

Abstract
Non-technical summary
The enteric nervous system (ENS) is an autonomous nervous system integrated along the gut that controls major gastrointestinal (GI) functions such as motility. Increasing data have demonstrated that nutritional factors can modulate the ENS phenotype and consequently impact upon GI functions. Western diet is central in the development of obesity but surprisingly no study has characterized its impact upon ENS phenotype and functions. We show that Western diet-induced obesity (DIO) prevented age-associated loss in a specific population of enteric neurons leading to an acceleration of gastric emptying. In addition, we showed that neuroprotective effects of DIO likely involved leptin and glial cell line-derived neurotrophic factor (GDNF). This is the first study demonstrating an impact of DIO upon the ENS. These DIO-induced neuroplastic changes in the ENS could be involved in the physiopathology of obesity.
Abstract
Nutritional factors can induce profound neuroplastic changes in the enteric nervous system (ENS), responsible for changes in gastrointestinal (GI) motility. However, long-term effects of a nutritional imbalance leading to obesity, such as Western diet (WD), upon ENS phenotype and control of GI motility remain unknown. Therefore, we investigated the effects of WD-induced obesity (DIO) on ENS phenotype and function as well as factors involved in functional plasticity. Mice were fed with normal diet (ND) or WD for 12 weeks. GI motility was assessed in vivo and ex vivo. Myenteric neurons and glia were analysed with immunohistochemical methods using antibodies against Hu, neuronal nitric oxide synthase (nNOS), Sox-10 and with calcium imaging techniques. Leptin and glial cell line-derived neurotrophic factor (GDNF) were studied using immunohistochemical, biochemical or PCR methods in mice and primary culture of ENS. DIO prevented the age-associated decrease in antral nitrergic neurons observed in ND mice. Nerve stimulation evoked a stronger neuronal Ca2+ response in WD compared to ND mice. DIO induced an NO-dependent increase in gastric emptying and neuromuscular transmission in the antrum without any change in small intestinal transit. During WD but not ND, a time-dependent increase in leptin and GDNF occurred in the antrum. Finally, we showed that leptin increased GDNF production in the ENS and induced neuroprotective effects mediated in part by GDNF. These results demonstrate that DIO induces neuroplastic changes in the antrum leading to an NO-dependent acceleration of gastric emptying. In addition, DIO induced neuroplasticity in the ENS is likely to involve leptin and GDNF.
Introduction
The enteric nervous system (ENS) plays a central role in the control of gastrointestinal (GI) functions both in health and in diseases. While submucosal neurons are major regulators of mucosal functions, myenteric neurons are involved in the control of GI motility (Kunze & Furness, 1999)). The enteric circuit responsible for peristalsis is characterized by a polarized innervation involving ascending excitatory neurons and descending inhibitory neurons. Excitatory neurons are cholinergic and colocalize often substance P while inhibitory neurons synthesize neuronal nitric oxide (nNOS) and also vasoactive intestinal peptide or ATP (Kunze & Furness, 1999)). These mediators are central in controlling motility and changes in their expression are often responsible for or associated with GI dysmotility (Di Nardo et al. 2008)).
Changes in the expression of neuromediators can occur under physiological conditions such as growth or ageing. In particular, during the postnatal period, age-associated increase in the proportion of choline acetyltransferase (ChAT) immunoreactivity in myenteric neurons and in the vesicular acetylcholine transporter immunoreactivity in fibres occurred and was associated with the development of colonic motility in mouse and rats (Roberts et al. 2007; de Vries et al. 2010)). Conversely, during ageing, loss of nitrergic and cholinergic neurons has been reported (Takahashi et al. 2000; Phillips, 2003)). ENS phenotype can also be modulated by environmental factors of both endogenous or exogenous (luminal) origin. In particular, cellular constituents of the neuronal environment such as immune cells, enteric glia or even intestinal epithelial cells can directly modulate the expression of key neuromediators or enzymes in enteric neurons and impact on GI motility (Schemann et al. 2005; Aubéet al. 2006; Moriez et al. 2009)).
In contrast, much less is known about the role of luminal factors, in particular of nutritional origin, in the control of ENS neurochemical coding. A recent study demonstrated that such an influence as butyrate increased the proportion of cholinergic neurons and cholinergic neuromuscular transmission, leading to enhanced colonic transit (Soret et al. 2010)). However, effects of long-term exposure to other dietary factors on ENS phenotype remain poorly documented. In particular, whether diet leading to obesity, such as Western diet (WD), high in saturated fatty acid and in simple carbohydrate, can lead to neuroplastic changes in the ENS and whether these changes could impact on GI motility remains unknown.
Sensing of dietary fat in the gut has been shown to regulate GI functions. The modulation of this sensing by diet could favour GI dysfunctions observed during obesity and also play a role in the development of obesity (Little & Feinle-Bisset, 2010)). In particular, long-term consumption of high-fat diet has been shown to enhance gastric emptying of solid meals. In healthy volunteers, gastric emptying of a high-fat test meal is faster after 14 days of high-fat diet than before this diet (Castiglione et al. 2002)). Similarly, gastric emptying is increased in patients receiving a high-fat diet as compared to patients eating a low-fat diet (Cunningham et al. 1991)). However, these data are still scarce and only a few animal studies describing the impact of high-fat diet upon gastric functions are available. For example, in rats, after exposure to high-fat diet for 14 days, the inhibitory effect of small intestinal infusion of oleate on gastric emptying is attenuated when compared with rats consuming a low-fat diet (Covasa & Ritter, 2000)). Furthermore, the mechanisms and factors responsible for putative functional changes induced by WD remain to be identified.
Leptin is a likely candidate involved in mediating these functional changes (Martínez et al. 1999)). Furthermore, circulating leptin levels are elevated during obesity and originate mainly from white adipose tissue (Considine et al. 1996)). However, whether changes in leptin also occur in the stomach during diet-induced obesity (DIO) remains unknown. In addition, although leptin-induced changes in gastric functions occur in part via a modulation of vagal afferent (Cakir et al. 2007)), the impact of leptin upon the ENS remains unknown. Although leptin has been shown to increase activity in intestinal enteric neurons (Liu et al. 1999; Reichardt et al. 2011)), its effects upon ENS phenotype and function in the stomach remain to be identified.
In the present study, we sought to characterize the impact of DIO upon the phenotype of myenteric neurons in the antrum and the jejunum and its functional impact upon motility as well as to identify factors involved in these modifications.
Methods
Ethical approval
Experiments were performed in accordance with an experimental protocol for animal study approved by the Inserm Institutional Animal Care and Use Committee.
