. [DOI] [PubMed] [Google Scholar]
- Schatteman GC, Hanlon HD, Jiao C, Dodds SG, Christy BA. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest. 2000;106:571–578. doi: 10.1172/JCI9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Lucke C, Rossig L, Fichtlscherer S, Vasa M, Britten M, Kamper U, et al. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005;111:2981–2987. doi: 10.1161/CIRCULATIONAHA.104.504340. [DOI] [PubMed] [Google Scholar]
- Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- Sirker AA, Astroulakis ZMJ, Hill JM. Vascular progenitor cells and translational research: the role of endothelial and smooth muscle progenitor cells in endogenous arterial remodelling in the adult. Clin Sci. 2009;116:283–299. doi: 10.1042/CS20080001. [DOI] [PubMed] [Google Scholar]
- Taddeo A, Presicce P, Brambilla L, Bellinvia M, Villa ML, Della Bella S. Circulating endothelial progenitor cells are increased in patients with classic Kaposi's sarcoma. J Invest Dermatol. 2008;128:2125–2128. doi: 10.1038/jid.2008.23. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, et al. Erythromycin-induced CXCR4 expression on microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L420–L431. doi: 10.1152/ajplung.90477.2008. [DOI] [PubMed] [Google Scholar]
- Theiss HD, Adam M, Greie S, Schobersberger W, Humpeler E, Franz WM. Increased levels of circulating progenitor cells after 1-week sojourn at moderate altitude. Respir Physiol Neurobiol. 2008;160:232–238. doi: 10.1016/j.resp.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- Valgimigli M, Rigolin GM, Fucili A, Porta MD, Soukhomovskaia O, Malagutti P, et al. CD34+ and endothelial progenitor cells in patients with various degrees of congestive heart failure. Circulation. 2004;110:1209–1212. doi: 10.1161/01.CIR.0000136813.89036.21. [DOI] [PubMed] [Google Scholar]
- Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:e1–e7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- Verma S, Kuliszewski MA, Li SH, Szmitko PE, Zucco L, Wang CH, et al. C-reactive protein attenuates endothelial progenitor cell survival, differentiation, and function: further evidence of a mechanistic link between C-reactive protein and cardiovascular disease. Circulation. 2004;109:2058–2067. doi: 10.1161/01.CIR.0000127577.63323.24. [DOI] [PubMed] [Google Scholar]
- Viscor G, Javierre C, Pagès T, Ventura JL, Ricart A, Martin-Henao G, et al. Combined intermittent hypoxia and surface muscle electrostimulation as a method to increase peripheral blood progenitor cell concentration. J Transl Med. 2009;7:91. doi: 10.1186/1479-5876-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmann S, Werner N, Czech T, Nickenig G. Improvement of endothelial function by systemic transfusion of vascular progenitor cells. Circ Res. 2006;99:e74–e83. doi: 10.1161/01.RES.0000246095.90247.d4. [DOI] [PubMed] [Google Scholar]
- Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- Xiao Q, Ye S, Oberhollenzer F, Mayr A, Jahangiri M, Willeit J, et al. SDF1 gene variation is associated with circulating SDF1α level and endothelial progenitor cell number – the Bruneck Study. PloS ONE. 2008;3:e4061. doi: 10.1371/journal.pone.0004061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentilin L, Tafuro S, Zacchigna S, Arsic N, Pattarini L, Sinigaglia M, Giacca M. Bone marrow mononuclear cells are recruited to the sites of VEGF-induced neovascularization but are not incorporated into the newly formed vessels. Blood. 2006;107:3546–3554. doi: 10.1182/blood-2005-08-3215. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Foudi A, Geay J-F, Berthebaud M, Buet D, Jarrier P, et al. Intracellular localization and constitutive endocytosis of CXCR4 in human CD34+ hematopoietic progenitor cells. Stem Cells. 2004;22:1015–1029. doi: 10.1634/stemcells.22-6-1015. [DOI] [PubMed] [Google Scholar]
- Zuo L, Clanton TL. Reactive oxygen species formation in the transition to hypoxia in skeletal muscle. Am J Physiol Cell Physiol. 2005;289:C207–C216. doi: 10.1152/ajpcell.00449.2004. [DOI] [PubMed] [Google Scholar]