Experimental protocol
Male C57BL/6J Rj mice aged 4 weeks (Janvier Laboratory, Le Genest-Saint-Isle, France) were maintained on a 12 h light–12 h dark cycle (22°C) and had free access to food and water. As described previously (Begriche et al. 2008)), after a 1 week adaptation period, animals were randomly assigned to receive for 12 weeks either a normal chow (ND group, Purified diet 210, SAFE, Augy, France) or a Western diet (WD group, Purified diet 230 HF, SAFE). Body weight and food intake were measured weekly. Fasting glycaemia was measured in tail vein blood samples using a glucometer (Roche, Paris, France). An oral glucose tolerance test (OGTT) was performed as previously described (Andrikopoulos et al. 2008)). Plasma insulin and leptin levels were measured using radioimmunoassay kits (RI-13K and ML-82K, respectively, Linco Research, St Charles, MO, USA). db/db mice (12 weeks old, Janvier) were also used. Mice were killed by cervical dislocation.
In vivo and ex vivo measurement of gastrointestinal transit and contractile activity
Gastric emptying (GE) and small intestinal transit were measured in vivo as described in the online Supplemental Material. For pharmacological characterization of GE, mice were intraperitonealy (i.p.) injected with either 0.9% sodium chloride (saline) or the nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester (l-NAME, 50 mg kg−1; Sigma), as previously described (De Winter et al. 2002)). Ex vivo neuromuscular transmission in circular muscle of antrum was measured in an organ bath chamber (Supplemental Material).
Calcium imaging of antral myenteric neurons
Longitudinal muscle myenteric plexus (LMMP; 10 × 20 mm) preparations of antrum were continuously perfused with ice cold Krebs solution following staining with the calcium-sensitive probe Fluo-4 AM (Supplemental Material). The impact of electrical stimulation of interganglionic fibre tract on intracellular calcium concentration in ganglionic structures was recorded by an ultra-fast neuroimaging technique (Supplemental Material). After the neuroimaging experiments, tissues were fixed and immunohistochemistry was performed to characterize whether responding cells were neurons or glial cells (Supplemental Material).
Immunohistochemical analysis
The stomach and about 2 cm of mid-jejunum were detubulized, stretched and fixed overnight in phosphate buffered saline (PBS) containing 4% paraformaldehyde. After microdissection, whole-mounts were permeabilized for 1 h in PBS/NaN3 containing 1% Triton X-100 and 10% donkey serum and incubated overnight at 4°C with primary antibodies (Supplemental Table 1)). After a washing step, whole-mounts were incubated for 1 h at room temperature with secondary antibodies (Supplemental Table 1)). Preparations were mounted on glass slides and viewed under a fluorescence microscope (Olympus IX50, France). Images were acquired with a digital camera (Olympus DP71) and analysed with the Cell B software. As previously described (Hoff et al. 2008)), two different ganglia were defined as two separate entities if their borders were separated by a gap of at least two neurons in width. The number of Hu- and nNOS-immunoreactive (IR) neurons as well as Sox-10-IR glial cells was counted in at least 20 randomly selected ganglia/tissue/mouse. In order to exclude that the difference between groups could be influenced by tissue compliance, we showed in preliminary experiments that neither ganglionic area nor number of neurons per ganglion was modified by the amplitude of stretch applied to the tissue prior fixation.
Determination of acetylcholine, GDNF and leptin levels
Specimens of antrum were placed in Lysing Matrix D tubes (MPBio, Thüringer, Germany) containing 500 μl of RIPA (Millipore, Temecula, CA, USA) with protease inhibitors (Roche Diagnostics, Indianapolis, IN, USA) and homogenized with a Precellys 24 (Bertin Technologies, Montigny-le-Bretonneux, France). The homogenate was centrifuged and the supernatant collected and stored at −80°C. Acetylcholine, glial cell line-derived neurotrophic factor (GDNF) or leptin levels were measured using Amplex Red (Molecular Probes, Eugene, OR, USA), GDNF Emax (Promega, Madison, WI, USA) or leptin Quantikine (R&D Systems, Mineapolis, MN, USA), respectively and normalized to protein concentration.
Role of leptin in modulation of GDNF production and in neuroprotection in the ENS
Effects of leptin on GDNF mRNA levels and release Primary cultures of rat ENS, obtained as previously described, were used at 12–13 days after culture (Chevalier et al. 2008)). Primary cultures were incubated for 24 h with various concentrations of recombinant rat leptin (R&D Systems). GDNF levels were measured in supernatants of culture using the Emax ImmunoAssay System (Promega). Quantitative PCR of GDNF mRNA expression was performed, as previously described (Soret et al. 2010)) using the following primers:
S6 (forward 5′-CCAAGCTTATTCAGCGTCTTGTTAACTCC-3′; reverse 5′-CCCTCGAGTCCTTCATTCTCTTGGC-3′);
GDNF (forward 5′-GCTGCCCGCCGGTAAGAG-3′; reverse 5′-TGGTGGCTTGAATAAAATCCATGAC-3′).
mRNA levels were quantified using the 2−ΔΔCt method (Livak & Schmittgen, 2001)) with S6 as the internal control.
Neuroprotective effects of leptin Primary cultures of ENS were exposed for 24 h to recombinant rat leptin (100 ng ml−1) in the presence or absence of neutralizing antibody against rat GDNF (1 μg ml−1; R&D Systems) or to recombinant rat GDNF (100 ng ml−1, R&D Systems). Primary cultures were fixed and stained with antibodies against Hu C/D and active caspase-3 (Supplemental Table 1)), as previously described (Abdo et al. 2010)). The number of active caspase-3- IR cells and Hu-IR cells was counted in 15 random ganglia for each well.
Statistical analysis
Values are expressed as means ± standard error of the mean (SEM). The Mann–Whitney U test, the Kruskall–Wallis test followed by Dunn's multiple comparison test or two-way ANOVA followed by a Bonferonni post hoc test was performed using GraphPad Prism 5.00 (GraphPad Software Inc., La Jolla, CA, USA). Results were considered statistically significant when P< 0.05.
Results
Characterization of the diet-induced obesity model
WD mice had a significant greater caloric intake than ND mice (+43%, n =26, P < 0.0001) (Table 1)). This caloric intake resulted in a significant increase in weight gain as early as 2 weeks after the beginning of WD as compared to ND. After 12 weeks, WD mice had a 61% higher weight (n= 37–38, P < 0.0001) than ND mice (Table 1)). At the end of the diet, WD mice had increased fat mass (+487%, n= 35–37, P < 0.0001), leptinaemia (+129%, n= 6, P= 0.002), and insulinaemia (+154%, n= 6, P= 0.005) without any change in fasting glycaemia (n= 20, P= 0.09) as compared to ND mice (Table 1)). However, after 4 and 8 weeks of diet, OGTT showed that the area under the glucose response curve was significantly higher in WD as compared to ND mice (Table 1, +24%, n= 19–20, P < 0.0001 and +25%, n= 9–10, P= 0.003, respectively).
Table 1.
Characterization of the diet-induced obesity model
| ND | WD | ||
|---|---|---|---|
| Daily food intake | (g) | 19.4 ± 0.6 | 17.9 ± 0.5 |
| (kcal) | 66.6 ± 1.9 | 95.5 ± 2.8 | |
| Body weight (g) | 0 week of diet | 18.3 ± 0.2 | 18.2 ± 0.2 |
| 12 weeks of diet | 24.4 ± 0.25 | 39.2 ± 0.8 | |
| Epididymal WAT (g) | 0.24 ± 0.01 | 1.41 ± 0.1 | |
| Plasma glucose (mg dl−1) | 108.2 ± 1.9 | 102.8 ± 2.3 | |
| Plasma leptin (ng ml−1) | 0.75 ± 0.07 | 1.72 ± 0.19 | |
| Plasma insulin (ng ml−1) | 0.28 ± 0.05 | 0.71 ± 0.06 | |
| OGTT: AUC glucose (mg dl−1× 120 min) | 4 weeks of diet | 22422 ± 753 | 27707 ± 614 |
| 8 weeks of diet | 23430 ± 1283 | 29407 ± 1207 |
WAT: white adipose tissue; OGTT: oral glucose tolerance test; AUC glucose: area under the glucose response curve. Values are expressed as means ± SEM. *P < 0.05, Mann–Whitney test.
Diet-induced obesity modulated ENS phenotype in the antrum
Changes in total neuronal population In order to determine the effects of DIO upon ENS phenotype, the number of Hu-IR neurons per ganglion was quantified before diet (T0) and after 12 weeks of WD or ND (Fig. 1A and B)). The total number of Hu-IR neurons per ganglion decreased with time between T0 and the end of ND (Fig. 1B, −36%, n= 5–7, P < 0.05). In contrast, DIO significantly prevented this decrease (Fig. 1A and B, n= 5–8, P > 0.05).
Figure 1. Impact of diet-induced obesity upon ENS phenotype in antrum and jejunum.
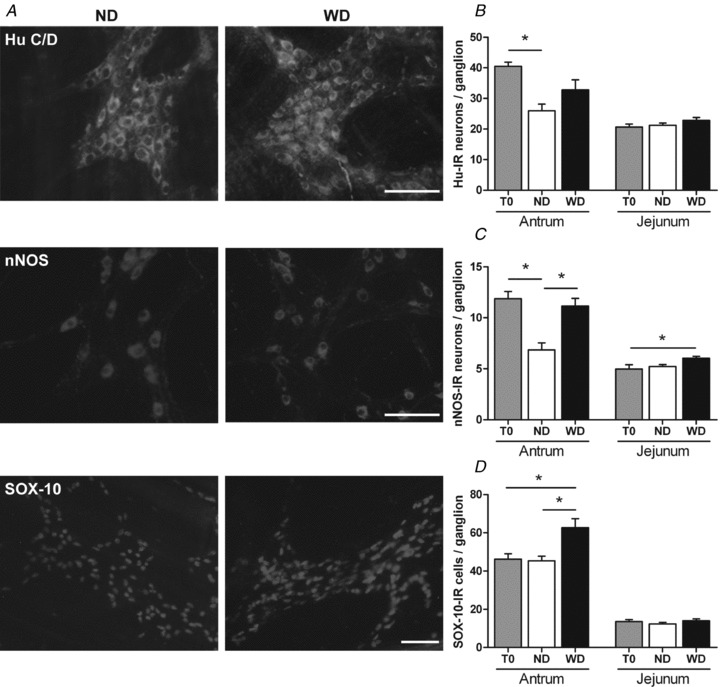
A, immunohistochemical labelling of antral myenteric plexus in mice after 12 weeks of normal diet (ND) and Western diet (WD). Myenteric neurons were stained with anti-Hu C/D and anti-nNOS antibodies. Enteric glial cells were stained with anti-Sox-10 antibody. Scale bar = 50 μm. B–D, quantitative analysis of the number of Hu-IR neurons (B), nNOS-IR neurons (C) and Sox-10-IR cells (D) per ganglion was performed in antrum and jejunum in mice before the beginning of the diet (T0, n= 5) and in mice after 12 weeks of ND or WD (n= 7–11). Values are expressed as means ± SEM. *P < 0.05, Kruskall–Wallis test.
Changes in nitrergic population The number of nNOS-IR neurons per ganglion decreased between T0 and the end of ND (Fig. 1C, −42%, n= 5–7, P < 0.05). DIO prevented this decrease in nNOS-IR neurons (Fig. 1C, n= 5–8, P > 0.05). This resulted in a significantly larger number of nitrergic neurons per ganglion at the end of WD than ND (Fig. 1A and C, +63%, n= 7–8, P < 0.05). Concerning the cholinergic population, the faint staining of ChAT-IR neurons and the dense innervation of ChAT-IR fibres in ganglia prevented the precise analysis of this neuronal population (Supplemental Fig. 1)). Therefore, acetylcholine content was measured in the antrum. At the end of diet, acetylcholine level was similar between ND and WD mice (8.9 ± 1.8 and 12.7 ± 2.8 μmol (g of protein)−1, respectively, n= 10, P= 0.45).
µCa2+½i transients in antral myenteric neurons In order to assess differences in the degree of activation, we recorded µCa2+½i transients in response to electrical train pulse stimulation of interganglionic fibre tracts. The stimulation evoked a slowly developing increase in µCa2+½i with superimposed individual peaks (Fig. 2A)). These peaks correspond to action potential discharge (Shuttleworth & Smith, 1999; Michel et al. 2011)). The number of peaks was similar in ND and WD mice (ND: 9.7 ± 0.1 in 30 cells, 8 ganglia and 4 animals vs. WD: 9.8 ± 0.2 in 19 cells, 7 ganglia and 4 animals, P= 0.28). The maximal amplitude of the slow increase in µCa2+½i was 53% higher in myenteric cells of WD as compared to ND mice (P < 0.001) (Fig. 2B)). Immunohistochemical staining of PGP 9.5 and S-100 revealed immunoreactivity in 12 out of 35 cells from 5 tissues (3 ND and 2 WD mice). In the remaining 23 cells, the immunoreactivity was too faint to perform reliable analysis. All cells which responded to fibre tract stimulation with an increase in µCa2+½i associated with peaks were identified as neurons (10 PGP 9.5 positive cells).
Figure 2. Impact of diet-induced obesity upon µCa2+½i in response to electric stimulation of interganglionic fibre tracts in myenteric neurons.
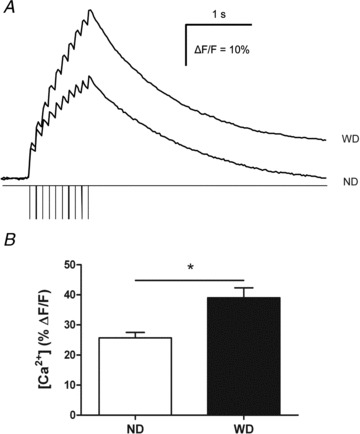
A, typical recordings showing calcium responses in a myenteric neuron from normal diet (ND) mice and from Western diet (WD) mice during and following interganglionic fibre tract train pulse stimulation. Calcium peaks can be observed during each electrical pulse. B, percentage of changes of resting fluorescence (ΔF/F) induced by train pulse stimulations of interganglionic fibre tracts in cells of the myenteric plexus (ND: n= 30 cells, 8 ganglia, 4 mice and WD: n =19 cells, 7 ganglia, 4 mice). Values are expressed as means ± SEM. *P < 0.05, Mann–Whitney test.
Changes in enteric glial cells Lastly, the number of Sox-10-IR cells per ganglion in the antrum was similar between T0 and the end of ND (Fig. 1D, n= 4–7, P > 0.05). However, after WD, the number of Sox-10-IR cells was significantly increased as compared to ND and T0 (Fig. 1A and D, +36% and +38%, respectively, n= 4–7, P < 0.05).
Changes in ENS phenotype in the jejunum In jejunum, no difference was observed in the number of Hu-IR neurons between the different groups (Fig. 1B, n= 5–11, P > 0.05). However, there was an increase in the number of nNOS-IR neurons per ganglion at the end of WD as compared to T0 (Fig. 1C, +21%, n= 5–11, P < 0.05). Lastly, the number of Sox-10-IR cells was similar in the different groups of mice (Fig. 1D, n= 5–11, P > 0.05).
Diet-induced obesity increased gastric emptying and nitrergic neuromuscular transmission in antrum
In order to determine the putative functional impact of DIO-associated neuroplastic changes in the antrum, gastric emptying and neuromuscular transmission in the antrum were evaluated.
In vivo study At the end of diet, GE was significantly increased by 39% (n= 16–17, P < 0.05) in WD as compared to ND mice (Fig. 3A)). l-NAME significantly reduced GE in WD (–34%, n= 7, P < 0.05) but not in ND mice (n= 9, P > 0.05) (Fig. 3A)). Saline did not modify GE (data not shown). In contrast, small intestinal transit was similar in ND and WD mice (Fig. 3B, n= 10, P= 0.36).
Figure 3. Impact of diet-induced obesity upon gastric emptying, small intestinal transit and neuromuscular transmission in antrum.
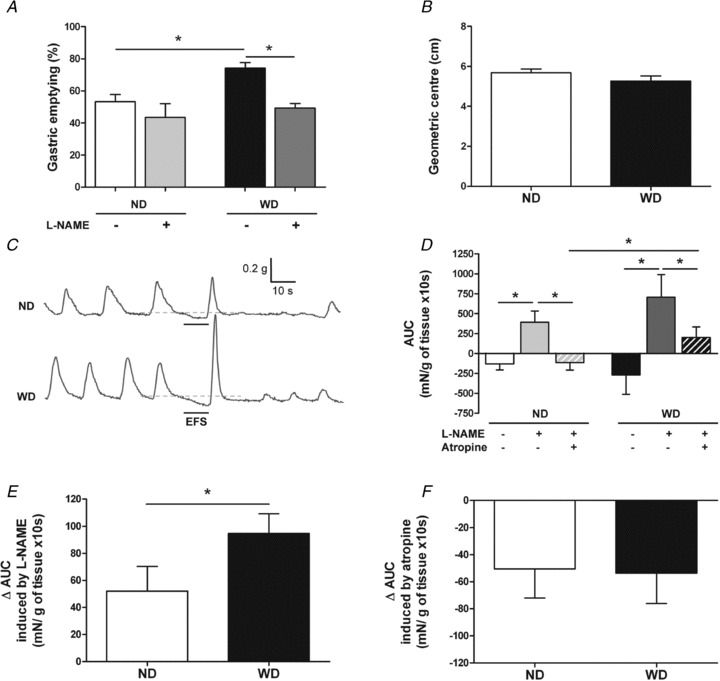
A, gastric emptying of solids in mice after 12 weeks of ND or WD following intraperitoneal injection of saline (n= 16–17) or l-NAME solution (n= 7–9). Values are expressed as means ± SEM. *P < 0.05, Kruskall–Wallis test. B, small intestinal transit in mice after 12 weeks of ND or WD (n= 10). Values are expressed as means ± SEM. *P < 0.05, Mann–Whitney test. C, typical recordings showing EFS-mediated contractile responses in ND and WD mice in basal conditions. D, quantitative analysis of the area under the curve (AUC) measured during EFS in ND and WD mice in basal conditions, in the presence of l-NAME and in the presence of l-NAME and atropine (n= 25–27). Values are expressed as means ± SEM. *P < 0.05, Kruskall–Wallis test. E, quantitative analysis of l-NAME sensitive AUC (ΔAUC; difference between AUC in the presence of l-NAME and AUC without drug) in ND and WD mice (n= 25–27). Values are expressed as means ± SEM. *P < 0.05, Mann–Whitney test. F, quantitative analysis of atropine sensitive AUC (ΔAUC; difference between AUC in the presence of l-NAME and atropine and AUC in the presence of l-NAME) in ND and WD mice (n= 25–27). Values are expressed as means ± SEM. *P < 0.05, Mann–Whitney test.
Ex vivo study In basal conditions, electrical field stimulation (EFS) induced a small decrease in basal tone in antral circular muscle strips (Fig. 3C)). The area under the curve (AUC) of the EFS-induced response showed no significant difference between ND and WD mice (Fig. 3C and D, n= 25–27, P= 0.24). In both groups, EFS-induced decrease in basal tone was significantly inhibited by l-NAME (Fig. 3D)). However, the l-NAME sensitive component of EFS-induced AUC was significantly larger in WD as compared to ND mice (Fig. 3E)). Further addition of atropine significantly reduced EFS-induced AUC in both groups. In this latter condition, EFS-induced AUC was significantly larger in WD as compared to ND (Fig. 3D)). However, the atropine sensitive component of EFS-induced AUC was similar in both groups (Fig. 3F)).
Responses to carbachol and sodium nitroprusside (SNP) were similar in both groups (Supplemental Fig. 2, n= 25, P > 0.05 and n= 7–10, P > 0.05, respectively).
Diet-induced obesity increased expression of GDNF in the antrum
We next determined whether DIO-induced prevention of age-associated changes in the ENS could be associated with increased expression of neuroprotective factors such as GDNF. In ND mice, antral GDNF concentration remained constant over the duration of diet (Fig. 4A, n= 5–10, P > 0.05). In contrast, DIO induced a time-dependent increase in GDNF concentration as early as after 8 weeks of diet (Fig. 4A)). At the end of diet, GDNF concentration was significantly increased by 65% (n= 10, P < 0.05) in WD as compared to ND mice (Fig. 4A)). Immunohistochemical analysis revealed GDNF and Ret expression at the level of neurons while GFRα1 expression was found both in neurons and glial cells (Supplemental Fig. 3)).
Figure 4. Impact of diet-induced obesity upon GDNF and leptin content in antrum. Involvement of leptin in the regulation of GDNF production.
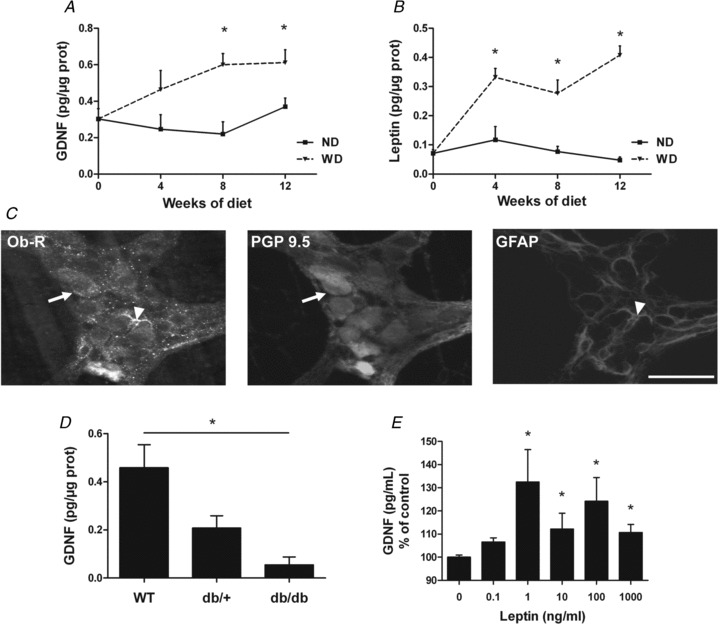
A, evolution of GDNF concentration in antrum in mice before diet (n= 5), and after 4 weeks (n= 5), 8 weeks (n= 5) and 12 weeks (n= 10) of normal diet (ND) or Western diet (WD). Values are expressed as means ± SEM. *P < 0.05, two-way ANOVA. B, evolution of leptin concentration in antrum in mice before diet (n= 5), and after 4 weeks (n= 5), 8 weeks (n= 5) and 12 weeks (n= 10) of ND or WD. Values are expressed as means ± SEM. *P < 0.05, two-way ANOVA. C, immunohistochemical labelling of antral myenteric plexus with anti-ObR, anti-PGP 9.5 and anti-GFAP antibodies. ObR-IR cells coexpressed PGP 9.5 (arrows) and GFAP (arrowheads). Scale bar = 50 μm. D, GDNF concentration in antrum in 12 week-old WT, db/+ and db/db mice (n= 4–7). Values are expressed as means ± SEM. *P < 0.05, Kruskall–Wallis test. E, GDNF concentration in supernatants of primary cultures of ENS treated with different doses of leptin for 24 h, normalized to control (n= 14–23; control 69.5 ± 5.4 pg ml−1). Values are expressed as means ± SEM. *P < 0.05 vs. control, Kruskall–Wallis test.
Leptin was involved in the regulation of GDNF production
In parallel to changes in GDNF concentration, we also characterized the impact of DIO upon leptin. There was no time-dependent change in leptin concentration in the antrum during ND (Fig. 4B, n= 5–10, P > 0.05). In contrast, DIO induced a time-dependent increase in leptin concentration as early as after 4 weeks of diet (Fig. 4B)). At the end of diet, leptin concentration was increased by 753% (n= 10, P < 0.001) in WD as compared to ND mice (Fig. 4B)).
Consequently, we tested the hypothesis that leptin could be involved in the regulation of GDNF production. Leptin receptors were expressed both in neurons and in some glial cells in antrum (Fig. 4C)) and in primary culture of ENS. In db/db mice (devoid of leptin receptors), we showed that antral GDNF concentration was significantly reduced as compared to WT mice (Fig. 4D, −88%, n= 4–7, P < 0.05). Furthermore, leptin induced a significant increase in GDNF content in supernatants of primary culture of ENS (Fig. 4E, n =14–23, P < 0.05). Leptin (1 ng ml−1) also induced a significant increase in GDNF mRNA expression as compared to control (3.4 ± 0.7 vs. 1.2 ± 0.3, respectively, n =5, P =0.03).
Leptin had neuroprotective effects via GDNF
Finally, neuroprotective effects of leptin, and GDNF involvement in these effects, were investigated. Leptin significantly reduced by 86% (n= 10–12, P < 0.05) the proportion of active caspase-3-IR neurons in primary culture of ENS as compared to control conditions (Fig. 5A)). The effect of leptin was abolished with anti-GDNF neutralizing antibody (Fig. 5A, n= 10–13, P > 0.05). Finally, we showed that GDNF significantly reduced by 91% (n= 5–7, P= 0.03) the proportion of active caspase-3-IR neurons as compared to control (Fig. 5B)).
Figure 5. Involvement of GDNF in neuroprotective effects of leptin.
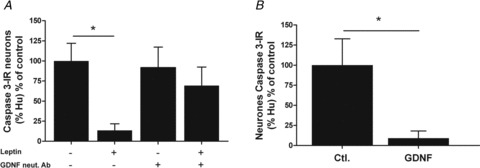
A, percentage of active caspase-3-IR neurons (of total Hu-IR neurons) in primary cultures of ENS treated with leptin and anti-GDNF neutralizing antibody (GDNF neut. Ab) and normalized to control (n= 10–13; control: 0.48 ± 0.23%). Values are expressed as means ± SEM. *P < 0.05, Kruskall–Wallis test. B, percentage of active caspase-3-IR neurons (of total Hu-IR neurons) in primary cultures of ENS treated with GDNF and normalized to control (n= 5–7; control: 0.25 ± 0.08%). Values are expressed as means ± SEM. *P < 0.05, Mann–Whitney test.
Discussion
This study demonstrated that DIO induced neuroplastic changes in the antrum leading to an NO-dependent acceleration of GE. In particular, DIO prevented age-associated loss of gastric myenteric neurons, especially nitrergic ones, observed in ND mice. In addition, during WD but not ND, a time-dependent increase in leptin and GDNF concentrations occurred in the antrum. Finally, leptin increased GDNF production in the ENS and induced neuroprotective effects mediated in part by GDNF.
An important finding of our study was that DIO prevented age-associated neuroplastic changes in the antrum. Indeed, in the antrum of ND mice, the number of neurons per ganglion decreased by 36% during the course of the diet (i.e. between 5 weeks and 17 weeks of age) while they were preserved in obese mice. Age-associated loss of myenteric neurons has already been reported in the antrum of mice during the first year of life (El-Salhy et al. 1999)). Similar to our results, loss of neurons started after 12 weeks of age and reached 30% at 52 weeks as compared to 4-week-old mice (El-Salhy et al. 1999)). However, as mice used in our study are still young (i.e. 5 weeks), the observed effects of DIO could also result from the interference of the diet with the normal maturation process of the ENS occurring during this period. Furthermore, we also identified nitrergic neurons as being specifically affected by age. Although no data exist in the antrum, various studies have reported age-associated loss of nitrergic myenteric neurons in the colon of rats and mice (Takahashi et al. 2000; Liu et al. 2009)). This increase in neuronal density and in particular of NOS-IR neurons could be responsible for the larger Ca2+ concentration induced by fibre tract nerve stimulation in the ENS in WD as compared to ND mice. Indeed, NO has been shown to increase intracellular calcium concentration in myenteric neurons (Sitmo et al. 2007)).
Another finding of this study was that the effects of DIO upon nitrergic neurons had functional impact upon GE and neuromuscular transmission in the antrum. Indeed, after 12 weeks of diet, the number of nNOS-IR neurons was significantly larger in the antrum of WD than in ND mice. Consequently, the amplitude of both NO-dependent neuromuscular transmission and NO-dependent GE was increased in WD as compared to ND mice. In addition, no change in cholinergic neuromuscular transmission was observed in our study, which is consistent with the absence of effects of DIO upon acetylcholine content in the antrum. Our data are in accordance with the key role played by NO in the control of GE. Indeed, in nNOS KO mice, GE was reduced (Mashimo et al. 2000)) and administration of NO biosynthesis inhibitors delayed GE in rats and mice (Plourde et al. 1994; Orihata & Sarna, 1994)). Similarly, the WD-induced increase in GE observed in mice is in agreement with studies in patients demonstrating an acceleration of GE consecutively to high-fat diet (Cunningham et al. 1991; Castiglione et al. 2002)) or during obesity (Cardoso-Júnior et al. 2007)). This increase in GE could also participate in the development of obesity by inhibiting satiety and by enhancing food intake. This further supports the key role of gastric motility in the control of food intake and in the long-term regulation of body weight (Janssen et al. 2011)).
We also demonstrated that the effects of DIO were mediated in part by leptin and GDNF. We first showed that DIO increased leptin content in the antrum as early as after 4 weeks of diet. This time course of leptin changes in the stomach is similar to the one observed in the plasma during high-fat diet (Collin et al. 2006; Sutherland et al. 2008)). We next demonstrated that leptin could induce GDNF synthesis and release by the ENS. We first identified leptin receptors in neuronal and glial structures of the antrum, which is in agreement with a previous study showing the expression of leptin receptors on ileal myenteric neurons (Liu et al. 1999)). Although a role of leptin in the regulation of GDNF production in the central nervous system (CNS) and the ENS has never been described, leptin can increase the secretion of other neurotrophins such as brain derived-neurotrophic factor in the CNS (Komori et al. 2006)). Mechanisms involved in the regulation of GDNF secretion by leptin remain unknown but could involve STAT3 as (1) IL-1β increased GDNF release by rat glioma cells via a STAT3-dependent mechanism (Tanabe et al. 2009)) and (2) leptin increased STAT3 phosphorylation in neurons of the nodose ganglia (de Lartigue et al. 2011)).
We finally demonstrated that leptin exerted neuroprotective effects in part via GDNF, although we cannot exclude a direct effect of leptin. Neuroprotective effects of leptin have never been identified in the central or peripheral nervous system. However, leptin has been shown to exert cytoprotective effects upon neuronal cell lines during oxidative stress (Lu et al. 2006; Weng et al. 2007)). Furthermore, the neuroprotective role of GDNF that we observed is consistent with previous studies showing that GDNF increased survival of postnatal myenteric neurons (Rodrigues et al. 2011)). GDNF also prevented hyperglycaemia-induced loss of myenteric neurons, especially nitrergic ones, in streptozotocine-induced diabetic mice (Anitha et al. 2006)). These results are also consistent with data showing that GDNF+/– mice have fewer myenteric neurons than wild-type animals in the stomach (Shen et al. 2002)). Although in our study different animal models were used to characterize the effects of leptin and GDNF, leptin signalling in the ENS and GDNF neuroprotective effects have been described in various species such as mice, rat and guinea pig (Liu et al. 1999; Anitha et al. 2006; Reichardt et al. 2011)). Our study also extends the protective effects of leptin described in the stomach to the ENS. Indeed, in the stomach, leptin exerts mucosal protective effects and favours repair processes (Brzozowski et al. 1999; Konturek et al. 2001)). Interestingly, GDNF has also been shown to protect intestinal epithelial barrier (Zhang et al. 2010)). Therefore, it is tempting to speculate for a central role of leptin and GDNF in the regulation of neuronal and barrier homeostasis in the stomach.
The overall protective effects of DIO observed in our study might be surprising with regard to the long-term adverse effects of obesity. However, our model probably reflects the earlier phases of obesity. In particular, our results strongly suggest that DIO can exert a beneficial effect upon gastric functions via upregulation of leptin. However, loss of leptin sensitivity, as observed in the latter phases of obesity after 20 weeks of high-fat diet in mice (Lin et al. 2000)), would lead to loss of neuroprotective effects of DIO and favour the development of enteric neuropathies and GI dysfunctions.
In conclusion, our study demonstrated that DIO induced changes in gastric functions characterized by increasing gastric emptying. These functional changes were associated with a prevention of age-associated neuronal cell death likely to involve the production of leptin and GDNF. Nutritional intervention taking advantage of the WD-beneficial factors (such as leptin or GNDF) could be used as a new therapeutic approach in the treatment of gastric enteric neuropathies and associated dysfunctions. In addition, this study identifies the ENS as a new actor involved in obesity-associated gastrointestinal dysfunctions which could favour the development of obesity.
Acknowledgments
We thank Birgit Kuch and Mandy Biraud for their technical support. This research was supported by grants from the Fondation pour la Recherche Médicale, the Agence Nationale de la Recherche (ALIA 2009), Deutsche Forschungsgemeinschaft (DFG Sche 267/8-1) and the Nycomed pharmaceutical company. Charlotte Baudry is a recipient of a research grant from the French Ministère de l’Enseignement Supérieur.
Glossary
Abbreviations
- ChAT
choline actetyltransferase
- DIO
diet-induced obesity
- EFS
electrical field stimulation
- ENS
enteric nervous system
- GDNF
glial cell line-derived neurotrophic factor
- GE
gastric emptying
- GI
gastrointestinal
- ND
normal diet
- nNOS
neuronal nitric oxide synthase
- OGTT
oral glucose tolerance test
- WD
Western diet
Authors contributions
C.B. and R.M. contributed to conception and design of the experiments, collection, analysis and interpretation of data and to drafting of the article and critical revision for important intellectual content. S.B.V., M.S. and M.N. contributed to conception and design of the experiments and revision of the article for important intellectual content. F.R., J.M. and A.B. contributed to conception and design of the experiments, and collection, analysis and interpretation of data. We confirm that all of the authors approved the final version of the manuscript and had no conflicts of interest to disclose.
Supplementary Material
Supplementary Table 1
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Abdo H, Derkinderen P, Gomes P, Chevalier J, Aubert P, Masson D, Galmiche J-P, Vanden Berghe P, Neunlist M, Lardeux B. Enteric glial cells protect neurons from oxidative stress in part via reduced glutathione. FASEB J. 2010;24:1082–1094. doi: 10.1096/fj.09-139519. [DOI] [PubMed] [Google Scholar]
- Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab. 2008;295:E1323–E1332. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- Anitha M, Gondha C, Sutliff R, Parsadanian A, Mwangi S, Sitaraman SV, Srinivasan S. GDNF rescues hyperglycemia-induced diabetic enteric neuropathy through activation of the PI3K/Akt pathway. J Clin Invest. 2006;116:344–356. doi: 10.1172/JCI26295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubé A-C, Cabarrocas J, Bauer J, Philippe D, Aubert P, Doulay F, Liblau R, Galmiche JP, Neunlist M. Changes in enteric neurone phenotype and intestinal functions in a transgenic mouse model of enteric glia disruption. Gut. 2006;55:630–637. doi: 10.1136/gut.2005.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begriche K, Lettéron P, Abbey-Toby A, Vadrot N, Robin M-A, Bado A, Pessayre D, Fromenty B. Partial leptin deficiency favors diet-induced obesity and related metabolic disorders in mice. Am J Physiol Endocrinol Metab. 2008;294:E939–E951. doi: 10.1152/ajpendo.00379.2007. [DOI] [PubMed] [Google Scholar]
- Brzozowski T, Konturek PC, Konturek SJ, Pajdo R, Duda A, Pierzchalski P, Bielański W, Hahn EG. Leptin in gastroprotection induced by cholecystokinin or by a meal. Role of vagal and sensory nerves and nitric oxide. Eur J Pharmacol. 1999;374:263–276. doi: 10.1016/s0014-2999(99)00314-3. [DOI] [PubMed] [Google Scholar]
- Cakir B, Kasimay O, Devseren E, Yeğen BC. Leptin inhibits gastric emptying in rats: role of CCK receptors and vagal afferent fibers. Physiol Res. 2007;56:315–322. doi: 10.33549/physiolres.930865. [DOI] [PubMed] [Google Scholar]
- Cardoso-Júnior A, Coelho LGV, Savassi-Rocha PR, Vignolo MC, Abrantes MM, de Almeida AM, Dias EE, Vieira Júnior G, de Castro MM, Lemos YV. Gastric emptying of solids and semi-solids in morbidly obese and non-obese subjects: an assessment using the 13C-octanoic acid and 13C-acetic acid breath tests. Obes Surg. 2007;17:236–241. doi: 10.1007/s11695-007-9031-4. [DOI] [PubMed] [Google Scholar]
- Castiglione KE, Read NW, French SJ. Adaptation to high-fat diet accelerates emptying of fat but not carbohydrate test meals in humans. Am J Physiol Regul Integr Comp Physiol. 2002;282:R366–R371. doi: 10.1152/ajpregu.00190.2001. [DOI] [PubMed] [Google Scholar]
- Chevalier J, Derkinderen P, Gomes P, Thinard R, Naveilhan P, Vanden Berghe P, Neunlist M. Activity-dependent regulation of tyrosine hydroxylase expression in the enteric nervous system. J Physiol. 2008;586:1963–1975. doi: 10.1113/jphysiol.2007.149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin P, Chapados N, Dufresne E, Corriveau P, Imbeault P, Lavoie J-M. Time course of changes in in vitro lipolysis of intra-abdominal fat depots in relation to high-fat diet-induced hepatic steatosis in rats. Br J Nutr. 2006;96:268–275. doi: 10.1079/bjn20061775. [DOI] [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- Covasa M, Ritter RC. Adaptation to high-fat diet reduces inhibition of gastric emptying by CCK and intestinal oleate. Am J Physiol Regul Integr Comp Physiol. 2000;278:R166–R170. doi: 10.1152/ajpregu.2000.278.1.R166. [DOI] [PubMed] [Google Scholar]
- Cunningham KM, Daly J, Horowitz M, Read NW. Gastrointestinal adaptation to diets of differing fat composition in human volunteers. Gut. 1991;32:483–486. doi: 10.1136/gut.32.5.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Salhy M, Sandström O, Holmlund F. Age-induced changes in the enteric nervous system in the mouse. Mech Ageing Dev. 1999;107:93–103. doi: 10.1016/s0047-6374(98)00142-0. [DOI] [PubMed] [Google Scholar]
- Hoff S, Zeller F, von Weyhern CWH, Wegner M, Schemann M, Michel K, Rühl A. Quantitative assessment of glial cells in the human and guinea pig enteric nervous system with an anti-Sox8/9/10 antibody. J Comp Neurol. 2008;509:356–371. doi: 10.1002/cne.21769. [DOI] [PubMed] [Google Scholar]
- Janssen P, Vanden Berghe P, Verschueren S, Lehmann A, Depoortere I, Tack J. Review article: the role of gastric motility in the control of food intake. Aliment Pharmacol Ther. 2011;33:880–894. doi: 10.1111/j.1365-2036.2011.04609.x. [DOI] [PubMed] [Google Scholar]
- Komori T, Morikawa Y, Nanjo K, Senba E. Induction of brain-derived neurotrophic factor by leptin in the ventromedial hypothalamus. Neuroscience. 2006;139:1107–1115. doi: 10.1016/j.neuroscience.2005.12.066. [DOI] [PubMed] [Google Scholar]
- Konturek PC, Brzozowski T, Sulekova Z, Brzozowska I, Duda A, Meixner H, Hahn EG, Konturek SJ. Role of leptin in ulcer healing. Eur J Pharmacol. 2001;414:87–97. doi: 10.1016/s0014-2999(01)00748-8. [DOI] [PubMed] [Google Scholar]
- Kunze WA, Furness JB. The enteric nervous system and regulation of intestinal motility. Annu Rev Physiol. 1999;61:117–142. doi: 10.1146/annurev.physiol.61.1.117. [DOI] [PubMed] [Google Scholar]
- de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Diet-induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab. 2011;301:E187–E195. doi: 10.1152/ajpendo.00056.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Thomas TC, Storlien LH, Huang XF. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int J Obes Relat Metab Disord. 2000;24:639–646. doi: 10.1038/sj.ijo.0801209. [DOI] [PubMed] [Google Scholar]
- Little TJ, Feinle-Bisset C. Oral and gastrointestinal sensing of dietary fat and appetite regulation in humans: modification by diet and obesity. Front Neurosci. 2010;4:178. doi: 10.3389/fnins.2010.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Seino S, Kirchgessner AL. Identification and characterization of glucoresponsive neurons in the enteric nervous system. J Neurosci. 1999;19:10305–10317. doi: 10.1523/JNEUROSCI.19-23-10305.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M-T, Kuan Y-H, Wang J, Hen R, Gershon MD. 5-HT4 receptor-mediated neuroprotection and neurogenesis in the enteric nervous system of adult mice. J Neurosci. 2009;29:9683–9699. doi: 10.1523/JNEUROSCI.1145-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔC(T) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lu J, Park C-S, Lee S-K, Shin DW, Kang J-H. Leptin inhibits 1-methyl-4-phenylpyridinium-induced cell death in SH-SY5Y cells. Neurosci Lett. 2006;407:240–243. doi: 10.1016/j.neulet.2006.08.053. [DOI] [PubMed] [Google Scholar]
- Martínez V, Barrachina MD, Wang L, Taché Y. Intracerebroventricular leptin inhibits gastric emptying of a solid nutrient meal in rats. Neuroreport. 1999;10:3217–3221. doi: 10.1097/00001756-199910190-00017. [DOI] [PubMed] [Google Scholar]
- Mashimo H, Kjellin A, Goyal RK. Gastric stasis in neuronal nitric oxide synthase-deficient knockout mice. Gastroenterology. 2000;119:766–773. doi: 10.1053/gast.2000.16509. [DOI] [PubMed] [Google Scholar]
- Michel K, Michaelis M, Mazzuoli G, Mueller K, VandenBerghe P, Schemann M. Fast calcium and voltage sensitive dye imaging in enteric neurons reveal calcium peaks associated with single action potential discharge. J Physiol. 2011;589:5941–5947. doi: 10.1113/jphysiol.2011.219550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriez R, Abdo H, Chaumette T, Faure M, Lardeux B, Neunlist M. Neuroplasticity and neuroprotection in enteric neurons: Role of epithelial cells. Biochem Biophys Res Commun. 2009;382:577–582. doi: 10.1016/j.bbrc.2009.03.073. [DOI] [PubMed] [Google Scholar]
- Di Nardo G, Blandizzi C, Volta U, Colucci R, Stanghellini V, Barbara G, Del Tacca M, Tonini M, Corinaldesi R, De Giorgio R. Review article: molecular, pathological and therapeutic features of human enteric neuropathies. Aliment Pharmacol Ther. 2008;28:25–42. doi: 10.1111/j.1365-2036.2008.03707.x. [DOI] [PubMed] [Google Scholar]
- Orihata M, Sarna SK. Inhibition of nitric oxide synthase delays gastric emptying of solid meals. J Pharmacol Exp Ther. 1994;271:660–670. [PubMed] [Google Scholar]
- Phillips R. Aging of the myenteric plexus: neuronal loss is specific to cholinergic neurons. Auton Neurosci. 2003;106:69–83. doi: 10.1016/S1566-0702(03)00072-9. [DOI] [PubMed] [Google Scholar]
- Plourde V, Quintero E, Suto G, Coimbra C, Taché Y. Delayed gastric emptying induced by inhibitors of nitric oxide synthase in rats. Eur J Pharmacol. 1994;256:125–129. doi: 10.1016/0014-2999(94)90236-4. [DOI] [PubMed] [Google Scholar]
- Reichardt F, Krueger D, Schemann M. Leptin excites enteric neurons of guinea-pig submucous and myenteric plexus. Neurogastroenterol Motil. 2011;23:e165–e170. doi: 10.1111/j.1365-2982.2010.01665.x. [DOI] [PubMed] [Google Scholar]
- Roberts RR, Murphy JF, Young HM, Bornstein JC. Development of colonic motility in the neonatal mouse-studies using spatiotemporal maps. Am J Physiol Gastrointest Liver Physiol. 2007;292:G930–G938. doi: 10.1152/ajpgi.00444.2006. [DOI] [PubMed] [Google Scholar]
- Rodrigues DM, Li AY, Nair DG, Blennerhassett MG. Glial cell line-derived neurotrophic factor is a key neurotrophin in the postnatal enteric nervous system. Neurogastroenterol Motil. 2011;23:e44–e56. doi: 10.1111/j.1365-2982.2010.01626.x. [DOI] [PubMed] [Google Scholar]
- Schemann M, Michel K, Ceregrzyn M, Zeller F, Seidl S, Bischoff SC. Human mast cell mediator cocktail excites neurons in human and guinea-pig enteric nervous system. Neurogastroenterol Motil. 2005;17:281–289. doi: 10.1111/j.1365-2982.2004.00591.x. [DOI] [PubMed] [Google Scholar]
- Shen L, Pichel JG, Mayeli T, Sariola H, Lu B, Westphal H. GDNF haploinsufficiency causes Hirschsprung-like intestinal obstruction and early-onset lethality in mice. Am J Hum Genet. 2002;70:435–447. doi: 10.1086/338712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth CW, Smith TK. Action potential-dependent calcium transients in myenteric S neurons of the guinea-pig ileum. Neuroscience. 1999;92:751–762. doi: 10.1016/s0306-4522(99)00012-3. [DOI] [PubMed] [Google Scholar]
- Sitmo M, Rehn M, Diener M. Stimulation of voltage-dependent Ca2+ channels by NO at rat myenteric neurons. Am J Physiol Gastrointest Liver Physiol. 2007;293:G886–G893. doi: 10.1152/ajpgi.00124.2007. [DOI] [PubMed] [Google Scholar]
- Soret R, Chevalier J, De Coppet P, Poupeau G, Derkinderen P, Segain JP, Neunlist M. Short-chain fatty acids regulate the enteric neurons and control gastrointestinal motility in rats. Gastroenterology. 2010;138:1772–1782.e4. doi: 10.1053/j.gastro.2010.01.053. [DOI] [PubMed] [Google Scholar]
- Sutherland LN, Capozzi LC, Turchinsky NJ, Bell RC, Wright DC. Time course of high-fat diet-induced reductions in adipose tissue mitochondrial proteins: potential mechanisms and the relationship to glucose intolerance. Am J Physiol Endocrinol Metab. 2008;295:E1076–E1083. doi: 10.1152/ajpendo.90408.2008. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Qoubaitary A, Owyang C, Wiley JW. Decreased expression of nitric oxide synthase in the colonic myenteric plexus of aged rats. Brain Res. 2000;883:15–21. doi: 10.1016/s0006-8993(00)02867-5. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Nishimura K, Dohi S, Kozawa O. Mechanisms of interleukin-1β-induced GDNF release from rat glioma cells. Brain Res. 2009;1274:11–20. doi: 10.1016/j.brainres.2009.03.063. [DOI] [PubMed] [Google Scholar]
- de Vries P, Soret R, Suply E, Heloury Y, Neunlist M. Postnatal development of myenteric neurochemical phenotype and impact on neuromuscular transmission in the rat colon. Am J Physiol Gastrointest Liver Physiol. 2010;299:G539–G547. doi: 10.1152/ajpgi.00092.2010. [DOI] [PubMed] [Google Scholar]
- Weng Z, Signore AP, Gao Y, Wang S, Zhang F, Hastings T, Yin X-M, Chen J. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J Biol Chem. 2007;282:34 479–34 491. doi: 10.1074/jbc.M705426200. [DOI] [PubMed] [Google Scholar]
- De Winter BY, Bredenoord AJ, De Man JG, Moreels TG, Herman AG, Pelckmans PA. Effect of inhibition of inducible nitric oxide synthase and guanylyl cyclase on endotoxin-induced delay in gastric emptying and intestinal transit in mice. Shock. 2002;18:125–131. doi: 10.1097/00024382-200208000-00006. [DOI] [PubMed] [Google Scholar]
- Zhang DK, He FQ, Li TK, Pang XH, Cui DJ, Xie Q, Huang XL, Gan HT. Glial-derived neurotrophic factor regulates intestinal epithelial barrier function and inflammation and is therapeutic for murine colitis. J Pathol. 2010;222:213–222. doi: 10.1002/path.2749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.